Abstract
Flavone derivatives have emerged as promising antiviral agents, with baicalein demonstrating the potent inhibition of the SARS-CoV-2 main protease (Mpro). In this study, the unique electronic and structural properties of 3-hydroxyflavone (3-HF) were investigated using the density functional theory (B3PW91/cc-pVTZ), providing insights into its potential as a bioactive scaffold. Among mono-hydroxyflavone (n-HF) isomers, 3-HF exhibits an extensive intramolecular hydrogen-bonding network linking the phenyl B-ring to the A- and γ-pyrone C-rings, enabled by the distinctive C3-OH substitution. Despite a slight non-planarity (dihedral angle: 15.4°), this hydrogen-bonding network enhances rigidity and influences the electronic environment. A 13C-NMR chemical shift analysis revealed pronounced quantum mechanical effects of the C3-OH group, diverging from the trends observed in other flavones. A natural bond orbital (NBO) analysis highlighted an unusual charge distribution, with predominantly positive charges on the γ-pyrone C-ring carbons, in contrast to the typical negative charges in flavones. These effects impact C1s orbital energies, suggesting that the electronic structure plays a more significant role in 13C-NMR shifts than simple ring assignments. Given the established antiviral activity of hydroxylated flavones, the distinct electronic properties of 3-HF may enhance its interaction with SARS-CoV-2 Mpro, making it a potential candidate for further investigation. This study underscores the importance of quantum mechanical methods in elucidating the structure–activity relationships of flavones and highlights 3-HF as a promising scaffold for future antiviral drug development.
1. Introduction
It is well recognized that conjugated organic systems possessing π-electrons, which are typically delocalized, exhibit varying degrees of stability, influenced by their structural and electronic characteristics. In heterocyclic compounds, factors such as the nature and position of substituents, intra- and intermolecular interactions, and ionization and protonation/deprotonation processes play a pivotal role in modulating electron delocalization and, in turn, the stability and reactivity of these molecules. Flavonoid compounds, a prominent class of polyphenolic substances, have gained significant attention due to their diverse biological activities, including antioxidant, anti-inflammatory, and anticancer properties [1]. Recent studies have also identified several flavonoids as inhibitors of viral replication, making them promising candidates for therapeutic intervention in diseases such as COVID-19 [2]. Notably, compounds like 3-hydroxyflavone (3-HF) have demonstrated a unique combination of chemical features and bioactivity, meriting further exploration of their electronic structures.
Flavonoids are a structurally diverse group of naturally occurring compounds, widely distributed in plants and dietary sources [3]. They share a common 15-carbon skeleton comprising two benzene rings (A- and B-rings) connected by a heterocyclic pyran ring (C-ring) (see Figure 1). Flavonoids are classified into subclasses, including flavones, flavanones, flavonols, isoflavones, and anthocyanins, based on their structural variations, such as substitutions and the degree of oxidation in the C-ring [3]. Among these, flavones are characterized by the absence of oxygenation at the C3 position [4], a double bond between C2 and C3, and a ketone group at C4 [5]. The position and number of hydroxyl groups on the flavone backbone are critical determinants of their electron delocalization, hydrogen-bonding capabilities, and radical-scavenging activity, which underpin their biological function. In mono-hydroxyflavones, subtle changes in hydroxyl placement can lead to pronounced differences in chemical reactivity and stability—highlighted by the unique behavior of 3-HF, which features intramolecular hydrogen bonding. Due to hydroxylation at the C3 position of the C-ring, 3-HF is neither the most stable nor the least stable mono-hydroxyflavone. Previous studies have found that most naturally occurring flavones have a hydroxyl group at the C5 position of the A-ring, while hydroxylation at other positions, such as C7 (A-ring) or C3′ and C4′ (B-ring), varies across different plant species [5].
While experimental spectroscopic techniques offer foundational insight into the structural properties of flavonoids, they often depend on empirical or semi-empirical models fitted to natural derivatives, potentially overlooking subtle electronic effects in synthetic or non-naturally occurring isomers [6,7]. In this context, quantum mechanical (QM) methods, such as density functional theory (DFT), provide a robust framework for elucidating the structure–property relationships of flavones and their analogues [8,9,10]. These methods allow for a first-principle analysis of the electron density distribution, conjugation, orbital energetics, and solvation effects—essential for understanding how positional isomerism influences molecular properties. This is particularly relevant for non-natural derivatives like 8-hydroxyflavone [7], where the hydroxyl substitution at an atypical position significantly alters the electronic structure and intramolecular interactions compared to biosynthetically preferred isomers. A QM approach thus enables predictive insight beyond experimental limitations, offering a deeper understanding of how structural variations in flavone derivatives impact their stability and functional potential.
In this study, we explored the electronic structure of flavone and its eight mono-hydroxyflavone derivatives (n-HFs) using density functional theory (DFT)-calculated NMR chemical shifts. By analyzing and comparing validated DFT-calculated 13C-NMR chemical shifts, we aimed to elucidate the electronic properties that set 3-HF apart from other mono-hydroxyflavones, emphasizing its unique potential for antioxidant and antiviral applications. This computational framework not only reveals key aspects of their electronic environments, but also lays the groundwork for linking structural modifications to potential bioactivity in antioxidant and antiviral contexts.
2. Computational Details
The geometry optimization of flavone (0-HF) and mono-hydroxyflavones (n-HF, n = 3, 5, 6, 7, 8, 2′/6′, 3′/5′, and 4′) were performed using the density functional theory (DFT)-based B3PW91/cc-pVTZ model. Both optimization and nuclear magnetic resonance (NMR) calculations were conducted in dimethyl sulfoxide (DMSO, ε = 46.7) solvent using the polarizable continuum model (PCM). The 13C-NMR isotropic shielding values were calculated based on the obtained geometries of flavone and n-hydroxyflavones using the B3PW91/cc-pVTZ //B3PW91/cc-pVTZ model. In the calculations, the gauge invariant atomic orbital (GAIO) method [11] was employed [11,12]. The results obtained were compared with the internal reference standard, i.e., tetramethylsilane (TMS) for carbon and hydrogen (σC = 182.4656 ppm, and σH = 31.8821 ppm) and water for oxygen (σO = 320.0 ppm). This DFT method, together with the PCM solvent model, has been proven to produce reliable NMR chemical shifts for biomolecules such as resveratrol [13]. The electronic charges by the natural bond orbital (NBO) were calculated using the NBO version 3.1 program [14] embedded in the Gaussian 16 Program.
All the calculations were performed using the Gaussian 16 computational chemistry package [15]. The visualization was achieved using GaussView6.0 [16].
3. Results and Discussion
3.1. Stability of Mono-Substituted Flavones (n-Hydroxyflavones)
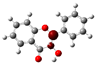
The structure of 3-HF (C15H10O3) consists of a phenyl benzopyrone skeleton with fifteen carbon atoms, in which two benzene rings (A and B) are linked through a γ-pyrone ring C, as shown in Figure 1, together with its nomenclature. All the hydrogens of the ten C-H bonds of the unsubstituted flavone (0-HF, C15H10O2) can be replaced by a hydroxyl group (-OH) as mono-hydroxyflavones. As a result, in addition to the unsubstituted flavone (0-HF), there are seven unique mono-hydroxyflavones, 3-HF, 5-HF, 6-HF, 7-HF, 8-HF, 2′/6′-HF, 3′/5′-HF, and 4′-HF. Here, 5-HF was the most stable mono-hydroxyflavone in the gas phase, in agreement with an earlier study [10], and it was also the most stable mono-hydroxyflavone in DMSO solvent obtained from the present study. 3-HF is unique because its -OH group replaces a single hydrogen atom on the γ-pyrone C-ring. Among the intramolecular hydrogen bonds (HBs) formed, the one within the γ-pyrone C-ring was the most significant, as highlighted in the structure shown at the bottom right of Figure 1.
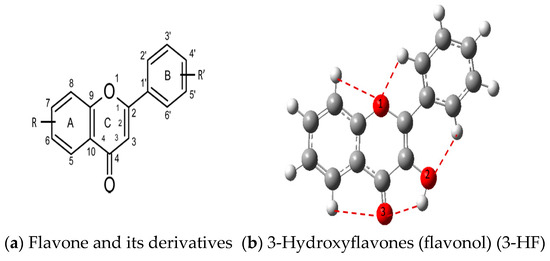
Figure 1.
(a) The structures of the flavone (0-HF: C15H10O2) and its derivatives by single substitutions for R, R’ = ˗OH and H in rings A, B, and C (all eight Hs can be substituted by -OH). (b) The optimized structure of 3-hydroxyflavones (3-HF: C15H10O3) in three-dimensional (3D) space. The possible intramolecular hydrogen bonding is marked on the optimized 3D structure of 3-HF.
The stability and electronic properties of mono-substituted hydroxyflavones (n-HFs) have been systematically calculated using DFT calculations. The ground electronic states of mono-substituted flavones (n-HF where n=0 for unsubstituted flavone, n = 2–8 for n-hydroxyflavones) were optimized in DMSO solvent. Due to the symmetry and the rotations around the C2-C1′ single bond at room temperature, the mono-substituted flavones on the phenyl B-ring would be practically the same in the measurements, although they are theoretically not identical [13]. For example, the 2′-HF and 6′-HF and the 3′-HF and 5′-HF pairs are hardly distinguished by room-temperature NMR measurements. As a result, we only studied 2′-HF (for 6′-HF) and 3′-HF (for 5′-HF). It was noted that the 2′-HF/6′-HF pair could be more different from the 3′-HF/5′-HF pair, as the former on the B-ring may engage with intramolecular interactions with the C-ring, forming certain hydrogen-bond networks, theoretically [10]. Figure 2 compares the stability of the n-HF compounds, such as the total energies and dipole moments. Here, the unsubstituted flavone is presented as a reference. The total electronic energies of these eight n-HFs provide insights into their relative stability, which follows the following order: 5-HF > 4′-HF ≈ 7-HF > 3′-HF > 6-HF > 2′-HF > 3-HF > 8-HF.
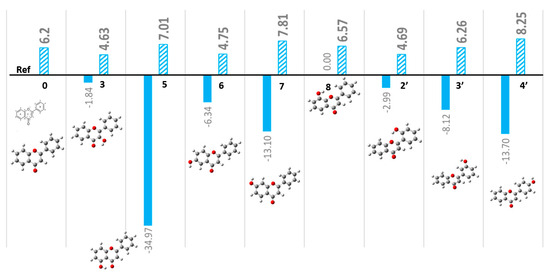
Figure 2.
Comparison of stability of single substituted n-hydroylflavones. The solid columns are for total energies in kJ/mol and the patterned columns are for dipole moments in Debye.
The most stable n-HF was 5-HF, with the hydroxyl group substitution in the benzopyrone system (specifically in the A- and C-rings). In contrast, 8-HF, which was the least stable, also resides in the same benzopyrone framework (opposite the C5 position (para) in the A-ring). This trend indicates that the stability of n-HFs is influenced by the position of the hydroxyl (-OH) substitution and the ability to form hydrogen bonds within the molecular framework. Notably, the differences in the total electronic energy between conformers are relatively small, especially for the n-HFs where hydroxyl rotation does not significantly affect intramolecular hydrogen bonding, as observed for 6-HF, 7-HF, 3′-HF, and 4′-HF. This stability trend is consistent with earlier gas-phase calculations by Meyer [10] (as shown in Figure S1) and correlates well with the distribution of dipole moments, which reflects the electronic environment and intermolecular interactions of these molecules. For example, the energy differences due to hydroxyl group rotation for 2′-HF and 6′-HF are small, calculated at ΔE ≈ 3.73 kJ/mol, while for 3′-HF and 5′-HF, the difference is as low as ΔE ≈ 0.29 kJ/mol (Meyer, HF/6-31G(d)) [10]. These small energy differences indicate the flexibility of certain hydroxyl groups in these n-HFs, suggesting that the stabilization effect of intramolecular hydrogen bonding can be a key factor in their conformational preferences. The results support the fact that most naturally occurring flavones have a hydroxyl group at the C5 position of the A-ring, while hydroxylation at other positions, such as C7 (A-ring) or C3′ and C4′ (B-ring), varies across different plant species [5].
The dipole moment distributions further support the stability trend. The dipole moments of n-HFs range from 4.61 Debye for 3-HF (the smallest) to 7.81 Debye for 7-HF (the largest) in DMSO solvent. This significant variation in dipole moments correlates with the molecular geometry and the position of the hydroxyl group, which affect the electronic distribution within the molecule. High dipole moments, such as in 7-HF, suggest stronger interactions with the solvent and other molecules, which can enhance stability in solution. Conversely, lower dipole moments, such as in 3-HF, reflect less-pronounced charge separation, potentially resulting in lower stabilization through dipole–dipole interactions.
The stability of n-HFs is primarily influenced by the position of the hydroxyl group and the ability to form intramolecular hydrogen bonds. The ability of specific hydroxyl positions to enhance the stability and dipole moments suggests opportunities for tuning molecular properties through selective substitutions. To further explore the intramolecular hydrogen-bonding networking associated with these n-HFs, Table 1 provides selected geometric properties of the flavones. While all the n-HFs are non-planar, with the phenyl B-ring deviating from the plane of the benzopyrone system (A- and C-rings), 3-HF and 4′-HF stand out by having the smallest dihedral angles between the B-ring and the benzopyrone, at 15.4° and 13.3°, respectively. More details of the geometric parameters obtained in the present study, with literature, for 0-HF and 3-HF are given in Tables S1 and S2, respectively.

Table 1.
Comparison of geometric properties of flavone and n-hydroxylflavones in DMSO solvent (Use color to indicate that 3-HF is special).
The results in Table 1 demonstrate that, among the mono-substituted n-HFs, 3-HF stood out due to its unique geometric and electronic properties. While other n-HFs, such as 5-HF, 8-HF, and 2′-HF, exhibited intramolecular hydrogen bonding, their structural arrangements do not support the formation of a two-ring intramolecular hydrogen-bonding network as observed in 3-HF (see Figure 1). For instance, 5-HF forms a single strong hydrogen bond between the hydroxyl group at the C5 position and the carbonyl oxygen, but the absence of a secondary hydrogen bond limits the molecule’s planarity. Similarly, 8-HF exhibits hydrogen bonding within the benzopyrone system; however, this interaction does not extend to the B-ring, reducing its overall stabilization (making 8-HF the most unstable mono-substituted n-HF). In the case of 2′-HF, the hydroxyl group on the B-ring primarily interacts within the B-ring itself, with minimal interaction with the benzopyrone system, further distinguishing it from 3-HF.
A distinctive feature of 3-HF is its ability to form two strong intramolecular hydrogen-bonding network connecting rings B and C. As shown in Figure 1b, the hydroxyl group at the C3 position of the γ-pyrone C-ring interacts with the carbonyl oxygen (O3), -C3-O2H···O3=C4, and the hydrogen atom on C6′ of the phenyl B-ring, -C3-(H)O2···HC6′. These interactions result in the formation of pentagonal and hexagonal hydrogen-bonded ring structures of -C3-O2H···O3=C4- and -C3-(H)O2··· HC6′-C1′-C2-, respectively, which stabilize the molecule. The hexagon ring in 3-HF also exhibits an additional H···O bond, which stops the phenyl ring B from free rotations along the C2-C1′ bond. The O2H···O3 distance of 3-HF was very short, underscoring their strength. Specifically, the O2H···O3 and H···O2H distances in 3-HF were among the shortest calculated, with the bond distances being 1.95 Å and 2.21 Å, respectively. This dual hydrogen-bonding network not only increases the geometric rigidity of the molecule but also plays a pivotal role in defining its electronic properties. It was also noted that 5-HF exhibited a dual hydrogen-bonding network, but the O2H···O3 and H···O2H distances in 5-HF were 1.64 Å and 2.61 Å, respectively. Thus, the dual hydrogen-bonding network of 5-HF may contribute to the total energy stabilization, and 5-HF was the most stable mono-flavone derivative, as indicated in Figure 2.
The near-planarity in 3-HF required for the hydrogen-bonding network, however, may impose geometric constraints, leading to an increased strain energy. The constrained arrangement around the hydroxyl group and the carbonyl oxygen introduces torsional and angular strain, which counterbalances the stabilizing effect of the strong hydrogen bonding. For instance, the relatively small dihedral angle, ∠321′6′, in 3-HF indicates a more planar structure compared to other n-HFs (except for 4′-HF). This increased planarity facilitates better conjugation between the B-ring and the benzopyrone system, thereby enhancing electronic delocalization. While 4′-HF also exhibits a small dihedral angle, its hydroxyl group at the C4′ position of the phenyl B-ring is more isolated and interacts minimally with other parts of the molecule. In contrast, the hydroxyl group in 3-HF, strategically positioned at the C3 site of the γ-pyrone C-ring, engages in critical intramolecular hydrogen-bonding interactions. This unique placement allows 3-HF to achieve a balance between electronic delocalization and structural stabilization, distinguishing it from other mono-hydroxyflavones.
Despite its extensive hydrogen-bonding network, the stability of 3-HF is determined by a delicate balance between geometric strain, electronic delocalization, and solvation effects. The strain imposed by maintaining a near-planar configuration, combined with the localization of electron density in specific regions, appears to counteract the stabilizing influence of the hydrogen bonds. This interplay explains why 3-HF, despite possessing the most intramolecular hydrogen bonding, ranks as the second least stable among mono-hydroxyflavones.
3.2. Validation of Calculated C-NMR Chemical Shifts of Flavone
The 13C NMR chemical shifts of unsubstituted flavone (0-HF) and 19 mono-substituted flavone derivatives have been experimentally measured in dimethyl sulfoxide-d₆ (DMSO) by Park et al. [7]. To validate the reliability of our computational approach, Table 2 presents a comparison between the DFT-calculated 13C chemical shifts for 0-HF (at the B3PW91/cc-pVTZ level in DMSO) and the experimental values reported by both Burns et al. [6] and Park et al. [7] in the same solvent. The calculated shifts exhibited a good overall agreement with the experimental data, with a root mean square deviation (RMSD) of 0.85 ppm relative to Burns et al. and 0.93 ppm relative to Park et al., indicating the robustness of the DFT method in reproducing experimental chemical shifts in polar solvents.
Table 2.
Comparison of the DFT (B3PW91/cc-pTVZ)-calculated 13C-NMR chemical shifts of flavone in DMSO solvent with experimental measurements (ppm) (Color is for readiness).
The largest deviation was observed at the C2 carbon, where the calculated value differed by 2.21 ppm from the measurement reported by Burns et al., and by 2.41 ppm from Park et al.’s data [6,7]. Such deviations at specific sites—especially those adjacent to the carbonyl and aromatic regions—may arise from subtle solvation effects, conformational flexibility, or limitations in the treatment of solvent–solute interactions within the implicit solvation model used. Nonetheless, the overall consistency affirms the suitability of this computational protocol for extending chemical shift predictions to substituted flavone systems, including the structurally and electronically complex 3-hydroxyflavone.
3.3. C-NMR Chemical Shifts Indicating Intermolecular Interactions of n-Hydroxyflavones
Table 3 reports the calculated 13C-NMR chemical shifts (δC) of the flavones. The accuracy of the calculated δC for n-HFs can be evaluated by comparing the root mean square deviation (RMSD) values between the calculated and experimental data [6] for each isomer. The RMSD values of the eight compounds ranged from 0.85 ppm (0-HF) to 1.56 ppm (5-HF). Note that the “experimental” δCs for 8-HF are the estimated averaged data from reference [17], as there are no available NMR measurements for this compound to our knowledge. As a result, it is no surprise that the error for this 8-HF was as large as 2.15 ppm. Other RMSD values for the n-HFs where a hydroxyl group replaced the Hs on the benzopyrone (A- and C-) rings exhibited smaller variations. The simulated individual H-NMR spectra of all the n-HFs are given in Figure S2 in the Supplementary Materials, which provides direct visualization of the patterns.
Table 3.
DFT-calculated 13C-NMR chemical shifts of flavone and mono-hydroxyflavones in DMSO solvent (ppm) * (color to emphases the pattern).
The real RMSD values for carbons in the phenyl B-ring are likely smaller than the tabulated values. A significant factor contributing to this discrepancy is the rotational flexibility of the C2–C1′ single bond. At room temperature, this bond allows for dynamic averaging of the chemical environments of carbons C2′/C6 and C3′/C5 in ring B [13]. Experimental NMR captures this averaged state, whereas computational models treat these environments as distinct [13]. Consequently, this difference inflates the RMSD values, suggesting larger deviations than may actually exist under experimental conditions. For example, the mono-substituted n-HFs on the B-ring exhibited both the smallest RMSD values, such as 0.92 ppm for 4′-HF and 0.98 ppm for 3′-HF, and the largest RMSD value of 2.13 ppm for 2′-HF. The notable RMSD for 2′-HF, which was significantly higher than those of 3′-HF and 4′-HF, can be attributed to the dynamic averaging of the carbon chemical shifts, δC, in the experimental measurements. Specifically, the δC for 2′-HF reflects an averaged state between 2′-HF and its isomer, 6′-HF, due to the “free” flip of the phenyl B-ring, while the computational calculations accounted only for 2′-HF. As illustrated in Figure 1, the structural differences between the 2′-HF and 6′-HF isomers were more pronounced compared to those of the 3′-HF and 5′-HF isomers. In contrast, 4′-HF, located at the para-position on the phenyl B-ring, has no isomer, resulting in a reduced variability and the smallest RMSD among the B-ring-substituted flavones.
Another key factor influencing the RMSD is the substitution pattern across the flavone structure. Unsubstituted flavone (0-HF) exhibited the lowest RMSD values, showing an excellent agreement with the experimental data. This was likely due to its structural rigidity, which minimizes conformational variability. In contrast, mono-substituted flavones on the A- and C-rings showed larger RMSD values. The flexibility of the hydroxyl group in these compounds introduces variability in the local chemical environment, as the hydroxyl group can rotate or “flip,” leading to deviations between the calculated and experimentally observed values [18]. Despite these discrepancies, an RMSD analysis provides a valuable tool for benchmarking computational predictions. The observed deviations emphasize the need to account for conformational flexibility, intramolecular interactions, and the inherent limitations of static computational models. These factors must be considered when interpreting RMSD values to ensure accuracy.
The systematically calculated δC values of the n-HFs presented in Table 3 reveal an intriguing pattern. The δC values for the flavones ranged from 100.64 ppm (C8 in 7-HF) to 182.96 ppm (C4 in 5-HF), spanning approximately 82 ppm. This wide range reflects the diverse chemical environments of the carbon atoms. Notably, the carbonyl carbon (C4) consistently exhibited the largest δC (>170 ppm) across all isomers, with 3-HF showing the smallest δC (C4) at 172.22 ppm and 5-HF exhibiting the largest δC (C4) at 182.96 ppm. The carbon chemical shifts also revealed three consistently prominent carbons—C4, C2, and C9—that displayed significantly higher δC values compared to the others. The fourth largest δC corresponded to the carbon bonded to the hydroxyl group in each n-HF isomer. For unsubstituted flavone, there were only three such carbons due to the absence of a hydroxyl group. Interestingly, the smallest δC was almost always associated with the C3 carbon in the n-HF isomers, except for 3-HF and 7-HF. In these two cases, the smallest δC was observed for C8. The unique position of C3 in the flavone structure contributes to its low chemical shift. It is the only carbon atom in the γ-pyrone C-ring that is not part of the phenyl ring (unlike C9 and C10) and does not directly bond with oxygen (unlike C2, C4, and C9), except in 3-HF. This structural uniqueness likely explains the consistently low δC values for C3 in most n-HF isomers. The remaining δC values corresponded to the phenyl carbons, which typically fell within the 110–140 ppm range, depending on their specific positions. Notably, 8-HF emerged as an outlier, as its experimental NMR values were based on literature estimates rather than direct measurements. This introduced additional uncertainty in the comparison and inflated the RMSD values.
Half of the n-HF isomers, such as 3-HF, 5-HF, 8-HF, and 2′-HF, were able to form intramolecular hydrogen bonding with oxygens (O1 or O3 on the γ-pyrone C-ring) on adjacent carbon atoms, which was reflected by the chemical shifts of the isomers. The environment of a 13C nucleus can also be affected in an inductive, through-space (field effect), or resonance-based manner by substituents that are bonded to its carbon framework (skeleton). These effects, which became apparent in the 13C substituent chemical shift analyses, caused the associated chemical shifts to vary in a systematic and predictable manner. Figure 3 plots the 13C-NMR spectra of these n-HFs with respect to 0-HF. The full width at half maximum (FWHF) of the spectra was given by 0.20 ppm. The 13C-NMR spectra of all the n-HF isomers are given in Figure S3 in the Supplementary Materials.
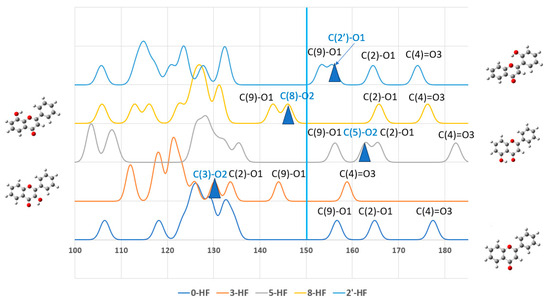
Figure 3.
13C-NMR spectra of the n-HFs that exhibited intramolecular hydrogen bonding and formed pentagon and/or hexagon rings, together with the unsubstituted flavone.
The carbons with the largest chemical shifts across all flavones were typically those forming C=O or C-O bonds, including C(4), C(2), and C(9), as well as the hydroxyl-substituted C(n) carbons. This indicates that chemical shifts are more influenced by the carbon–oxygen bonding environment than by their positions within the A-, B-, or C-rings. For the unsubstituted flavone (0-HF), only three chemical shifts occurred above 135 ppm, corresponding to its three C-O bonds. However, all the n-HF derivatives showed four chemical shifts above 135 ppm, which is attributable to the presence of an additional C-OH bond introduced by hydroxyl substitution. This further emphasizes the strong impact of oxygen bonding on the chemical shifts of carbons in flavones. As expected, all the flavones showed the C(4)=O carbonyl group as the one with the highest chemical shift, which aligns with its highly deshielded nature due to strong electron-withdrawing effects. The C-NMR chemical shifts (δC) of C4 in the most stable isomer, 5-HF, exhibited the largest δC for its carbonyl carbon C(4), consistent with its strong electron-withdrawing effects and stability and the hydrogen-bonding interaction of C5-O-H···O3=C4. The hydroxyl-substituted C-OH carbons exhibited δC values that were intermediate, but closer to the higher end of the chemical shift range, reflecting their partially deshielded environment. These carbons consistently had δC values above 135 ppm, which aligns with their attachment to electron-withdrawing oxygen atoms.
The specific δC ordering for various carbons provides insight into the electronic environments. For unsubstituted flavone, 0-HF, the chemical shift order is as follows:
δC (C(4)=O3) > δC (C(2)-O1) > δC (C(9)-O1)
For most n-HF derivatives, the order is slightly modified to the following:
where n = 5, 8, ad 2′. However, in 3-HF, the order is unique:
δC (C(4)=O3) > δC (C(2)-O1) > δC (C(n)-O2) > δC (C(9)-O1)
δC (C(4)=O3) > δC (C(9)-O1) > δC (C(2)-O1) > δC (C(3)-O2)
This unique δC ordering in 3-HF further highlights the distinct electronic effects of its extended hydrogen-bonding network and near-planar geometry. The deshielding effect on C(9) surpasses that on C(2) in 3-HF, reflecting the strong influence of hydrogen bonding and electronic delocalization in this isomer.
3.4. Quantum Mechanical Insights of the Uniqueness of 3-HF
The 13C-NMR chemical shifts of mono-hydroxyflavones are primarily influenced by their local electronic environments, including inductive effects, lone-pair resonance, and conjugation, which are accurately captured through DFT-based quantum mechanical calculations. This highlights the importance of computational approaches for reliably predicting 13C-NMR shifts. While empirical methods, such as curve fitting or machine learning (ML), provide estimates, they lack the ability to fully capture the fundamental quantum mechanical principles that dictate these chemical environments. For example, a recently developed ML model failed to accurately predict 13C-NMR chemical shifts for up to six classes of natural products, including flavonoids, lignans, sesquiterpenoids, steroids, alkaloids, and chromans [19]. These classes of natural products contain a delocalized conjugation framework.
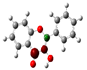
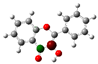
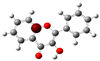
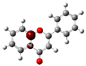
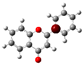
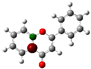
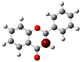
To highlight the unique electronic structure of 3-HF, Figure 4 presents the calculated electronic charge (natural bond orbital (NBO)) distributions of 3-HF compared to those of 5-HF, the most stable isomer, and 0-HF, the unsubstituted flavone. The analysis of NBO atomic charges and bonding in the flavones revealed significant variations in the electronic environments and roles of individual carbons and hydrogens. According to the NBO charge distribution, all hydrogens are positively charged (green), while carbons are generally negatively charged, except for those involved in C–O bonds, which exhibit positive charges (red). This distinction underscores the influence of oxygen’s strong electronegativity on neighboring carbons. In the case of 0-HF (unsubstituted flavone), the carbons C3 and C9 within the γ-pyrone C-ring are negatively charged. However, C3 is more negatively charged than C9 due to its position between two highly electronegative oxygen atoms. In contrast, C9 is part of the aromatic A-ring, where its negative charge is partially delocalized into the aromatic system, reducing its overall negative character. Notably, 0-HF lacks any C(n)–OH bonds, which sets its NBO charge distribution apart from its hydroxylated derivatives. An interesting observation is that the positively charged (green-colored) carbons bonded directly to oxygen atoms do not form C–H bonds. In 0-HF, C3 is the only carbon within the γ-pyrone C-ring that forms a C–H bond, further highlighting its unique electronic environment within the γ-pyrone C-ring of unsubstituted flavone.
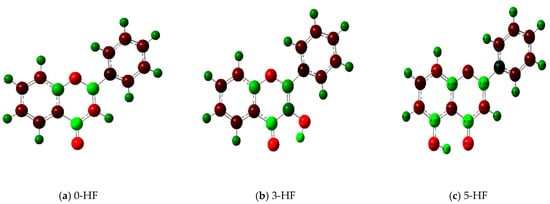
Figure 4.
Natural bond orbital (NBO) of 0-HF (a), 3-HF (b), and 5-HF (c). The carbons in the compounds that directly bond with oxygens are positively charged (in green color in the figure), such as C(4), C(9), C(2), and C(n)-OH, as the carbons are deshielded. The rest of the carbon atoms in the flavones/hydroylflavones are negatively charged (red color in the figure), and are shielded. The calculated NBO values are given in Table S3 in the Supplementary Materials.
In 3-HF and 5-HF, hydroxyl substitution introduces a fourth positively charged carbon, corresponding to the hydroxylated carbon (C3 in 3-HF and C5 in 5-HF, respectively). The substitution significantly alters the local electronic environment, highlighting the differences in the charge distribution compared to unsubstituted flavone. In 3-HF, the hydroxyl group at C3 enables the formation of a robust intramolecular hydrogen-bonding network. This group participates in multiple hydrogen bonds, including H···O(3)=C and C(6′)–H···O(2), forming a pentagonal ring, and C(2′)–H···O(1), constituting a hexagonal ring. These hydrogen-bonding interactions contribute to the geometric rigidity of 3-HF, although they also introduce additional electronic strain, as discussed earlier. The distinct charge distributions and bonding environments in flavones are primarily determined by the presence and positioning of hydroxyl groups and the resulting C–O bonds. Hydroxyl substitution not only influences the local electronic properties of flavones, but also enhances the negative charge on specific carbons. Furthermore, it enables hydrogen bonding, which plays a crucial role in determining their structural stability and energetic properties. Such quantum mechanical effects of flavones are well predicted using pattern-recognition methods such as machine learning [19]. The chemical shifts δC of the carbons with C-O bonding for these n-HFs are further compared in Figure S4. For the three δCs with single C-O bonding (that is, C2, C9, and Cn for n-HFs), they all follow the same trend that C9 has the smallest δC among these carbons, except for 3-HF, where C9 has the second largest δC after δC (C4=O). This is in good agreement with the measurement of Burns et al. [6].
From a structural point of view, the unsubstituted 0-HF and mono-substituted 3-HF share the same molecular skeleton with the same number carbon atoms, but 3-HF has one more oxygen atom. The uniqueness of 3-hydroxyflavone (3-HF) compared to flavone (0-HF) can be attributed to the significant changes in the carbon core electron density distributions (C1s) caused by the presence of the hydroxyl group at the C3 position. Table 4 compares the top six C1s orbitals of 3-HF and 0-HF (out of 15 C1s core orbitals). The DFT calculations reveal that the local energy C1s orbitals, 4a-12a for 3-HF and 3a-11a for 0-HF, exhibited minimal changes in their local density distributions. This is because these core C1s electrons are primarily dominated by the aromatic phenyl rings in the A-ring and B-ring, as shown in Figure S5. However, the top six C1s core electrons in Table 4 demonstrate noticeable reordering in 3-HF (13a-18a) compared to 0-HF (12a-17a). All these top six carbon C1s core orbitals are located in the γ-pyrone C-ring, plus the C1′ in the B-ring and C2 in the γ-pyrone C-ring. Specifically, the hydroxyl group at C3 introduces a significant electronic perturbation, causing a pair-swapping in the order of the six top C1s electron densities (see Table 4 by matching colors). For instance, the sixth highest C1s state in 0-HF (12a on C3, blue) shifts to become the third highest C1s state (17a on C3) in 3-HF. This reordering is directly attributed to the C3-OH group, which alters the local electronic environment and increases the electron density around the C3 carbon, as indicated in their 13C-NMR chemical shifts. These changes reflect the strong electronic influence of the hydroxyl group in 3-HF, distinguishing it from unsubstituted flavone and highlighting its unique electronic structure and reactivity.
Table 4.
DFT-calculated carbon core electron density distributions of flavone (0-HF) and mono-hydroxyflavones (3-HF) in DMSO solvent *.
The mono-substitution on C3 for 3-HF also caused the C1s electron density reordering of the highest C1s orbitals on C4 and C2 in 0-HF, which was switched in 3-HF as C2 and C4 in 3-HF. It was also noted that a certain amount of electron density sharing between C4 and C3 of 3-HF was observed, and this was not seen in 0-HF under the same conditions.
4. Conclusions
The unique intramolecular hydrogen-bonding network of 3-hydroxyflavone (3-HF) was analyzed relative to unsubstituted flavone (0-HF) and other mono-hydroxyflavones (n-HFs). Using the density functional theory (B3PW91/cc-pVTZ) in dimethyl sulfoxide (DMSO), 3-HF was found to be 1.85 kJ/mol more stable than the least stable (8-HF), but 33.13 kJ/mol less stable than the most stable (5-HF). This is consistent with earlier studies. Geometrically, 3-HF is unique, as its hydroxyl group resides in the γ-pyrone C-ring, unlike other n-HFs, where the hydroxyl groups are in the phenyl A- or B-rings. This positioning allows 3-HF to form an intramolecular hydrogen-bonding network connecting both the B-ring and the A-/C-rings. Although 3-HF is slightly non-planar, with a dihedral angle of 15.4°, this structure enhances the rigidity due to its extended hydrogen-bonding network. These unique properties are further reflected in the 13C-NMR chemical shift patterns, which are distinct from other n-HFs.
Contrary to previous studies suggesting that 13C-NMR shifts depend on the flavone’s ring structure, these findings indicate that the shifts are predominantly influenced by the electronic environment of carbons within C-O bonds. The natural bond orbital (NBO) analysis revealed that all the carbon atoms in the γ-pyrone C-ring of 3-HF, except C10, carry positive charges, with C1 in the B-ring also exhibiting higher-energy C1s orbitals. These quantum mechanical effects uniquely define the electronic properties of 3-HF, which may be challenging to predict with machine learning due to their dependence on quantum phenomena. In conclusion, 3-HF’s distinct geometry, electronic structure, and 13C-NMR pattern underscore the significant role of its C3 hydroxyl group, and quantum mechanical approaches can accurately capture the nuanced behaviors of such systems, offering insights that complement empirical methods.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/app15115928/s1, References [9,10,20,21] are cited in the Supplementary Materials.
Author Contributions
F.W.: conceptualization, methodology, validation, investigation, resources, formal analysis, writing—original draft, review and editing, and project administration. V.V.: data curation, calculation, and review and editing. F.W.: wrote the main manuscript text and prepared the figures. V.V.: conducted the calculations. All authors reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data are contained within the article and Supplementary Materials.
Acknowledgments
The authors thank the supercomputing facilities provided by the Swinburne University of Technology Supercomputing Facilities (OzSTAR and Ngarrgu Tindebeek (which means “Knowledge of the Void” in the Moondani Toombadool language)).
Conflicts of Interest
The authors declare no conflicts of interest in this research.
References
- Wang, X.; Han, J.; Chou, A.; Yang, J.; Pan, J.; Borchers, C.H. Hydroxyflavones as a New Family of Matrices for MALDI Tissue Imaging. Anal. Chem. 2013, 85, 7566–7573. [Google Scholar] [CrossRef] [PubMed]
- Sopjani, M.; Falco, F.; Impellitteri, F.; Guarrasi, V.; Nguyen Thi, X.; Dërmaku-Sopjani, M.; Faggio, C. Flavonoids derived from medicinal plants as a COVID-19 treatment. Phytother. Res. 2024, 38, 1589–1609. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as potential anti-inflammatory molecules: A review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef] [PubMed]
- Crozier, A.; Jaganath, I.B.; Clifford, M.N. Dietary phenolics: Chemistry, bioavailability and effects on health. Nat. Prod. Rep. 2009, 26, 1001–1043. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.C.; Ellis, D.A.; March, R.E. A predictive tool for assessing 13C NMR chemical shifts of flavonoids. Magn. Reson. Chem. 2007, 45, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Moon, B.-H.; Lee, E.; Lee, Y.; Yoon, Y.; Ahn, J.-H.; Lim, Y. 1H and 13C-NMR data of hydroxyflavone derivatives. Magn. Reson. Chem. 2007, 45, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; van Rhee, A.M.; Sanders, L.H.; Jacobson, K.A. Flavonoid derivatives as adenosine receptor antagonists: A comparison of the hypothetical receptor binding site based on a comparative molecular field analysis model. J. Med. Chem. 1998, 41, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Aparicio Martínez, S. A systematic computational study on flavonoids. Int. J. Mol. Sci. 2010, 11, 2017–2038. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M. Ab initio study of flavonoids. Int. J. Quantum Chem. 2000, 76, 724–732. [Google Scholar] [CrossRef]
- Ebrahimi, H.P.; Shaghaghi, H.; Tafazzoli, M. Gauge invariant atomic orbital-density functional theory prediction of accurate gas phase 1H and 13C NMR chemical shifts. Concepts Magn. Reson. Part A 2011, 38, 269–279. [Google Scholar] [CrossRef]
- Sarotti, A.M.; Pellegrinet, S.C. A multi-standard approach for GIAO 13C NMR calculations. J. Org. Chem. 2009, 74, 7254–7260. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chatterjee, S. Dominant carbons in trans-and cis-resveratrol isomerization. J. Phys. Chem. B 2017, 121, 4745–4755. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital theory: Discovering chemistry with NBO7. In Complementary Bonding Analysis; De Gruyter: Amsterdam, The Netherlands, 2021; pp. 129–156. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Fox, D.J.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J.G. GaussView 6.0. 16; Semichem Inc: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- March, R.E.; Burns, D.C.; Ellis, D.A. Empirically predicted 13C NMR chemical shifts for 8-hydroxyflavone starting from 7, 8, 4′-trihydroxyflavone and from 7, 8-dihydroxyflavone. Magn. Reson. Chem. 2008, 46, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Backler, F.; Wang, F. Impact of intramolecular hydrogen bonding of gallic acid conformers on chemical shift through NMR spectroscopy. J. Mol. Graph. Model. 2020, 95, 107486. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Song, J.; Yang, M.; Yao, L.; Zhang, J.; Shi, H.; Ji, X.; Deng, Y.; Wang, X. Cross-modal retrieval between 13C NMR spectra and structures for compound identification using deep contrastive learning. Anal. Chem. 2021, 93, 16947–16955. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.; Urbańczyk-Lipkowska, Z.; Baer, S.; Barbara, P. The crystal structures and hydrogen-bond properties of three 3-hydroxy-flavone derivatives. J. Mol. Struct. 1986, 144, 155–167. [Google Scholar] [CrossRef]
- Li, Y.; Siddique, F.; Aquino, A.J.A.; Lischka, H. Molecular Dynamics Simulation of the Excited-State Proton Transfer Mechanism in 3-Hydroxyflavone Using Explicit Hydration Models. J. Phys. Chem. A 2021, 125, 5765–5778. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).