1. Introduction
Inflammation is a complex biological response triggered by harmful stimuli such as injuries, infections, pathogens, and other foreign substances. The organism responds to these stimuli through a coordinated mechanism that involves the elimination of pathogenic factors and the activation of protective systems for self-repair and immune defense [
1].
At the tissue level, inflammation manifests through redness, swelling, heat, pain, and a loss of tissue function—hallmarks resulting from local immune, vascular, and cellular inflammatory responses to injury or infection [
2]. Microcirculatory events occurring during the inflammatory process include increased vascular permeability, the recruitment and accumulation of leukocytes, and the release of inflammatory mediators [
3,
4].
The inflammatory response is a highly coordinated physiological process that involves the activation of various signaling pathways aimed at regulating the levels of inflammatory mediators [
5]. These mediators are crucial for both tissue-resident cells and immune cells. Inflammation serves as a fundamental pathogenic mechanism in numerous chronic diseases, which can significantly impact quality of life [
6,
7].
While the specific characteristics of the inflammatory response may vary based on factors such as the type of stimulus and its anatomical location, there exists a general sequence of events that delineates the inflammatory process. This sequence can be articulated through four pivotal steps:
Cell surface receptors recognize harmful stimuli;
Inflammatory pathways are activated;
Inflammatory markers are released;
Immune cells are recruited to the affected site [
8,
9].
Despite the pivotal role that inflammation plays in the body’s defense mechanism, synthetic anti-inflammatory drugs have come to dominate the treatment landscape for alleviating inflammatory conditions [
10]. These pharmaceuticals are widely prescribed for their ability to suppress or inhibit pro-inflammatory mediators, providing symptomatic relief to patients suffering from inflammatory diseases. However, the use of such medications is frequently accompanied by a range of adverse side effects [
11]. Notably, gastrointestinal complications, including gastric and duodenal ulcerations, can arise from the prolonged use of these drugs. These complications can lead to significant discomfort and even necessitate the discontinuation of therapy, ultimately impacting patient adherence to prescribed regimens [
12]. As a result, there is a growing interest in exploring alternative therapeutic options that exhibit anti-inflammatory properties with reduced side effects [
13].
Given their widespread occurrence in nature and their incorporation into cosmetic and pharmaceutical preparations, as well as their roles in the food industry as flavoring and preservative agents, it becomes essential to thoroughly examine the pharmacological potential of monoterpenes, particularly their anti-inflammatory properties [
14]. Inflammation is a key factor in numerous health conditions, and the ability of certain monoterpenes to modulate inflammatory responses could offer valuable insights into new therapeutic strategies [
15].
In recent years, natural products have garnered significant attention as potential alternatives or adjuncts in the management of inflammation. Among these, terpenes represent a large and structurally diverse group of natural compounds synthesized by a wide variety of plants [
9]. This extensive family comprises approximately 55,000 unique compounds, each possessing varying chemical structures and, consequently, distinct biological functions [
16]. Emerging evidence from the scientific literature points to the ability of terpenoids to modulate the inflammatory process by targeting and inhibiting various molecular pathways associated with inflammation [
17].
Moreover, the anti-inflammatory activity of monoterpenes is supported by a growing body of research that explores their molecular mechanisms and biological effects. Studies have indicated that these compounds can inhibit pro-inflammatory mediators, modulate immune responses, and exert protective effects in various tissues [
18].
In vitro studies have shown that linalool effectively inhibits the production of pro-inflammatory cytokines, specifically TNF-α (tumor necrosis factor-alpha) and IL-1β (interleukin-1 beta), in macrophages. These cytokines play a crucial role in mediating inflammation, and their overproduction is linked to various inflammatory disorders [
19,
20].
Additionally, research involving a murine microglial cell line known as BV2 has further demonstrated linalool’s efficacy in diminishing not only TNF-α and IL-1β but also other inflammatory mediators such as nitric oxide and prostaglandin E2. The modulation of these inflammatory factors suggests that linalool may have broader implications for neuroinflammation and conditions affecting the central nervous system, providing a potential avenue for therapeutic exploration in neurodegenerative diseases [
21].
Moreover, in vivo studies conducted in animal models have provided compelling evidence of linalool’s protective effects in various inflammatory scenarios. Both intraperitoneal and subcutaneous administration of linalool have been shown to mitigate lung injury induced by endotoxins, as well as by the Gram-negative bacterium Pasteurella multocida, in mice. This is particularly noteworthy given the increasing prevalence of respiratory infections and related inflammatory conditions in both humans and animals [
22].
The therapeutic effects of linalool in these contexts are further reflected in the decreased concentrations of inflammatory markers such as TNF-α and IL-6 in lung tissue [
23]. These findings underscore the potential of linalool as a promising anti-inflammatory agent [
24].
Other significant terpenoids are pinenes—α-pinene and β-pinene—which are plant-derived compounds with various biological activities such as antifungal, antiviral, and antimicrobial properties [
25]. However, their bioavailability is limited due to their rapid metabolism and elimination from the body [
26]. While some studies have explored the biological effects of α-pinene and β-pinene, further research is needed to better understand their bioavailability and potential health benefits, including their antimicrobial, anticancer, anti-inflammatory, and antiallergic effects [
27].
This suggests a promising potential for terpenoids to be harnessed as effective anti-inflammatory agents in the quest for safer therapeutic options [
28].
The anti-inflammatory properties of monoterpenes, which demonstrate bioactivity in experimental models related to inflammation, have been thoroughly studied [
29]. The data suggest that this class of compounds has therapeutic potential as a source for developing new anti-inflammatory agents [
30]. Therefore, these substances deserve the attention of researchers and pharmaceutical companies for further clinical studies and potential applications in the treatment of diseases associated with inflammatory processes.
Understanding the mechanisms of inflammation can lead to more effective treatments. In this context, computer-assisted prediction plays a crucial role by enabling researchers to analyze the biological activity of compounds like terpenes [
31]. By utilizing advanced computational techniques, scientists can investigate the complex interactions between these natural products and biological targets, which accelerates drug discovery. This streamlined process not only enhances our understanding of the therapeutic potential of these compounds but also provides a safer and more efficient pathway to develop treatments for chronic diseases associated with inflammation.
Overall, the interplay between inflammation and chronic disease emphasizes the importance of understanding the underlying mechanisms of the inflammatory response and the potential of natural products such as terpenes in establishing effective, safer treatment strategies. In this context, computer-aided drug design (CADD) has emerged as an important strategy for accelerating the identification of bioactive natural compounds, as demonstrated by Schneider et al., who highlight its utility in exploring both structure- and ligand-based approaches to drug discovery [
32].
The computer-assisted prediction of the biological activity of specific chemical compounds, considering their intricate chemical structures, has become a standard and invaluable technique in the field of drug discovery. This approach not only enhances the understanding of how various compounds interact with biological targets but also streamlines the overall process, significantly reducing the time and costs associated with traditional experimental methods [
33,
34].
In silico drug discovery encompasses a range of advanced computer techniques, enabling researchers to identify, optimize, and design potential drug candidates effectively. By utilizing tools such as molecular modeling and CADD, scientists can simulate and predict how new compounds might behave in biological systems [
35]. This computational framework is revolutionizing the drug development landscape, allowing for a more targeted approach to finding therapeutic agents that can address complex diseases [
36].
The advantages of in silico methods are manifold. They facilitate high-throughput virtual screening, enabling researchers to evaluate thousands of compounds quickly against specific biological targets. Moreover, they assist in the rational design of molecules with enhanced efficacy and reduced toxicity, significantly improving the success rate of drug development [
37]. With these tools at their disposal, researchers can also predict potential side effects early in the drug development process, addressing safety concerns before clinical trials begin [
38]. This proactive approach not only enhances the efficiency of drug discovery but also raises ethical standards in the field by reducing the reliance on animal testing and experimental approaches that may be harmful to living subjects [
39].
Among the various in silico techniques, molecular docking plays a critical role in understanding molecular interactions. This computational method is employed to estimate the strength of the interactions between proteins and ligands, ultimately determining the optimal binding poses and free energy values associated with these interactions [
40,
41]. By simulating how a ligand binds to a receptor, molecular docking can elucidate the nature of non-covalent interactions, such as hydrogen bonds, hydrophobic interactions, and ionic bonds, which are crucial for ligand recognition and specificity [
42]. This insight is particularly valuable, as it allows scientists to explore the intricate mechanisms of action and binding affinities of new drug candidates, paving the way for innovative therapeutic developments [
43,
44].
As the field advances, the integration of machine learning and artificial intelligence into in silico drug discovery is expected to enhance predictive accuracy and model complexity further. These emerging technologies promise to aid in refining docking algorithms and pharmacokinetic predictions, ultimately leading to the discovery of more effective and safer drug candidates [
45,
46].
Given the limitations of conventional anti-inflammatory drugs, such as gastrointestinal toxicity and cardiovascular risks, there is a pressing need to explore naturally occurring compounds with safer profiles. Monoterpenes, widely found in essential oils, have shown promising anti-inflammatory activity in preliminary studies, yet their interactions with enzymes in the arachidonic acid pathway remain underexplored. This study employs a blind molecular docking approach to evaluate the inhibitory potential of six monoterpenes (α-pinene, β-pinene, menthol, camphor, limonene, and linalool) against COX-1, COX-2, 5-LOX, and PLA2, aiming to assess their suitability as lead compounds for novel anti-inflammatory agents. By comprehensively analyzing the binding affinities and interactions with catalytic residues across all four enzymes, this work contributes to the field by providing the first in silico evidence of monoterpenes’ multi-target inhibitory potential, offering a foundation for the rational design of safer, naturally derived therapeutics.
2. Materials and Methods
Ligand Dataset Preparation
Six terpenes—α-pinene, β-pinene, menthol, camphor, limonene, and linalool—are selected based on their reported anti-inflammatory potential [
47]. Arachidonic acid (AA) is included as a reference ligand for binding energy comparison. The 3D structures of all ligands are retrieved from the PubChem database in the SDF format.
Preparation of Compounds for Molecular Docking
Each ligand is imported into Avogadro [
48], where its geometry is optimized using the “Optimize Geometry” function. The structures are then saved in the MOL2 format. Subsequently, the MOL2 files are loaded into AutoDock Tools [
49], where polar hydrogens are added, Gasteiger charges are assigned, torsions are detected, and the ligands are saved in the PDBQT format for molecular docking.
Target Protein Preparation
The crystallographic structures of COX-1 (PDB ID: 6Y3C) [
50], COX-2 (PDB ID: 3HS5) [
51], 5-LOX (PDB ID: 3O8Y) [
52], and PLA
2 (PDB ID: 1KVO) [
53] are obtained from the RCSB Protein Data Bank [
54]. Initial preprocessing is performed in UCSF ChimeraX, where all non-standard residues are removed. The structures are then imported into AutoDock 4.2 [
55], where water molecules are deleted. Missing atoms are checked and repaired when necessary. Hydrogen atoms are added, with polar hydrogens retained. Kollman charges are assigned, atom types are set to AD4 specifications, and the proteins are saved in the PDBQT format for docking.
Molecular Docking Setup
Docking simulations are carried out using AutoDock 4.2 [
49] with the Lamarckian Genetic Algorithm (LGA) [
56]. A blind docking approach is applied by defining a grid box that includes all potential binding regions of the macromolecule. Docking parameters include 100 runs per ligand–enzyme pair, a maximum of 25,000,000 energy evaluations, a population size of 150, a mutation rate of 0.02, and a crossover rate of 0.8. The best binding pose is selected based on the lowest binding energy (ΔG). Docked conformations are clustered using a root-mean-square deviation (RMSD) tolerance of 2.0 Å to group similar poses, although higher RMSD values are expected in blind rigid-body docking due to the fixed protein conformation and the broad surface search space [
57].
Virtual Screening Protocol and Analysis
Ligand–enzyme interactions are analyzed using UCSF ChimeraX (Resource for Biocomputing, Visualization, and Informatics, University of California, San Francisco, CA, USA), focusing on the spatial localization of the ligand relative to catalytically important regions as reported in the literature [
58]. Specific attention is given to hydrogen bonds, hydrophobic contacts, and interactions with residues implicated in enzymatic function. Three-dimensional interactions are visualized in ChimeraX, while two-dimensional interaction diagrams are generated using BIOVIA Discovery Studio Visualizer v21.1.0.20298 (Dassault Systèmes, San Diego, CA, USA) to support residue-level interpretation [
59].
3. Results
3.1. COX-1 Interactions
The interaction parameters of the selected terpenes with COX-1, compared to arachidonic acid (AA), are summarized in
Table 1.
One of the fundamental parameters for evaluating ligand–enzyme interactions is the binding energy, expressed in kcal/mol. Lower binding energy values correlate with the formation of more stable ligand–enzyme complexes, which, in turn, indicate higher binding affinity.
In the present study, AA exhibits the lowest binding energy (−7.53 kcal/mol), reflecting its strong interaction with COX-1. Among the analyzed terpenes, menthol has the lowest binding energy (−6.56 kcal/mol), followed by camphor (−6.32 kcal/mol) and β-pinene (−6.31 kcal/mol), suggesting that these compounds possess relatively high affinity for COX-1.
The inhibition constant (Ki) represents another indicator of the potential inhibitory activity of the studied molecules, with lower Ki values corresponding to greater inhibitory potential. The lowest Ki value is observed for AA (3.02 µM), consistent with its role as the natural substrate of COX-1. Among the terpenes, menthol (15.50 µM) and camphor (23.37 µM) exhibit the lowest Ki values, suggesting that these compounds may exert inhibitory activity toward the enzyme.
The root-mean-square deviation (RMSD) values, relative to the reference structure, provide insights into the extent of conformational changes undergone by the ligands upon binding. High RMSD values (>50 Å) are recorded for all analyzed molecules, indicating significant spatial reorientation. The highest RMSD values are observed for camphor (67.64 Å) and β-pinene (67.775 Å), suggesting that these molecules undergo substantial conformational changes upon binding to COX-1.
The total internal energy and torsional free energy provide additional insight into the molecular stability of the ligands. The highest torsional energy values are recorded for linalool (+1.49 kcal/mol) and AA (+4.47 kcal/mol), suggesting increased flexibility and a greater tendency for conformational changes. In contrast, menthol exhibits the lowest torsional energy (+0.60 kcal/mol), indicating a relatively stable conformation upon binding to COX-1.
Unbound energy is a parameter reflecting the energetic state of the molecule in its free form. The lowest unbound energy is observed for AA (−1.61 kcal/mol), consistent with its high binding affinity.
In summary, AA exhibits the highest affinity for COX-1, consistent with its role as the enzyme’s natural substrate. Among the investigated terpenes, menthol and camphor demonstrate the most favorable parameters, identifying them as potential COX-1 inhibitors. In contrast, linalool and α-pinene show relatively weaker interactions, likely due to their higher torsional energy and conformational flexibility.
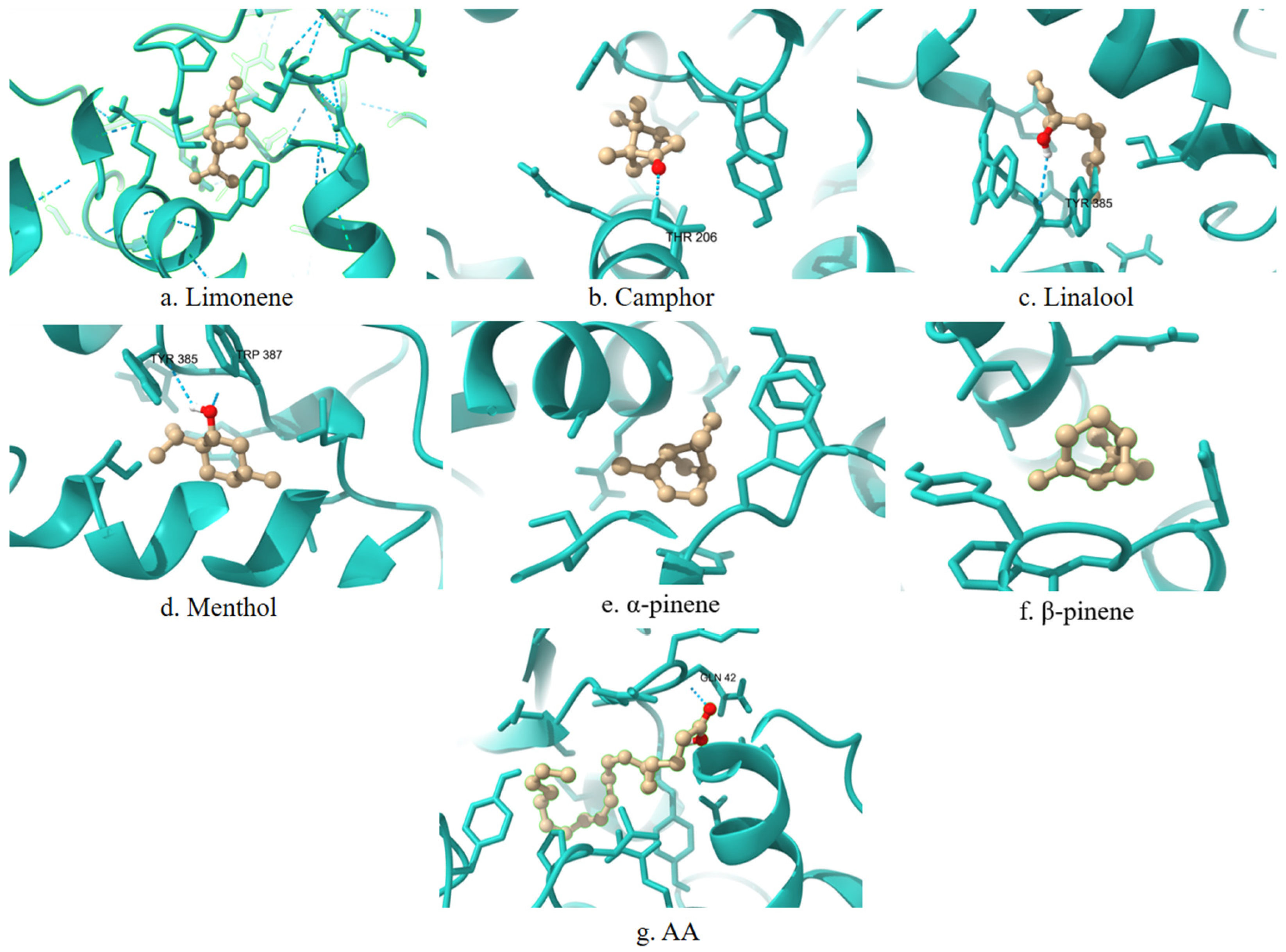
A 3D visualization of the interactions between the terpenes, AA, and the COX-1 enzyme is presented in
Figure 1. Ligands are shown in the ball-and-stick representation (light brown), and the enzyme is displayed as a cartoon structure (cyan). Hydrogen bonds and relevant interactions are indicated with dashed lines where applicable. The positioning of each ligand relative to key catalytic residues illustrates potential binding modes and differences in spatial orientation.
3.2. COX-2 Interactions
The interaction parameters of the selected terpenes with COX-2, compared to AA, are summarized in
Table 2.
AA exhibits the lowest binding energy (−7.07 kcal/mol), indicating strong interaction with COX-2. Among the analyzed terpenes, camphor has the lowest binding energy (−6.48 kcal/mol), followed by menthol (−6.42 kcal/mol).
The lowest Ki value is also observed for AA (6.55 µM), which is expected given its natural role in the COX-2 enzymatic reactions. Among the terpenes, camphor (17.84 µM) and menthol (19.80 µM) exhibit the lowest Ki values, identifying them as potential COX-2 inhibitors.
The observed high RMSD values (>50 Å) for all analyzed molecules indicate significant spatial reorientation upon binding. The highest RMSD values are recorded for β-pinene (67.77 Å) and menthol (67.75 Å), suggesting substantial conformational changes during interaction with the enzyme.
Among all studied terpenes, the highest torsional energy is observed for linalool (+1.49 kcal/mol) and AA (+4.47 kcal/mol), indicating greater molecular flexibility. In contrast, α-pinene and β-pinene exhibit zero torsional energy (+0.00 kcal/mol), suggesting conformational stability upon binding.
The lowest unbound energy is recorded for AA (−1.18 kcal/mol), consistent with its high binding affinity.
The results indicate that, among all compounds studied, AA exhibits the highest affinity for COX-2, consistent with its role as the enzyme’s natural substrate. Among the terpenes, camphor and menthol demonstrate the most favorable parameters, identifying them as potential COX-2 inhibitors. In contrast, linalool and α-pinene show comparatively weaker interactions, likely due to their higher torsional energy and conformational flexibility.
For comparison, previously published docking studies report a mean binding energy of −10.78 kcal/mol for selective COX-2 inhibitors and −6.69 kcal/mol for nonselective NSAIDs, as determined using AutoDock 4 [
60]. In this study, camphor (−6.48 kcal/mol) and menthol (−6.42 kcal/mol) show similar binding energies to the nonselective group, but remain significantly weaker than those observed for selective inhibitors.
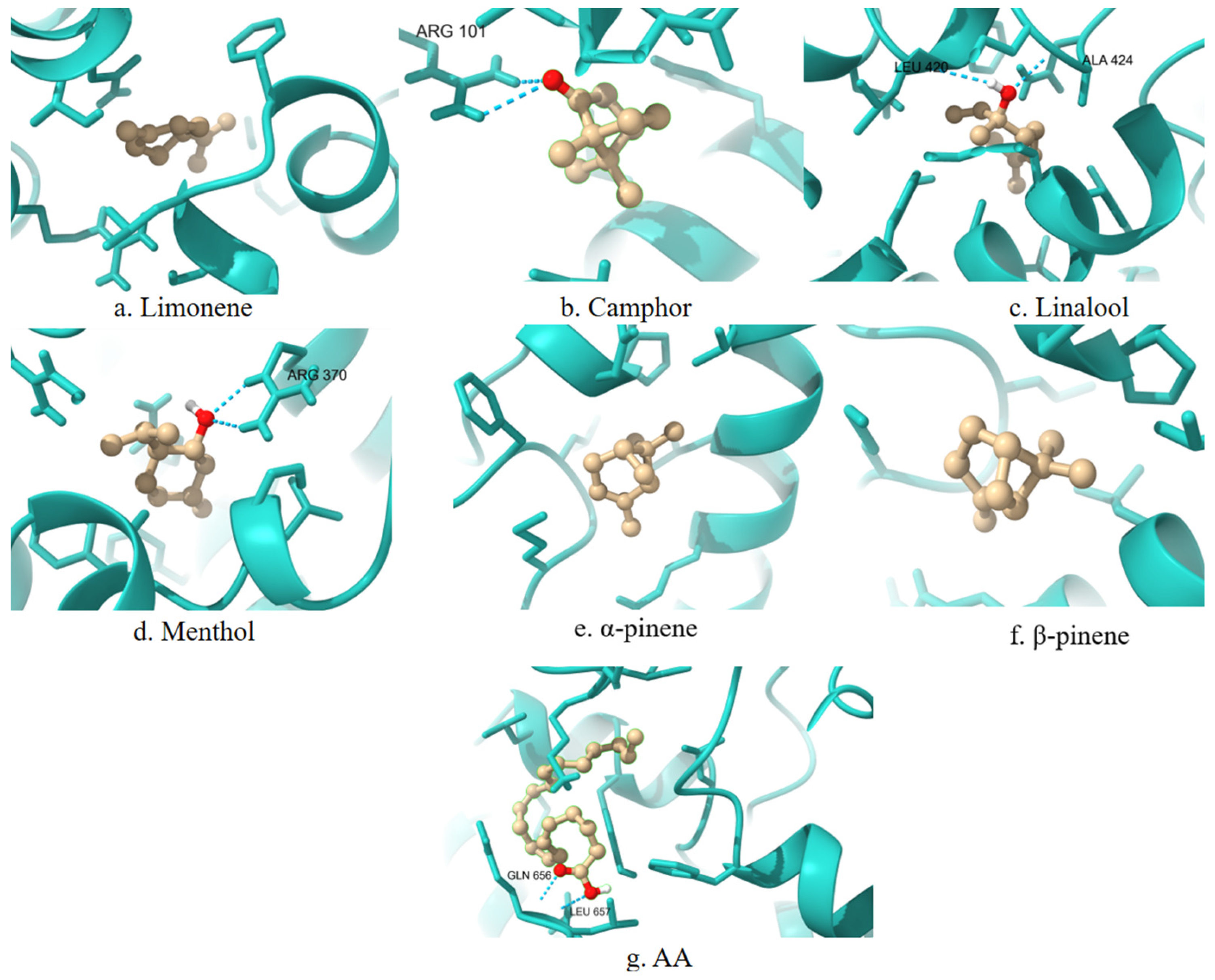
A 3D visualization of the interactions between the terpenes, AA, and the COX-2 enzyme is presented in
Figure 2. Ligands are shown in the ball-and-stick representation (light brown), and the enzyme is displayed as a cartoon structure (cyan). Hydrogen bonds and relevant interactions are indicated with dashed lines where applicable. The positioning of each ligand relative to key catalytic residues illustrates potential binding modes and differences in spatial orientation.
3.3. 5-LOX Interactions
The interaction parameters of the selected terpenes with 5-LOX, compared to AA, are summarized in
Table 3.
Limonene (−5.57 kcal/mol), menthol (−5.45 kcal/mol), and AA (−5.45 kcal/mol) exhibit the lowest binding energies, indicating stable interactions with 5-LOX. The remaining terpenes also demonstrate relatively favorable binding energies, with camphor displaying the highest value (−5.17 kcal/mol), which may suggest weaker interaction with the enzyme.
Limonene (82.28 µM), AA (101.13 µM), and menthol (102.00 µM) exhibit the lowest Ki values among the tested compounds, identifying them as the most promising potential 5-LOX inhibitors. In contrast, linalool shows a substantially higher Ki value (564.63 µM), indicating a significantly lower affinity for 5-LOX.
High RMSD values (>50 Å) are observed for camphor (77.022 Å) and limonene (64.854 Å), suggesting notable structural changes upon binding. Conversely, α-pinene (19.328 Å) and β-pinene (18.577 Å) demonstrate the lowest RMSD values, indicating minimal conformational changes during interaction with the enzyme.
Linalool (+1.49 kcal/mol) and AA (+4.47 kcal/mol) exhibit the highest torsional energies, suggesting greater conformational flexibility. In contrast, menthol (+0.60 kcal/mol) and α-pinene (+0.00 kcal/mol) show the lowest torsional energy values, indicating stable conformations upon binding to 5-LOX.
The lowest unbound energy is recorded for AA (−1.08 kcal/mol), supporting its high affinity for the enzyme.
The analysis reveals that limonene, menthol, and AA display the lowest binding energies, suggesting stable interactions with 5-LOX. Menthol and AA also demonstrate the lowest Ki values. In contrast, linalool exhibits the weakest affinity, suggesting lower inhibitory potential.
A 3D visualization of the interactions between the terpenes, AA, and the 5-LOX enzyme is presented in
Figure 3. Ligands are shown in the ball-and-stick representation (light brown), and the enzyme is displayed as a cartoon structure (cyan). Hydrogen bonds and relevant interactions are indicated with dashed lines where applicable. The positioning of each ligand relative to key catalytic residues illustrates potential binding modes and differences in spatial orientation.
3.4. PLA2 Interactions
The interaction parameters of the selected terpenes with PLA2, compared to AA, are summarized in
Table 4.
Limonene (−6.58 kcal/mol) exhibits the lowest binding energy, followed by menthol (−6.57 kcal/mol) and AA (−6.23 kcal/mol), indicating stable interactions with PLA2. The remaining terpenes also show good affinity, with β-pinene presenting the highest binding energy (−5.28 kcal/mol), which may indicate weaker interaction with the enzyme.
Among the terpenes, limonene (14.99 µM) and menthol (15.22 µM) exhibit the lowest Ki values, identifying them as the most promising potential inhibitors of PLA2. In contrast, β-pinene shows the highest Ki value (134.07 µM), indicating a weaker affinity for the enzyme.
High RMSD values are recorded for camphor (96.944 Å), linalool (96.905 Å), and menthol (98.205 Å), suggesting significant structural changes upon binding to PLA2. Notably, α-pinene (110.729 Å) and β-pinene (100.997 Å) exhibit the highest RMSD values, indicating pronounced flexibility during interaction.
The highest torsional energies are observed for α-pinene (+1.49 kcal/mol) and AA (+4.47 kcal/mol), suggesting greater conformational flexibility. In contrast, menthol (+0.60 kcal/mol) and limonene (+0.26 kcal/mol) display lower torsional energies, which may indicate more stable conformations when bound to PLA2.
AA exhibits the lowest unbound energy (−1.91 kcal/mol), consistent with its high affinity for PLA2. It is followed by α-pinene (−0.26 kcal/mol) and menthol (−0.17 kcal/mol).
The analysis reveals that limonene and menthol have the lowest binding energies, suggesting stable interactions with PLA2. These two compounds also demonstrate the lowest Ki values, identifying them as potential inhibitors of the enzyme. In contrast, β-pinene shows the weakest affinity, likely due to its high Ki value.
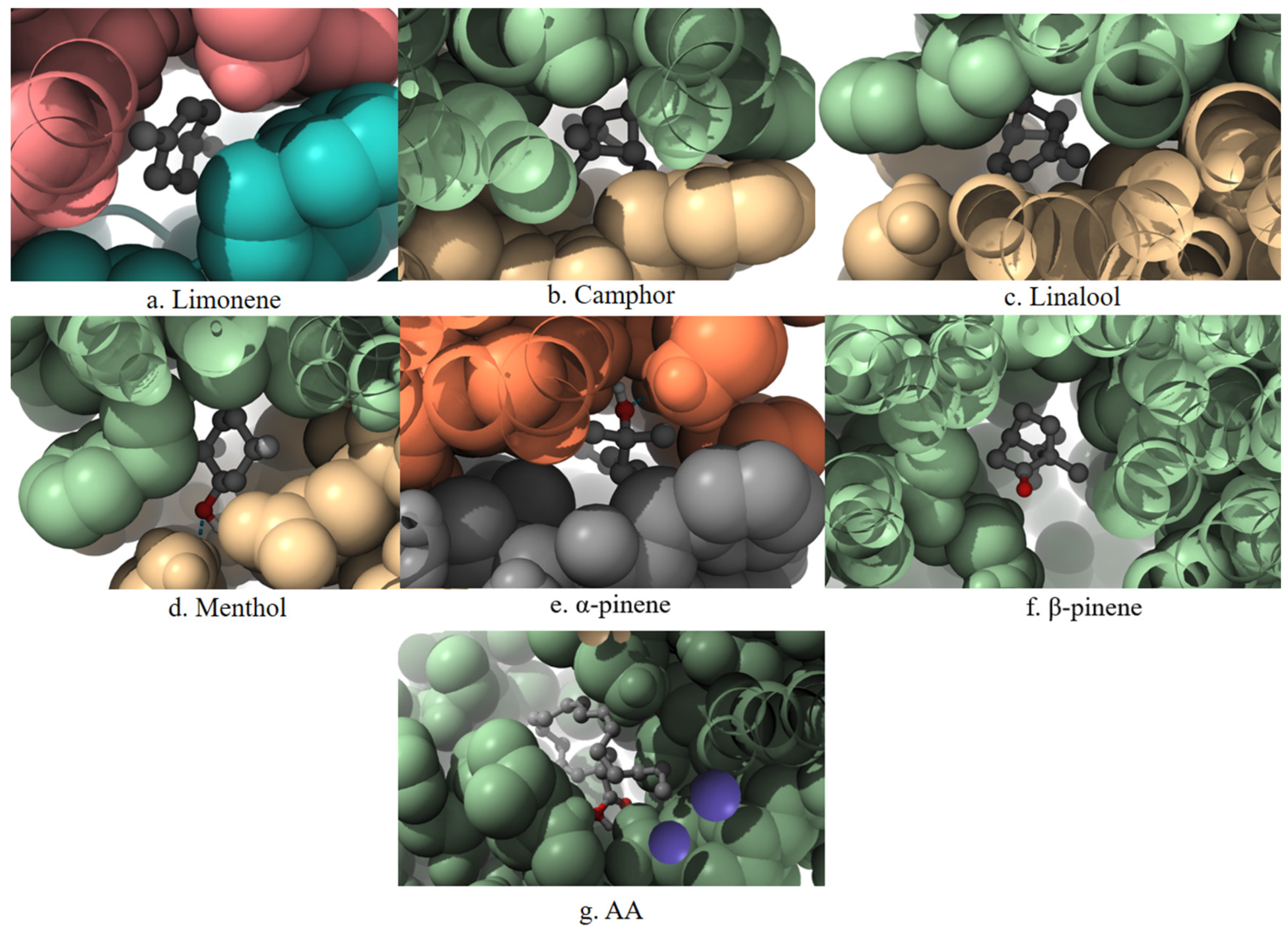
A 3D visualization of the interactions between the terpenes, AA, and the PLA2 enzyme is presented in
Figure 4. Ligands are shown in the ball-and-stick format, surrounded by a space-filling surface representation of the protein to illustrate spatial accommodation and surface contact. The visualization highlights the degree of penetration and positioning of each ligand within the catalytic pocket of PLA
2.
3.5. Blind Docking Results
Table 5 presents the amino acid sequences of the active sites of the enzymes investigated in this study.
Table 6 presents the results of the blind docking analysis of the interactions between the selected terpenes and the enzymes involved in arachidonic acid metabolism. For comparative evaluation, this table lists the amino acid residues involved in ligand–enzyme interactions as predicted by molecular docking, along with a comparison to known catalytic residues (“Match”). “Match” refers to overlap between contact residues and literature-defined catalytic residues for each enzyme, suggesting potential for functional inhibition.
For each ligand–enzyme complex, the determined parameters are as follows:
Active site sequence: a list of amino acid residues forming the catalytic site of the corresponding enzyme.
Binding residues: specific residues through which the terpene establishes contact with the enzyme during docking.
Matches: residues that appear most frequently across simulations, indicating potentially stable interactions and playing a key role in stabilizing the complex.
The data indicate that the terpenes interact selectively with the enzymes involved in arachidonic acid metabolism, establishing specific contacts within their active sites.
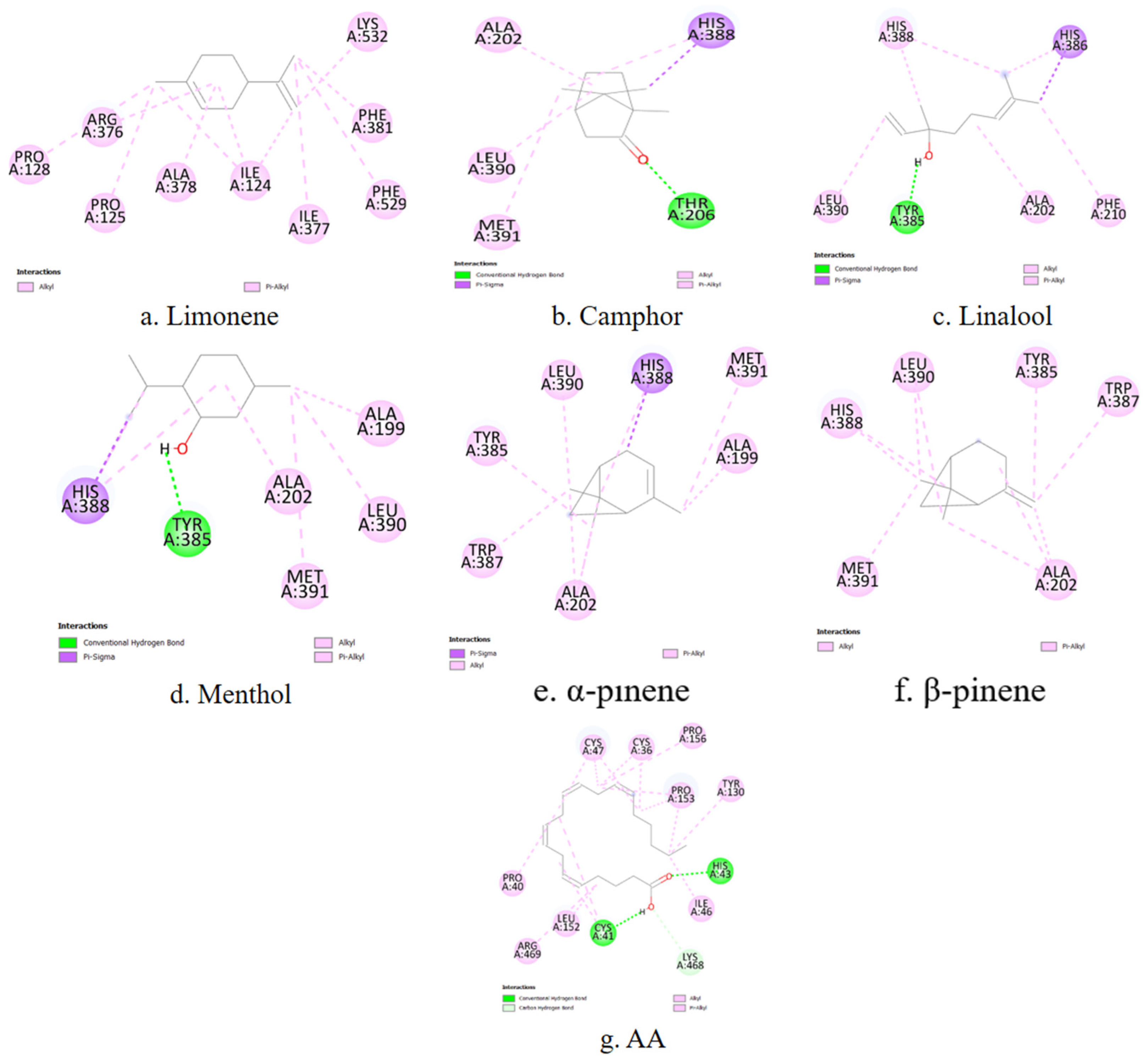
COX-1 is a constitutively expressed enzyme involved in the synthesis of prostaglandins essential for maintaining gastrointestinal and renal homeostasis. The analysis reveals that key contact residues within the active site include LEU117, ARG120, PHE205, PHE209, TYR348, PHE381, TYR385, and HIS388. Limonene forms a stable contact with PHE381, a residue within the active site. Camphor, linalool, menthol, α-pinene, and β-pinene exhibit strong interactions with HIS388 and TYR385, which may contribute to their affinity for the enzyme. Additionally, α-pinene and β-pinene interact with TRP387, which could play a stabilizing role in ligand binding.
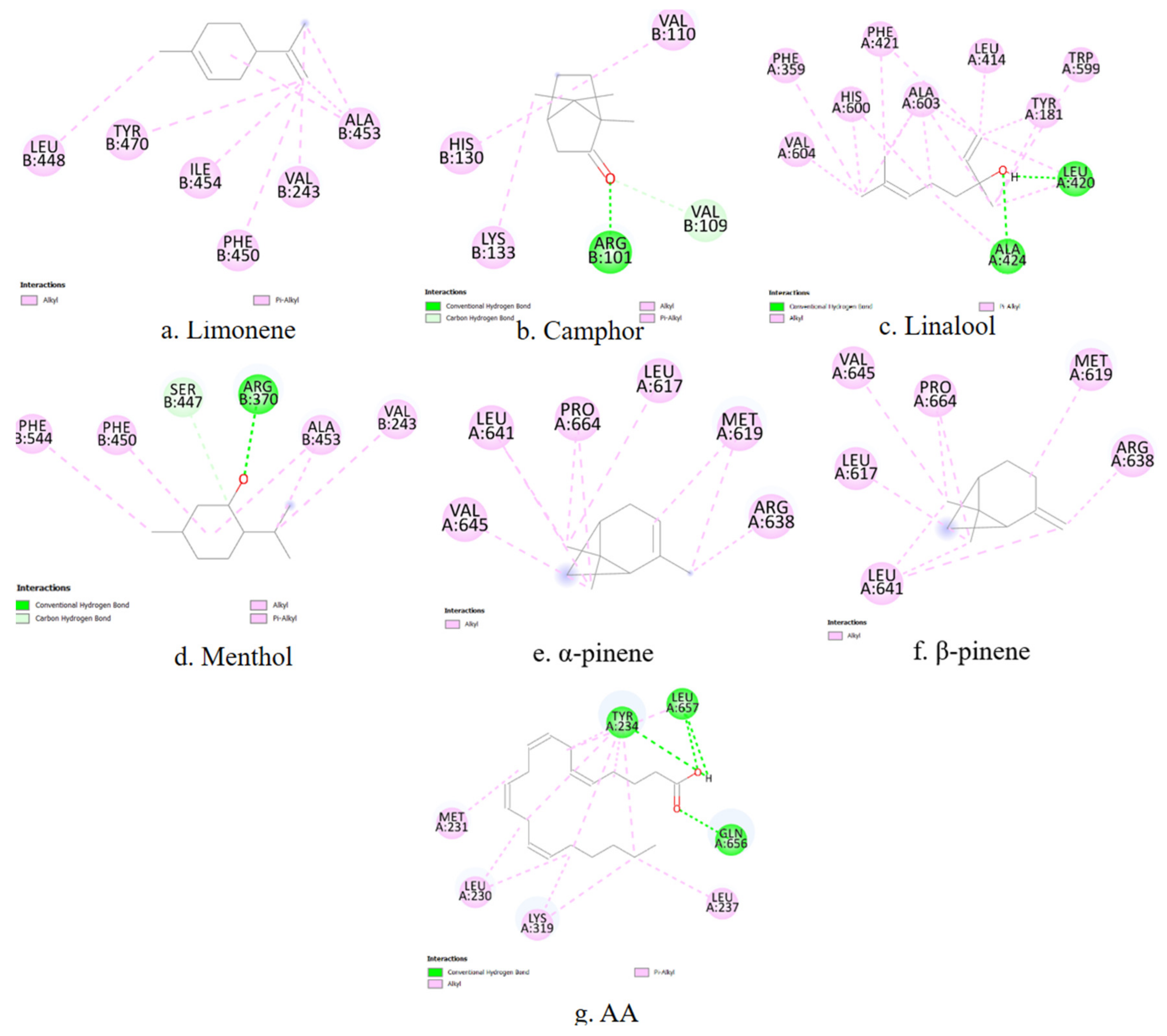
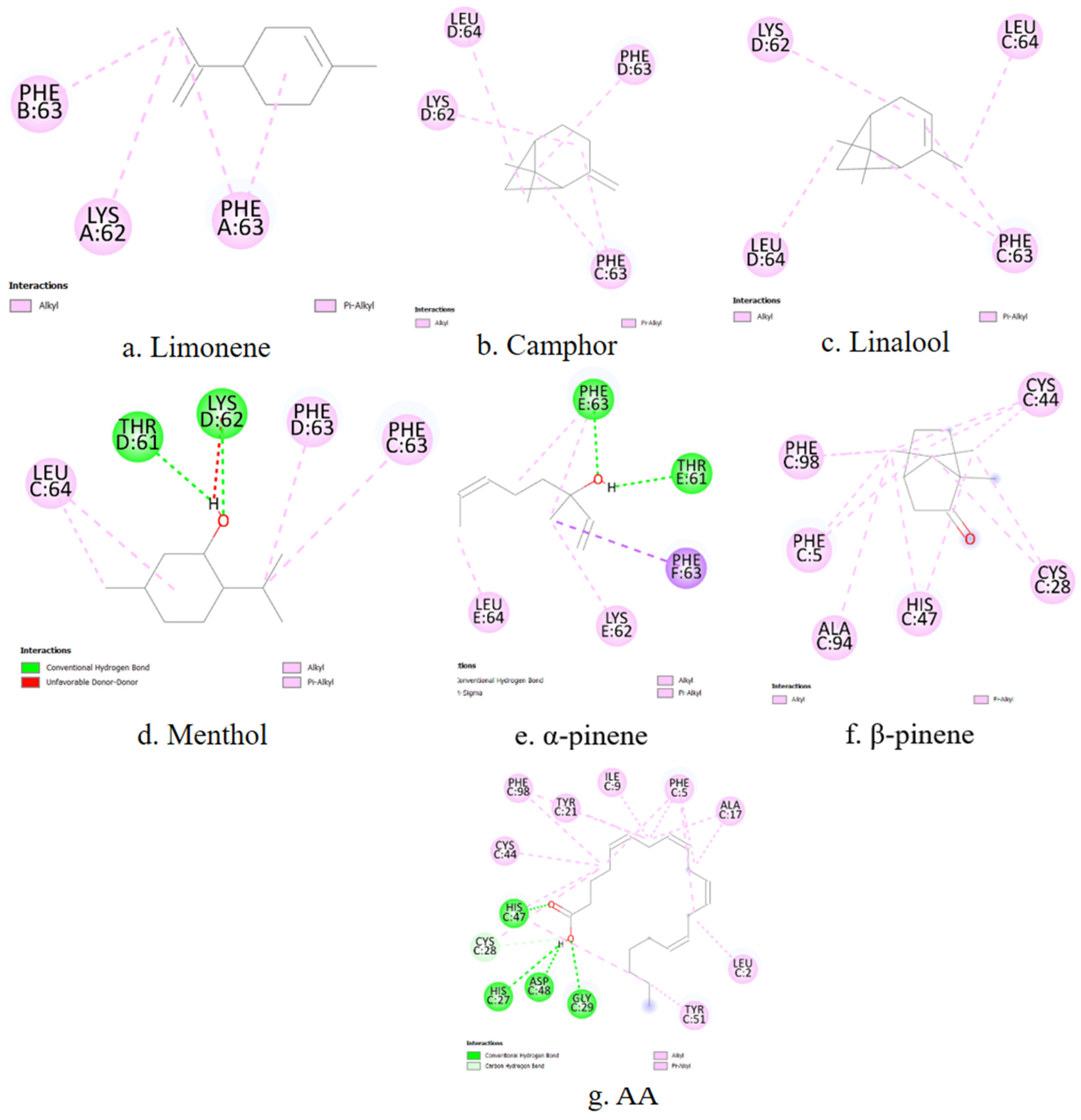
A representative 2D interaction map (
Figure 5) is generated to visualize the binding interactions between each terpene and the COX-1 active site. These diagrams illustrate the spatial arrangement and nature of non-covalent contacts, including conventional hydrogen bonds (green), π–σ interactions (purple), and hydrophobic contacts such as alkyl or π–alkyl interactions (pink). Notably, menthol and camphor form hydrogen bonds with TYR385 and HIS388, residues known to be essential for COX-1 catalytic activity. In contrast, α-pinene and β-pinene primarily establish hydrophobic interactions within the binding cavity. These 2D diagrams complement the numerical docking data and offer a structural rationale for the observed differences in binding energy and inhibition constants among the studied terpenes.
COX-2 is inducible under inflammatory conditions and is a preferred target for selective inhibitors. The primary amino acid residues involved in terpene interactions include TYR385, HIS388, TRP387, PHE381, and VAL349. Limonene again shows affinity for PHE381. In the case of camphor, no overlap can be observed between the ligand–enzyme interaction residues and the active site residues of COX-2. Linalool, menthol, α-pinene, and β-pinene demonstrate stable binding with HIS388 and TYR385, consistent with the patterns observed for COX-1. α-Pinene and β-pinene also exhibit specific affinity for TRP387, which may influence their selectivity toward COX-2.
To complement the numerical docking data, 2D interaction diagrams are generated to depict the binding modes of the investigated terpenes within the COX-2 active site (
Figure 6). These visualizations provide structural support for the observed affinity trends and help rationalize the inhibitory potential of each terpene based on its interaction fingerprint within the COX-2 enzyme.
5-LOX catalyzes the first step in leukotriene biosynthesis, which plays a critical role in chronic inflammatory diseases. The key amino acid residues involved in ligand binding include HIS367, HIS372, HIS550, TYR181, and TRP599. Among the tested compounds, only linalool shows stable contacts with LEU414, TRP599, ALA603 and TYR181, which overlap with its interaction profile with COX-2. For the remaining terpenes, no overlap is observed between the ligand–enzyme interaction residues and the amino acid sequence of the 5-LOX active site.
In addition to binding affinity data, the predicted interactions of the terpenes with 5-LOX are visualized using 2D ligand–enzyme diagrams (
Figure 7).
PLA2 is responsible for releasing AA from membrane phospholipids, representing the initial step in the inflammatory cascade. The main contact residues in the enzyme’s active site include HIS47, CYS44, TYR51, and PHE5. Among the terpenes, only β-pinene exhibits matching interactions, forming stable contacts with PHE5, CYS44, and HIS47, which are part of the active site of the phospholipase enzyme.
Figure 8 presents two-dimensional interaction schematics illustrating how the selected terpenes and the reference ligand AA engage with the PLA
2 active site.
4. Discussion
The present study aims to computationally evaluate the anti-inflammatory potential of six naturally occurring terpenes by investigating their binding interactions with four enzymes involved in AA metabolism. Arachidonic acid, as the natural substrate for these enzymes and a central precursor to pro-inflammatory mediators, serves as a reference ligand.
The selection of the six terpenes is guided by both pharmacological relevance and chemical diversity. These compounds are among the most widely distributed monoterpenes in nature, occurring in a broad range of essential oils derived from medicinal and aromatic plants [
61]. They have been historically used in traditional medicine for the treatment of inflammatory conditions, and several preclinical studies have reported anti-inflammatory, analgesic, or antioxidant properties for these molecules [
62,
63,
64,
65]. From a structural perspective, the selected terpenes represent a diverse subset of the monoterpene class, encompassing acyclic (linalool), monocyclic (limonene), and bicyclic (α- and β-pinene, camphor) skeletons, as well as different functional groups, including alcohols (menthol, linalool) and ketones (camphor). The focused nature of this compound set allows for a detailed comparative analysis of binding behavior while maintaining a manageable scope for computational evaluation. While broader screening libraries could provide a wider range of chemical scaffolds, our aim here is to explore the binding potential of well-characterized, biologically active terpenes with established use in natural medicine, as a proof-of-concept framework for further expansion in future work.
4.1. Comparative Analysis of Binding Affinities and Inhibition Constants
For both COX-1 and COX-2, AA consistently exhibits the lowest binding energies (−7.53 kcal/mol for COX-1 and −7.07 kcal/mol for COX-2) and inhibition constants (3.02 µM for COX-1 and 6.55 µM for COX-2), as summarized in
Table 2 and
Table 3. These results confirm AA’s strong and optimal positioning within the catalytic sites of both enzymes, aligning with its physiological role as the natural substrate.
Among the investigated terpenes, menthol and camphor consistently demonstrate the most favorable binding parameters for COX-1 and COX-2. For COX-1, menthol shows the lowest binding energy (−6.56 kcal/mol) and Ki (15.50 µM) among the terpenes, followed closely by camphor (−6.32 kcal/mol and 23.37 µM, respectively), according to
Table 1. Similarly, for COX−2, camphor exhibits the lowest binding energy (−6.48 kcal/mol) and Ki (17.84 µM) among the terpenes, followed by menthol (−6.42 kcal/mol and 19.80 µM), as presented in
Table 2. While these terpenes did not achieve the full interaction profile characteristic of AA, their relatively low binding energies and Ki values suggest a promising inhibitory potential against both cyclooxygenases.
A distinct binding pattern emerges for 5-LOX and PLA
2. In the case of 5-LOX, limonene (−5.57 kcal/mol) and menthol (−5.45 kcal/mol) exhibit binding energies comparable to that of AA (−5.45 kcal/mol). Furthermore, limonene (82.28 µM) shows an even lower Ki value than AA (101.13 µM) for 5-LOX, with menthol (102.00 µM) having a very similar Ki to AA, as reported in
Table 3. This suggests that limonene and menthol can interact effectively with 5-LOX, potentially exerting a competitive inhibitory effect.
Similarly, for PLA
2, limonene (−6.58 kcal/mol) and menthol (−6.57 kcal/mol) display even lower binding energies and significantly lower Ki values (14.99 µM and 15.22 µM, respectively) compared to AA (−6.23 kcal/mol and 27.27 µM), as shown in
Table 4. These findings strongly support their candidacy as potential inhibitors of 5-LOX and PLA
2, enzymes critical in the initial steps of the inflammatory cascade.
In contrast, linalool consistently shows weaker interactions across all enzymes, particularly with 5-LOX (Ki value of 564.63 µM; see
Table 3), indicating lower inhibitory potential.
4.2. Analysis of Conformational Parameters and Interaction Profiles
The high RMSD values (>50 Å) observed for all analyzed molecules across all enzymes indicate substantial spatial variation in the predicted binding poses. This is a typical feature of blind rigid-body docking simulations, where the entire protein surface is sampled rather than being restricted to a predefined active site, and the ligand is treated as a fully rigid structure without accounting for conformational flexibility. In this study, a 2 Å RMSD threshold was used solely for clustering geometrically similar poses. However, the high absolute RMSD values reflect the wide conformational space explored by the ligands. In blind rigid-body docking using AutoDock, RMSD is calculated relative to the reference pose—usually the lowest-energy conformation—and not relative to a known crystallographic binding site. Thus, elevated RMSD values in this context do not imply poor accuracy or instability, but instead indicate that ligands sampled diverse and spatially distinct regions of the protein surface. This behavior is expected when the docking grid encompasses the entire protein structure.
The torsional free energy provides additional insights into the conformational flexibility of the ligands upon binding. Linalool and AA consistently exhibit higher torsional energies (e.g., +1.49 kcal/mol for linalool and +4.47 kcal/mol for AA across all enzymes), suggesting a greater degree of conformational flexibility and the potential to adopt multiple orientations within the active site. Conversely, terpenes like menthol (e.g., +0.60 kcal/mol for COX-1) and limonene (e.g., +0.30 kcal/mol for COX-1) generally display lower torsional energies, pointing to more rigid and stable binding poses. Such stability can contribute to more effective enzymatic inhibition.
The most critical aspect of this study lies in the detailed analysis of specific amino acid residue interactions and their overlap with known catalytic sites, as shown in
Table 5 and
Table 6. This “match” provides strong evidence for the potential mechanism of inhibition.
For COX-1 and COX-2, both enzymes share considerable homology in their active sites, and this is reflected in the terpene interactions. The key active site residues for COX-1 include TYR385, HIS388, and TRP387. For COX-2, TYR385, SER530, and VAL116 are among the critical residues. Our results show that α-pinene and β-pinene consistently interact with TYR385, HIS388, and TRP387 in both COX-1 and COX-2 (
Table 6). Menthol and linalool also show strong interactions with HIS388 and TYR385 in both enzymes. This suggests a potential for direct interference with the catalytic machinery or with substrate access. These findings support the hypothesis that specific terpenes may modulate enzyme function by targeting residues essential for catalytic activity, even under the constraints of blind rigid-body docking.
5-LOX is a crucial enzyme for leukotriene biosynthesis, with catalytic residues including HIS367, HIS372, HIS550, TYR181, and TRP599. Our study uniquely identified linalool as the only terpene establishing stable contacts with multiple active site residues of 5-LOX, namely, LEU414, TRP599, ALA603, and TYR181. Despite its overall weaker binding affinity compared to some other terpenes (higher Ki), this specific interaction profile with the active site residues suggests a direct mode of action, potentially hinting at a more selective inhibitory mechanism for linalool on 5-LOX than other terpenes. The lack of direct matches for other terpenes, even with favorable binding energies (e.g., limonene and menthol), suggests that their inhibitory effect might be allosteric or involves residues not explicitly listed as “catalytic” in the current literature for 5-LOX active site definition, warranting further investigation.
PLA
2 is the enzyme that initiates the arachidonic acid cascade. Its key active site residues include HIS47, CYS44, TYR51, and PHE5. Remarkably, β-pinene was the only terpene found to interact with multiple matching catalytic residues of PLA
2: PHE5, CYS44, and HIS47. This direct engagement with the enzyme’s functional core positions β-pinene as a promising candidate for PLA
2 inhibition, despite its higher Ki value compared to limonene and menthol for this enzyme. For PLA
2, limonene and menthol, which show the lowest binding energies and Ki values, did not show direct “matches” with the listed catalytic residues, similar to AA for COX enzymes. This reinforces the idea that an optimal fit (low binding energy) can be achieved through interactions with residues forming the binding pocket, even if they are not explicitly defined as “catalytic” in the active site sequence list. However, their precise positioning within critical regions of PLA
2, as supported by their favorable thermodynamic parameters, further strengthens their candidacy as potential inhibitors. The space-filling representation in
Figure 4 visually supports the deep penetration and spatial accommodation of these ligands within the PLA
2 catalytic pocket.
Recent studies have employed virtual screening to identify natural products with inhibitory activity against enzymes involved in inflammation, such as COX-2 and 5-LOX. For instance, Bello-Vargas et al. (2023) conducted the molecular docking of COX-1/2 inhibitors, including celecoxib, reporting binding energies ranging from −6.5 to −9.0 kcal/mol for known inhibitors, indicating strong affinity to COX-2 [
66]. In comparison, the binding energies of menthol (−6.42 kcal/mol) and camphor (−6.48 kcal/mol) for COX-2, and limonene (−5.57 kcal/mol) for 5-LOX, observed in this study, suggest moderate-to-weak affinity. While these terpenes exhibit lower binding energies than synthetic inhibitors like celecoxib, their interactions with the key catalytic residues (e.g., TYR385 and HIS388 for COX-2; PHE177 and HIS372 for 5-LOX) indicate potential competitive inhibition. The smaller molecular size and simpler structure of monoterpenes compared to synthetic inhibitors may limit their potency but offer advantages in bioavailability and reduced toxicity, positioning them as viable lead compounds for further optimization through structural modifications, such as the addition of polar groups to enhance binding affinity.
4.3. Structure–Activity Relationship (SAR) Analysis of Terpenes
The differential binding affinities and interaction profiles observed across the six terpenes and four enzymes highlight crucial structure–activity relationships, emphasizing how subtle structural variations dictate their inhibitory potential.
Bicyclic Monoterpenes: Both pinenes are bicyclic monoterpenes with a rigid bicyclo [3.1.1]heptane core. The primary structural difference lies in the position of the double bond: α-pinene possesses an endocyclic double bond, while β-pinene has an exocyclic one. This structural rigidity likely contributes to their consistent interaction patterns with COX-1 and COX-2, particularly with residues like TYR385, HIS388, and TRP387. The compact, rigid structure of pinenes allows for stable hydrophobic interactions within the relatively narrow channels leading to the active sites of these cyclooxygenases. Their less favorable binding to 5-LOX and PLA2, compared to some other terpenes, suggests that their bicyclic framework might not optimally fit the active site geometries or hydrophobicity profiles of these latter enzymes, with the notable exception of β-pinene’s direct interaction with PLA2 catalytic residues (PHE5, CYS44, HIS47). This specific interaction of β-pinene might be facilitated by the slightly altered spatial presentation of its exocyclic double bond, allowing for precise contacts.
Cyclic Monoterpene Alcohols/Ketones: Menthol (a cyclic alcohol) and camphor (a bicyclic ketone) both contain a six-membered ring (cyclohexane in menthol, part of a bicyclic system in camphor) and oxygen-containing functional groups (-OH in menthol, C=O in camphor).
Menthol (5-Methyl-2-(propan-2-yl)cyclohexan-1-ol) is a cyclic monoterpene alcohol. Its hydroxyl group can participate in hydrogen bonding, while its bulky isopropyl and methyl groups contribute to hydrophobic interactions. Its consistently low binding energies and Ki values across all four enzymes, especially for COX-1, 5-LOX, and PLA2, indicate its versatile binding capabilities. The presence of the hydroxyl group provides an anchor point for polar interactions, while its overall compact, non-planar structure allows it to snugly fit into various binding pockets.
Camphor (1,7,7-trimethylbicyclo[2.2.1]heptan-2-one) is a bicyclic monoterpene ketone. The ketone group provides a site for hydrogen bonding (as an acceptor), and its rigid, compact structure with three methyl groups contributes significantly to hydrophobic interactions. Its favorable binding to COX-1 and COX-2 is likely due to its shape complementarity with the relatively confined active sites of these enzymes, similar to the pinenes, but with the added polar interaction from the carbonyl oxygen. The rigid bicyclic structure might limit its conformational adaptability for optimal binding to 5-LOX and PLA2 compared to more flexible terpenes.
Monocyclic Monoterpene: Limonene (1-Methyl-4-(prop-1-en-2-yl)cyclohex-1-ene) is a cyclic monoterpene characterized by a cyclohexene ring with two double bonds (one endocyclic and one exocyclic isopropenyl group). Its relative flexibility compared to the bicyclic pinenes, combined with its hydrophobic nature, appears to be highly advantageous for interactions with 5-LOX and PLA2. Its exceptional binding affinity to 5-LOX and PLA2, even surpassing AA in Ki for PLA2, suggests that its structure allows for an optimal fit into the active sites of these enzymes. The presence of two double bonds provides potential sites for π-stacking or hydrophobic interactions within the binding pocket.
Acyclic Monoterpene Alcohol: Linalool (3,7-dimethyl-1,6-octadien-3-ol) is an acyclic monoterpene alcohol, making it the most conformationally flexible among the investigated terpenes. Its acyclic nature allows for a wide range of conformations, as evidenced by its higher torsional free energy. While its overall binding affinities were generally weaker across most enzymes, it uniquely established direct interactions with multiple catalytic residues of 5-LOX (LEU414, TRP599, ALA603, and TYR181). This suggests that, despite its weaker overall affinity, its flexibility allows it to precisely orient its hydroxyl group and hydrocarbon chain to engage in critical interactions within the 5-LOX active site, potentially indicating a more specific, albeit less potent, inhibitory mechanism for this enzyme. For other enzymes, its high flexibility might prevent it from adopting a single, stable, and highly complementary conformation, leading to less favorable binding.
In summary, the SAR analysis reveals that the size, shape, the presence and position of double bonds, and the nature of oxygen-containing functional groups (hydroxyl vs. ketone) are critical determinants of the binding profiles of these terpenes. Bicyclic terpenes like pinenes and camphor tend to favor more constrained active sites like those of COX enzymes, while the more flexible monocyclic limonene and the acyclic linalool show distinct preferences and interaction modes for 5-LOX and PLA2, highlighting the importance of structural adaptability for engaging different enzymatic targets.
4.4. Limitations and Future Directions
While the in silico molecular docking approach provides valuable initial insights into potential ligand–enzyme interactions, it is essential to acknowledge its inherent limitations [
67,
68]. It is acknowledged that the rigid-body docking protocol does not account for receptor flexibility, which may influence binding site accessibility and interaction patterns. However, this approach enables the high-throughput comparison of ligand affinities and allows for the initial identification of compounds with the potential to modulate enzyme function. Future work may incorporate molecular dynamics simulations to refine the binding hypotheses and explore induced-fit effects [
69].
Furthermore, complex factors such as solvent effects, entropic contributions to binding, and the precise ionization states of catalytic residues in a physiological environment are not fully captured by current docking algorithms [
70]. These limitations necessitate caution when extrapolating purely in silico findings to actual biological activity.
The “Match” criterion in
Table 6, while indicative, should also be interpreted with care. The definition of “active site” residues can vary slightly between studies, and a ligand might inhibit an enzyme by interacting with residues adjacent to or within the binding pocket, even if they are not traditionally listed as “catalytic.” Thus, a “No match” does not definitively rule out inhibitory potential, especially given the favorable binding energies and Ki values.
To overcome the limitations of the present in silico approach and to validate the promising docking results, several follow-up studies are strongly recommended. One important step is the application of flexible-body docking, which allows for conformational changes in both the ligand and the protein. This is particularly relevant when investigating small molecules like terpenes that may induce subtle but significant shifts in the active site upon binding.
Another essential method is the use of molecular dynamics (MD) simulations. MD can provide a time-resolved view of ligand–protein interactions, allowing for the assessment of complex stability and conformational flexibility and the estimation of binding free energies using approaches such as MM/PBSA or MM/GBSA. This offers a more nuanced and accurate prediction of binding affinity beyond static docking results.
Beyond computational methods, experimental validation is crucial. In vitro enzymatic assays, such as those using recombinant COX or 5-LOX enzymes, can confirm inhibitory activity and enable the determination of IC50 values for the most promising terpene candidates.
Additionally, cell-based assays should be employed to assess anti-inflammatory activity in relevant biological systems, helping to bridge the gap between enzyme inhibition and biological effect. Finally, in vivo studies in animal models of inflammation are necessary to evaluate both the therapeutic potential and the safety profile of the selected terpenes, laying the groundwork for possible clinical development.
Despite these limitations, rigid-body docking remains a cost-effective and rapid method to prioritize ligands for further experimental validation. Its application in early-phase screening helps narrow down candidates for more resource-intensive dynamic modeling and biochemical testing.
Ultimately, this study serves as a strong foundation, identifying specific terpenes (menthol, camphor, limonene, α-pinene, β-pinene, and linalool) with varying degrees of potential to modulate arachidonic acid metabolism through interactions with the key inflammatory enzymes. The observed structure–activity relationships, particularly the direct engagement with catalytic residues for some terpenes, underscore their potential as novel anti-inflammatory agents.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}