
Flavone Hybrids and Derivatives as Bioactive Agents

Abstract
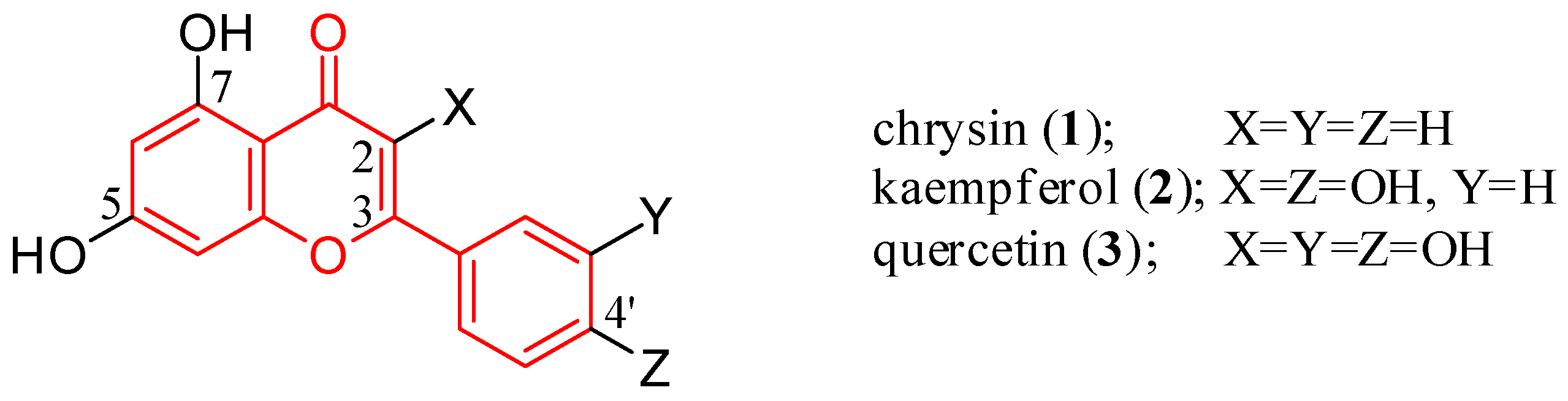
1. Introduction
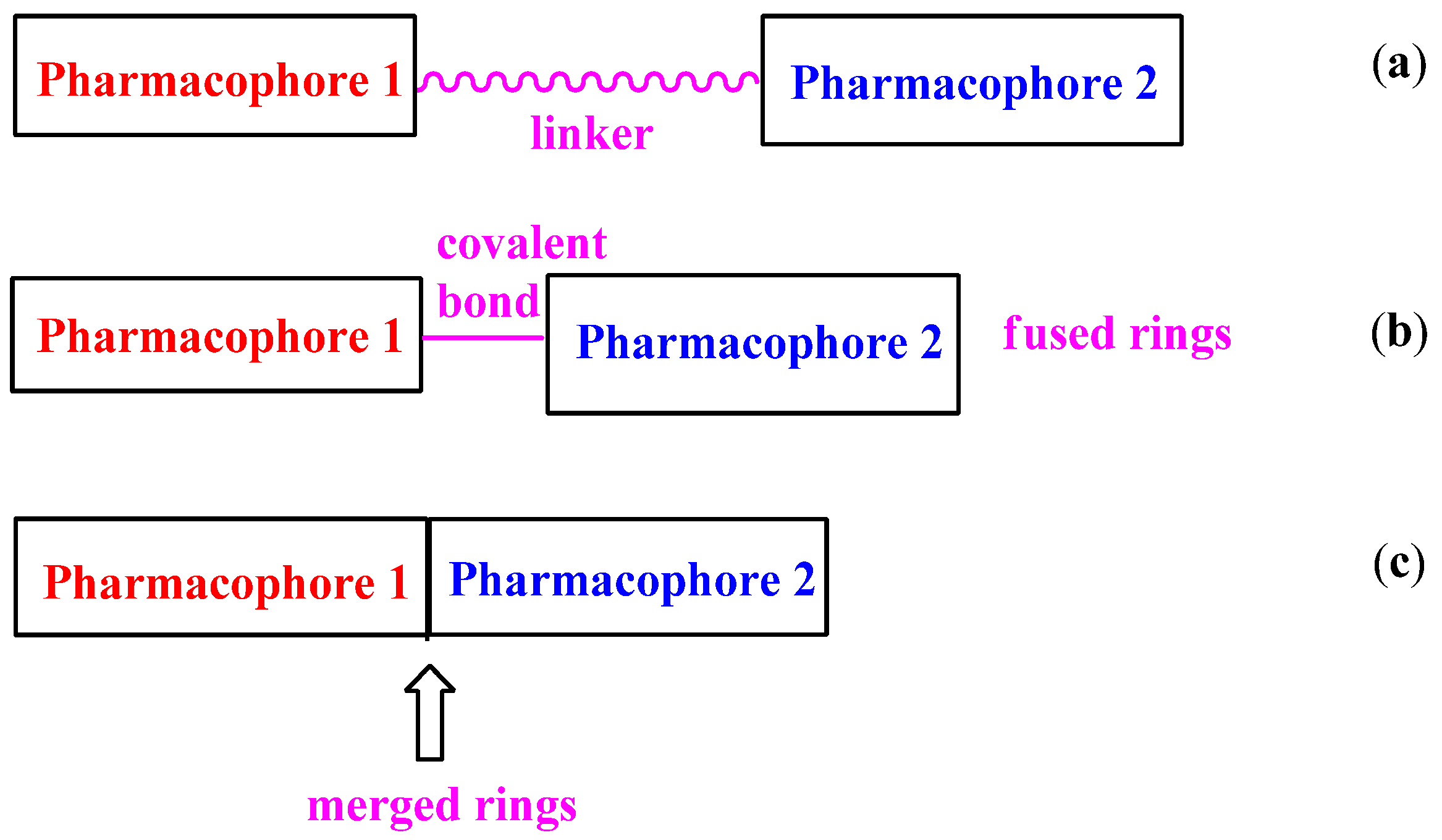
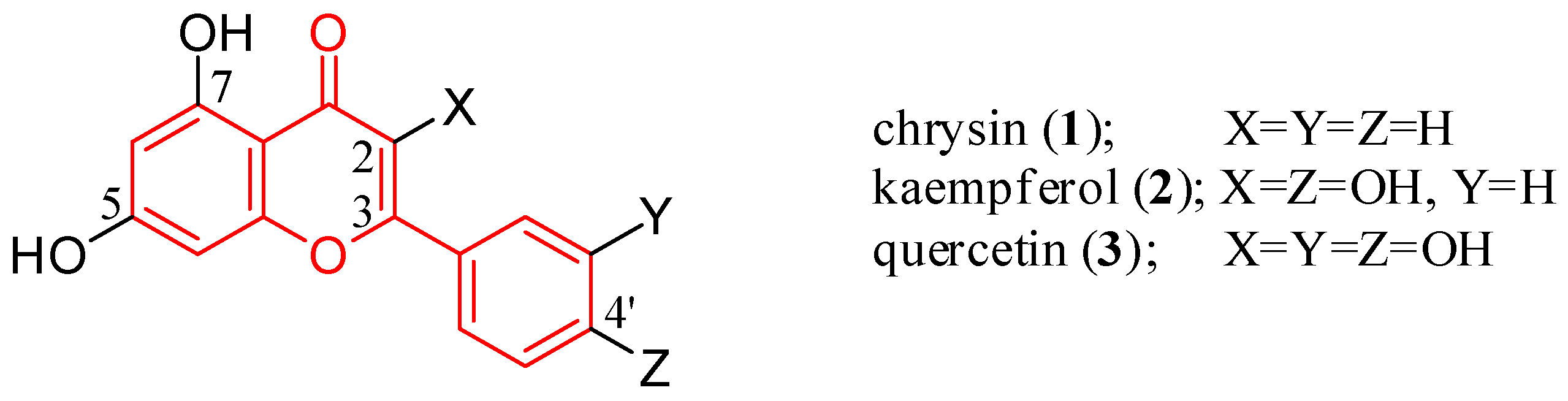
2. Flavone Hybrids with Different Pharmacophores
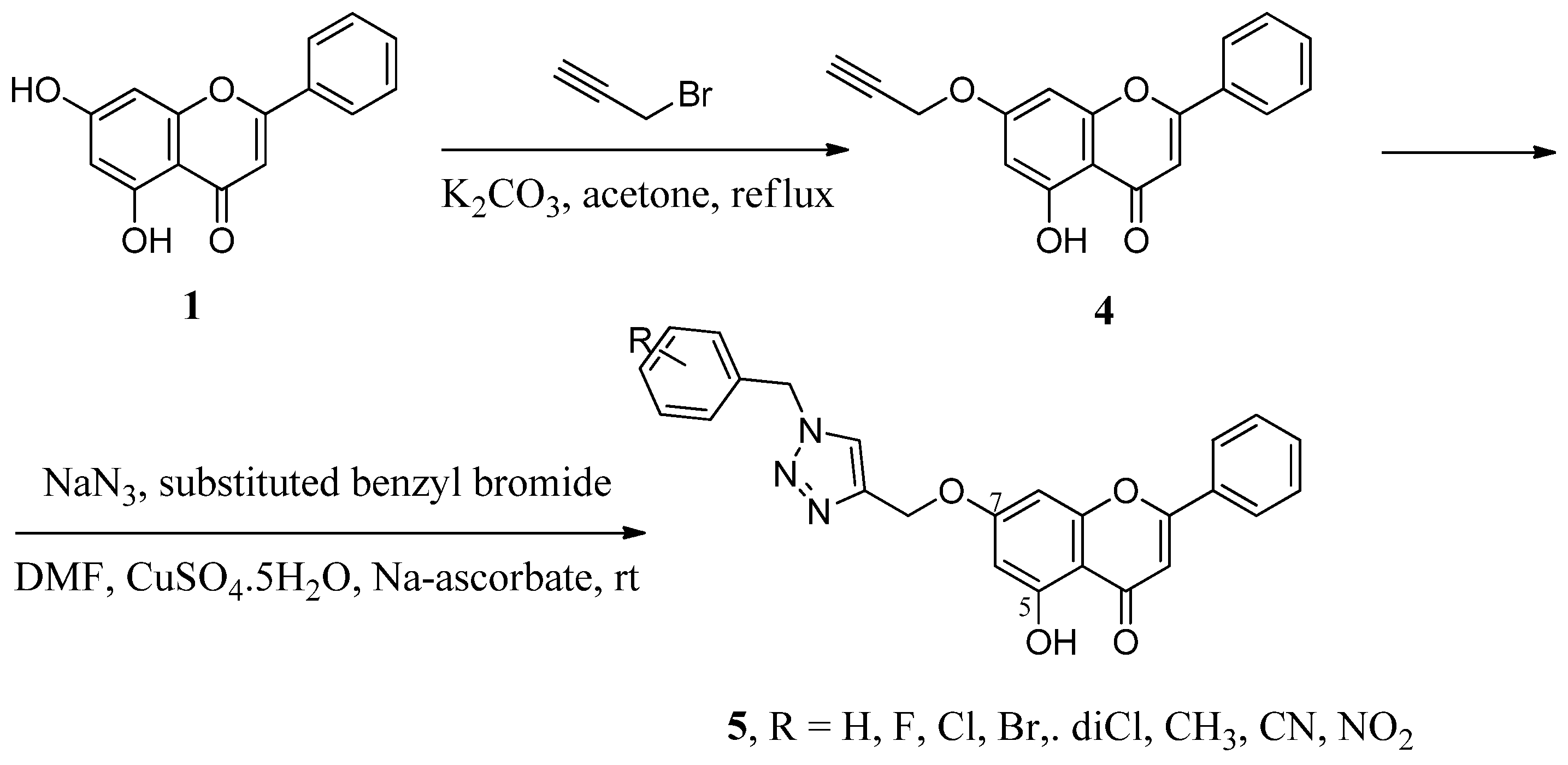
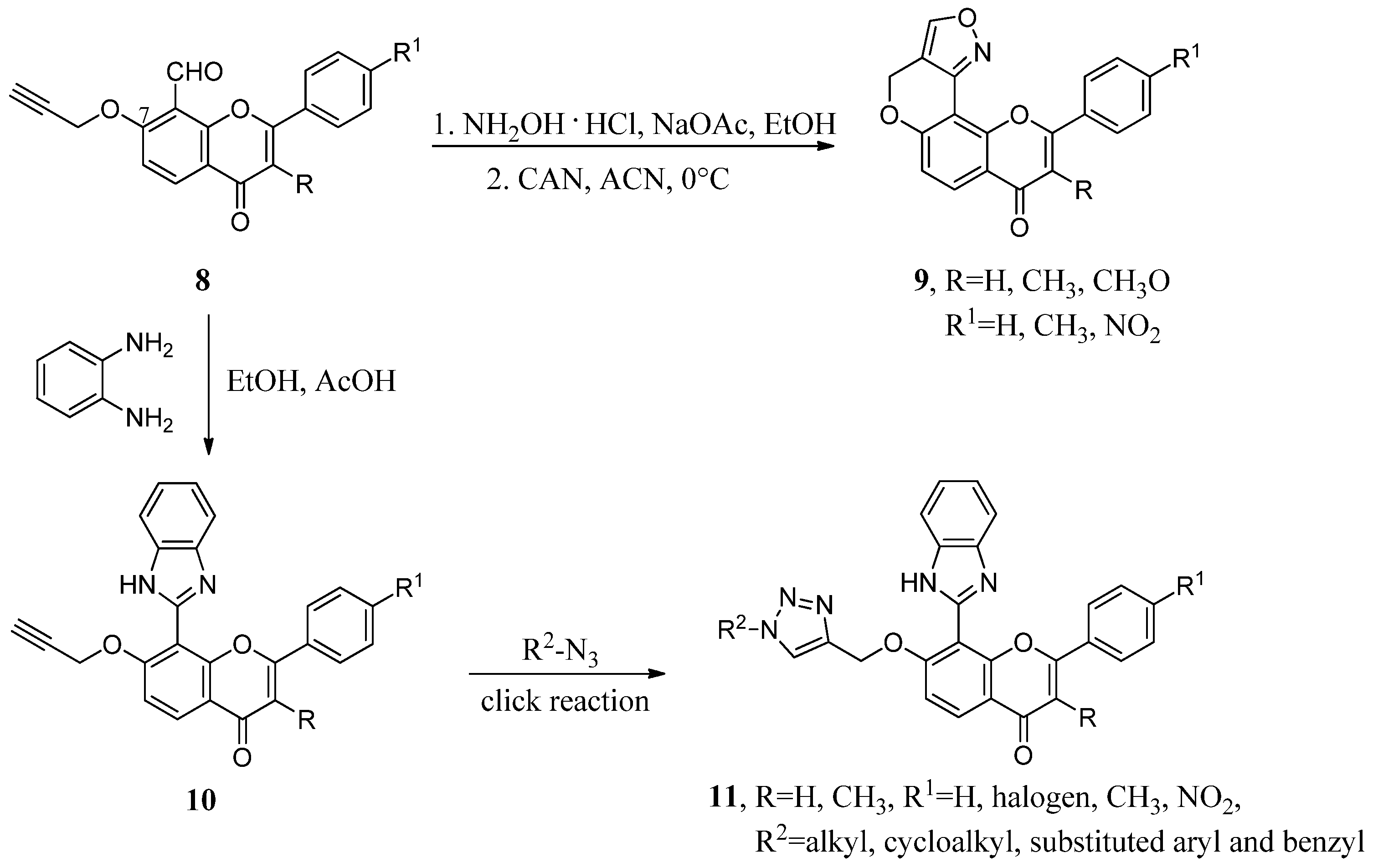
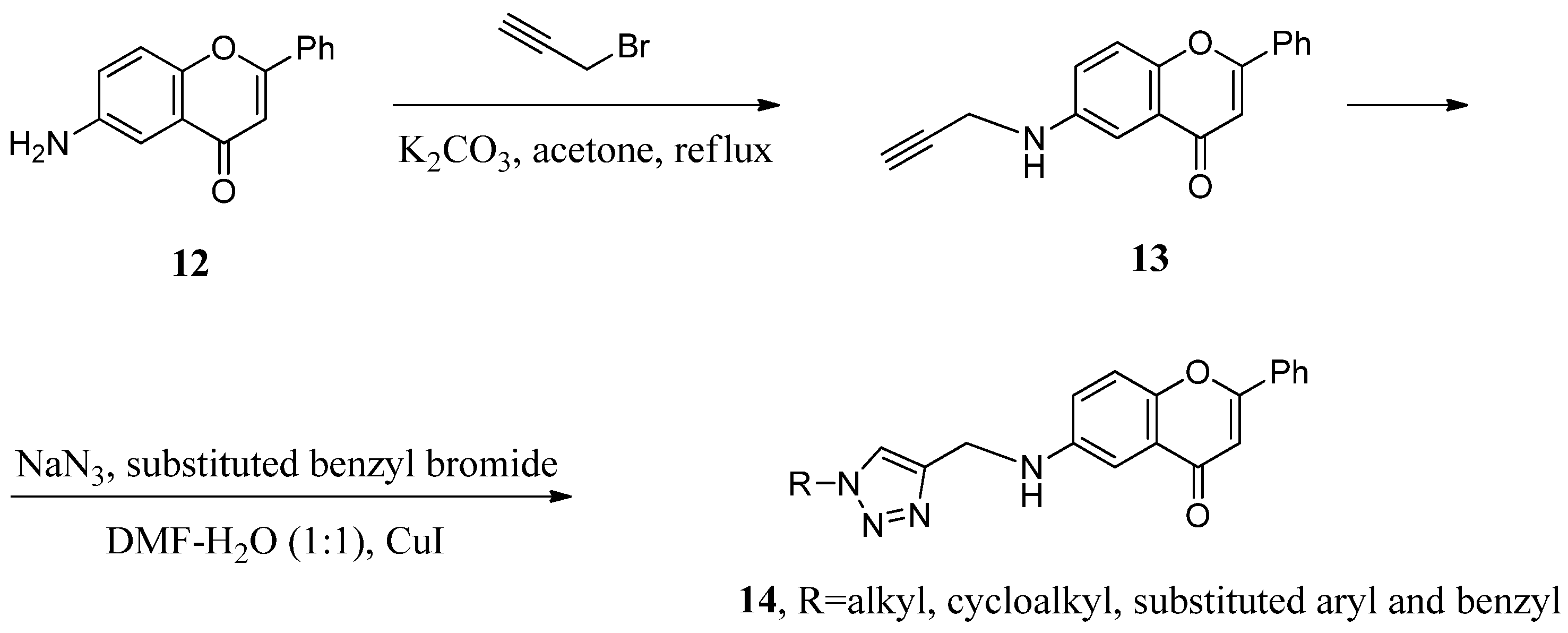
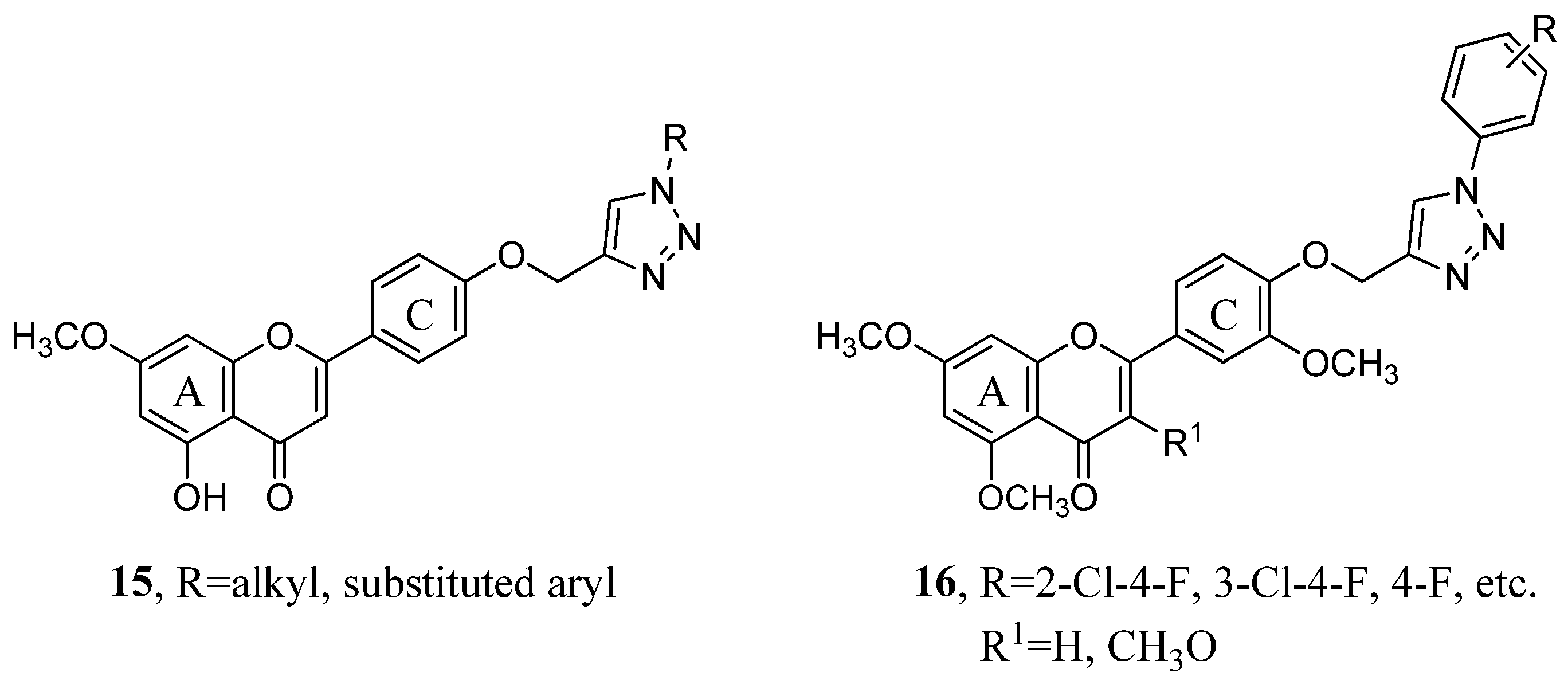
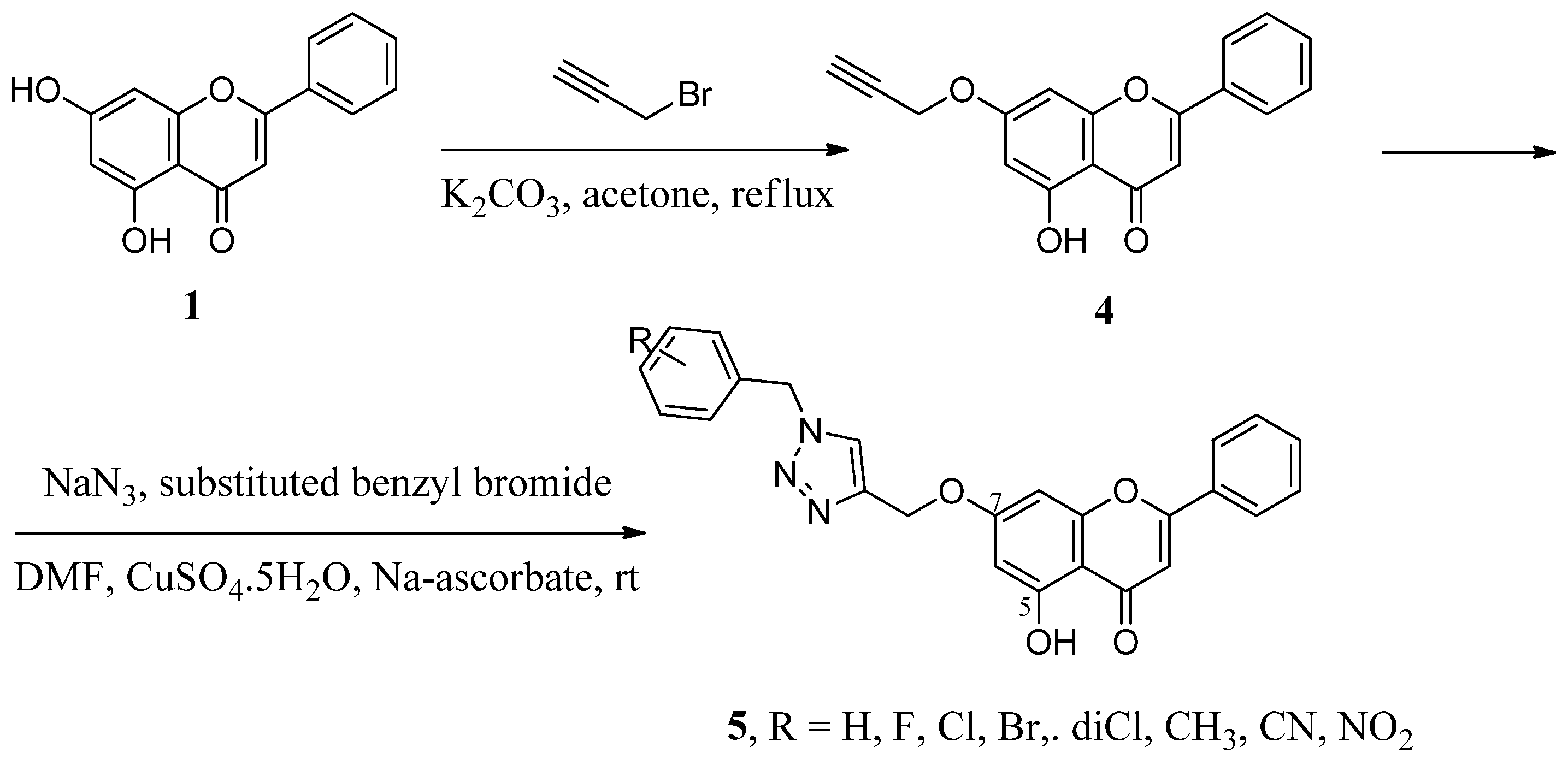
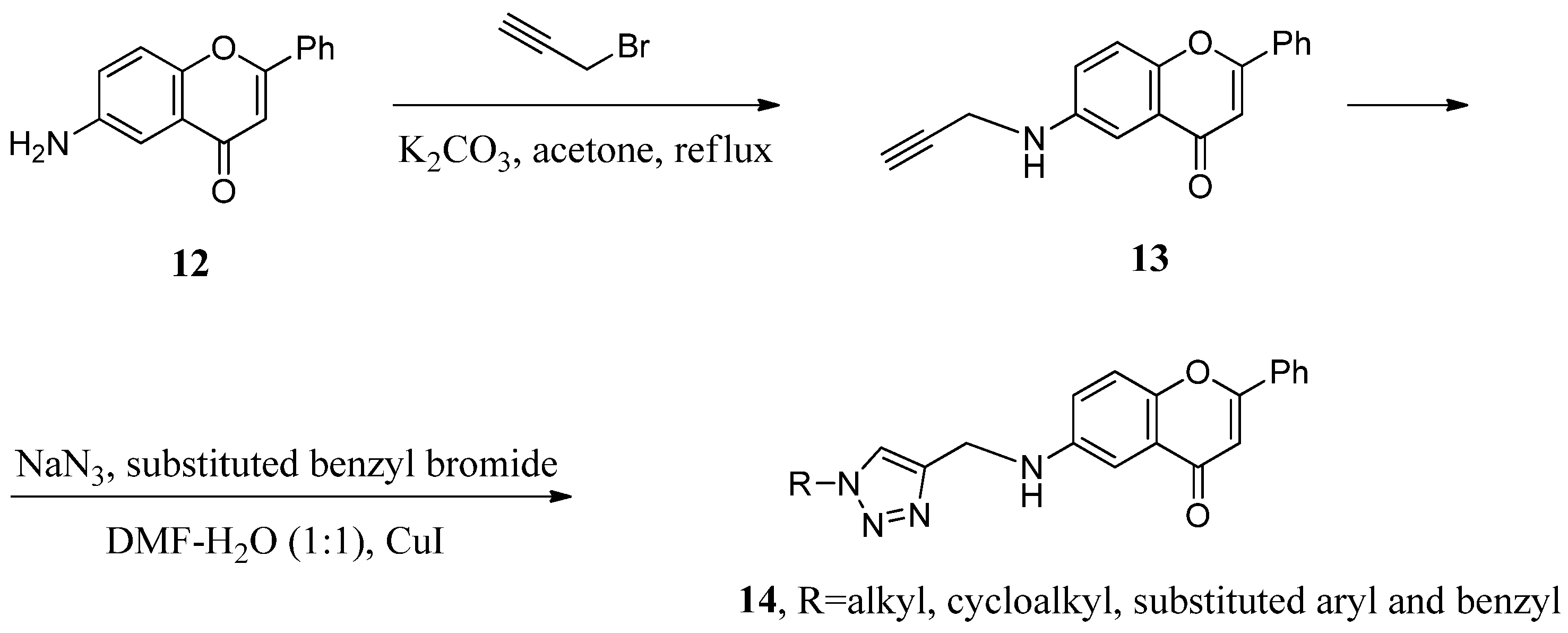
2.1. Hybrids with 1,2,3-Triazoles and Their Derivatives
2.2. Hybrids with Triphenylphosphine
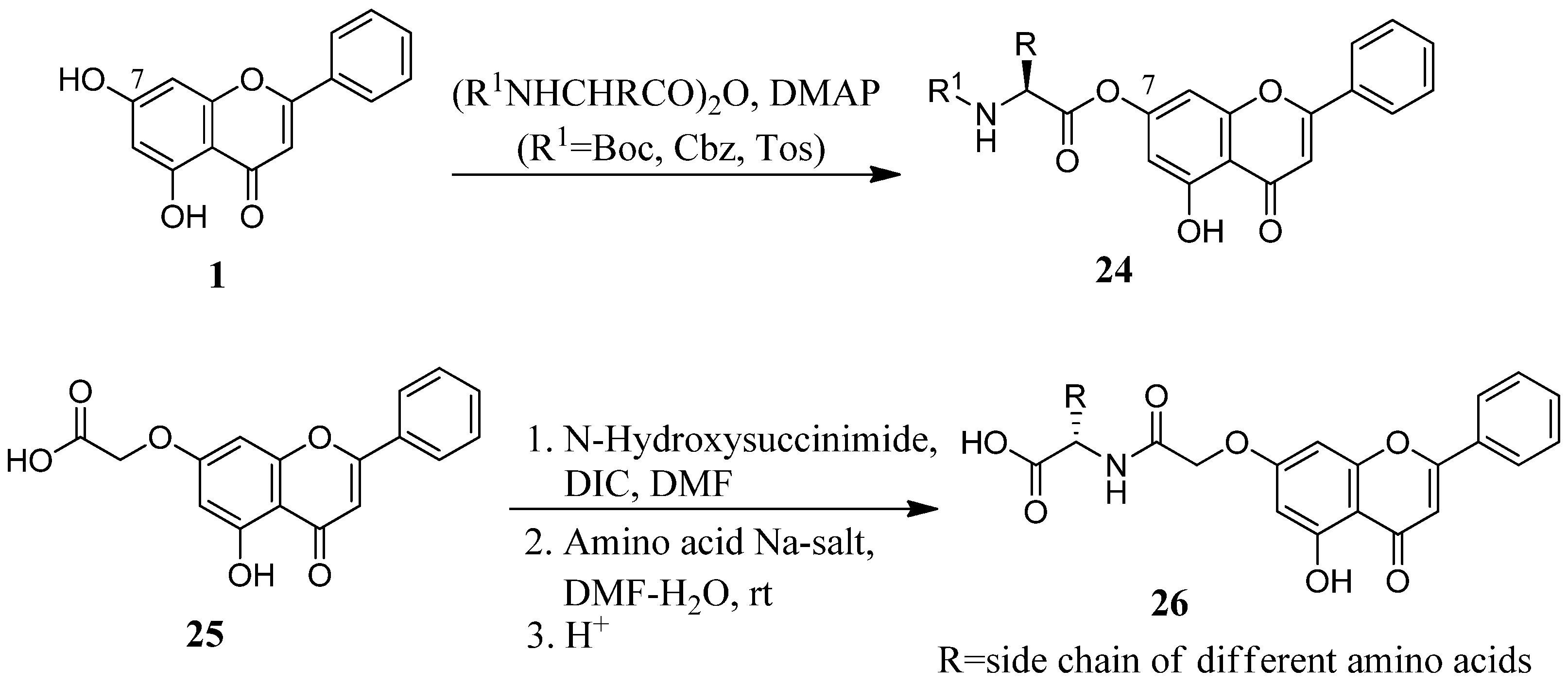
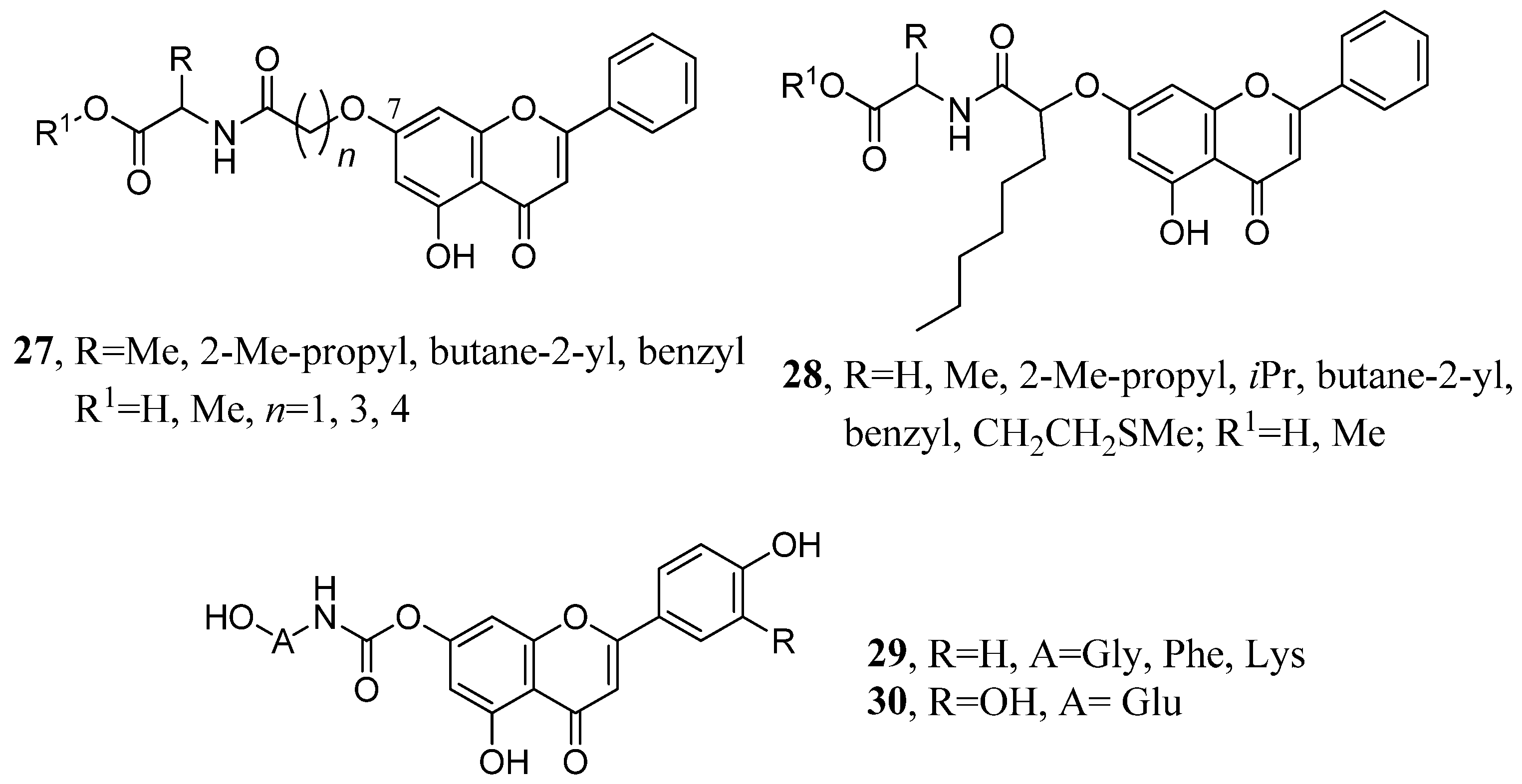
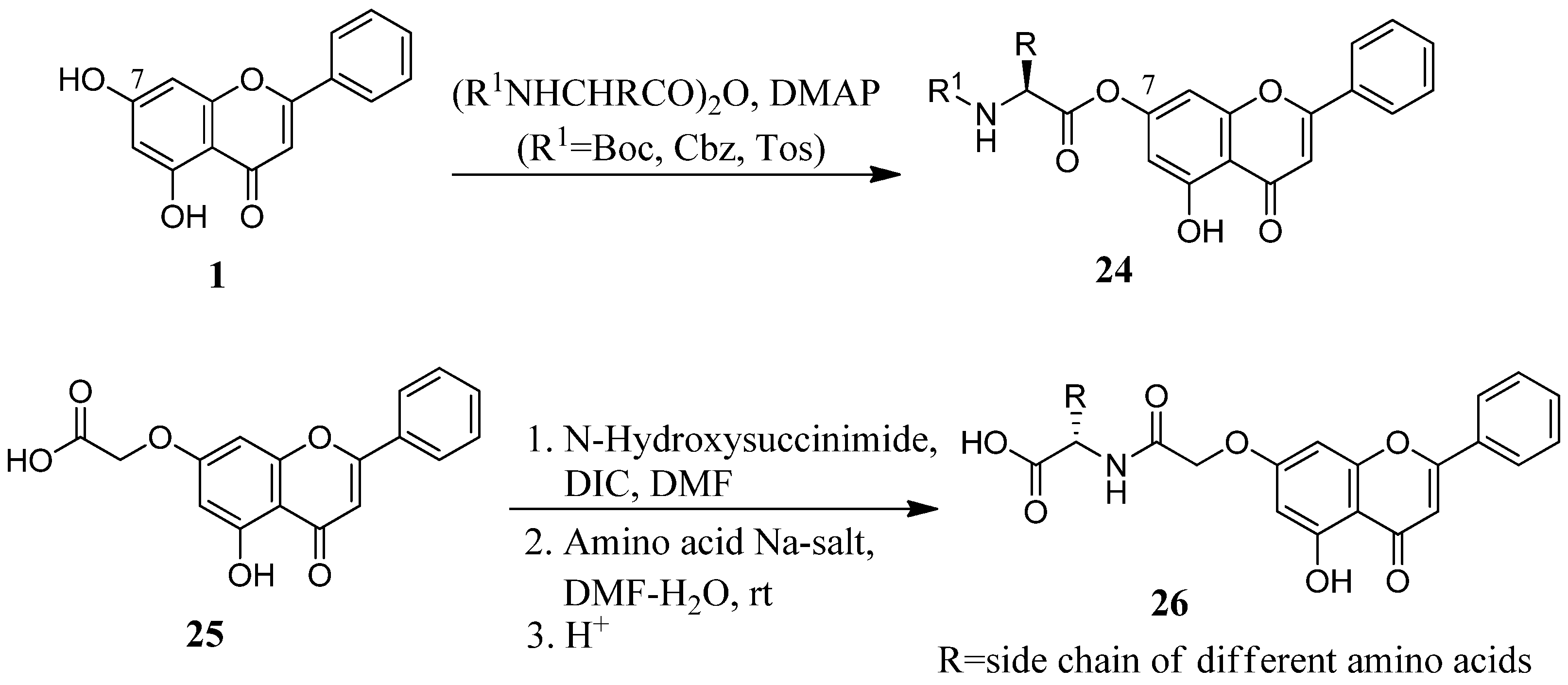
2.3. Flavone Hybrids with Amino Acids
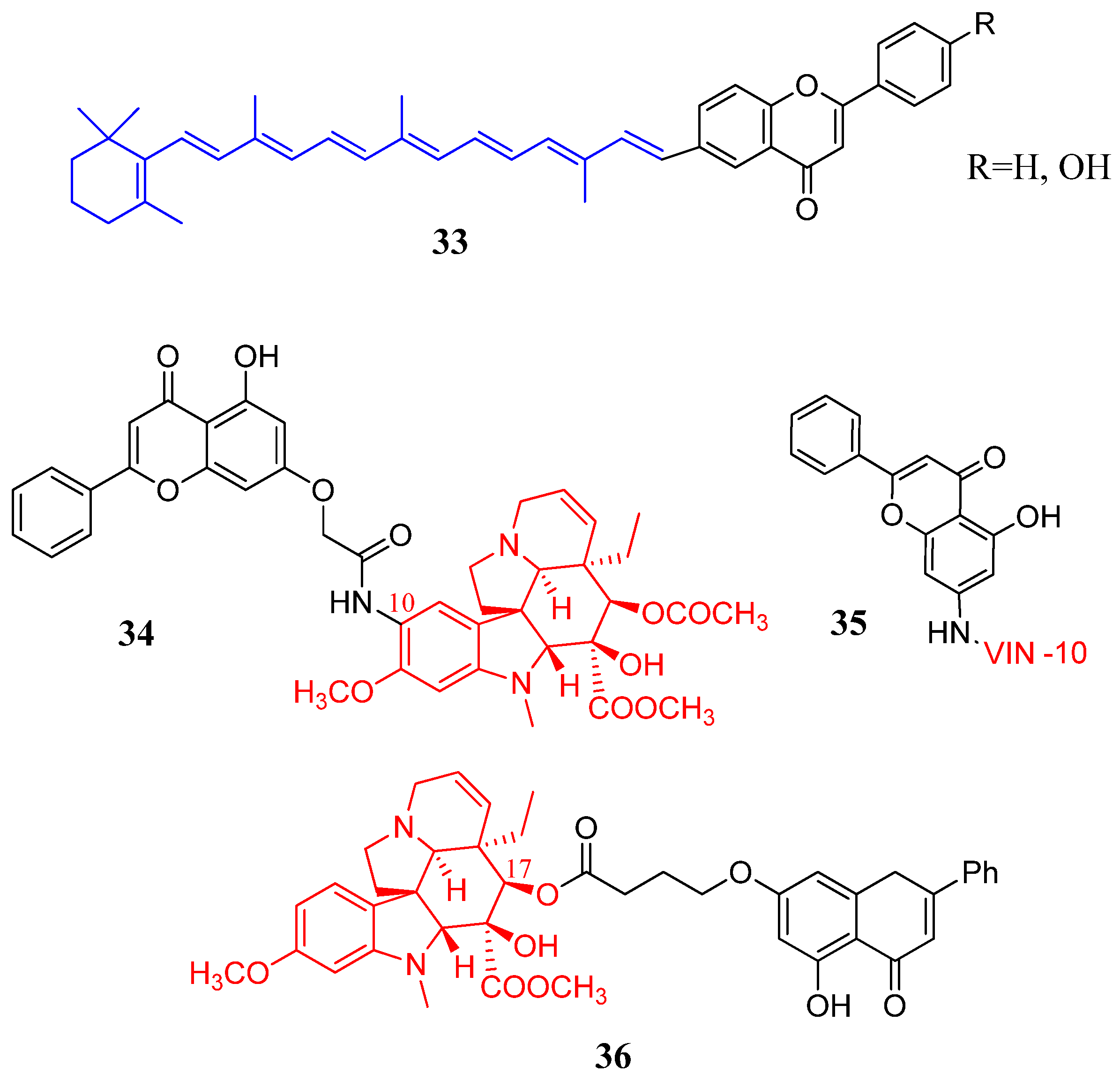
2.4. Flavones Coupled with Representative Natural Components
2.5. Cyclic Saturated Amines in Flavone Derivatives
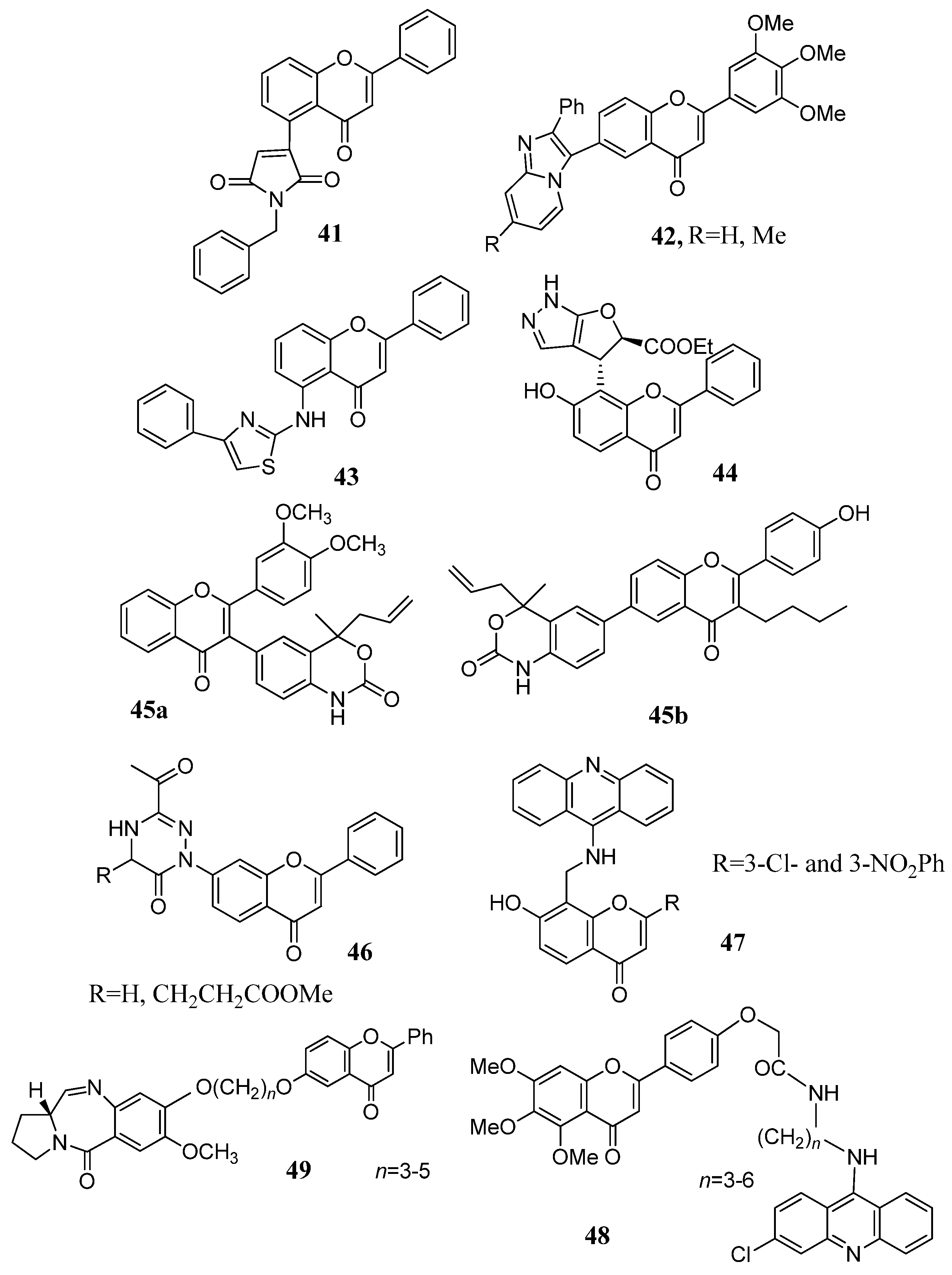
2.6. Heterocyclic Pharmacophores
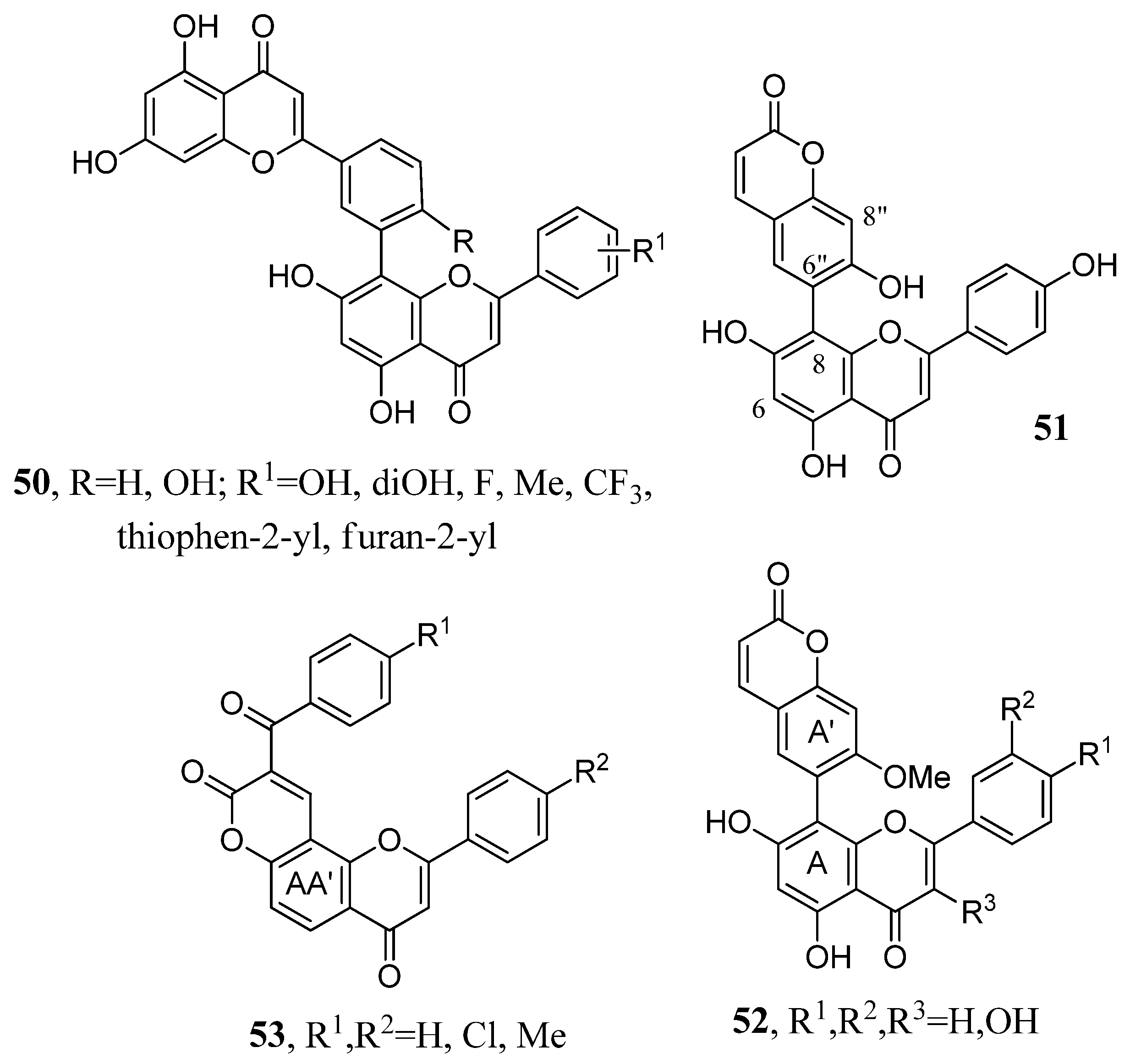
2.7. Flavone–Flavone and Flavone–Coumarin Hybrids
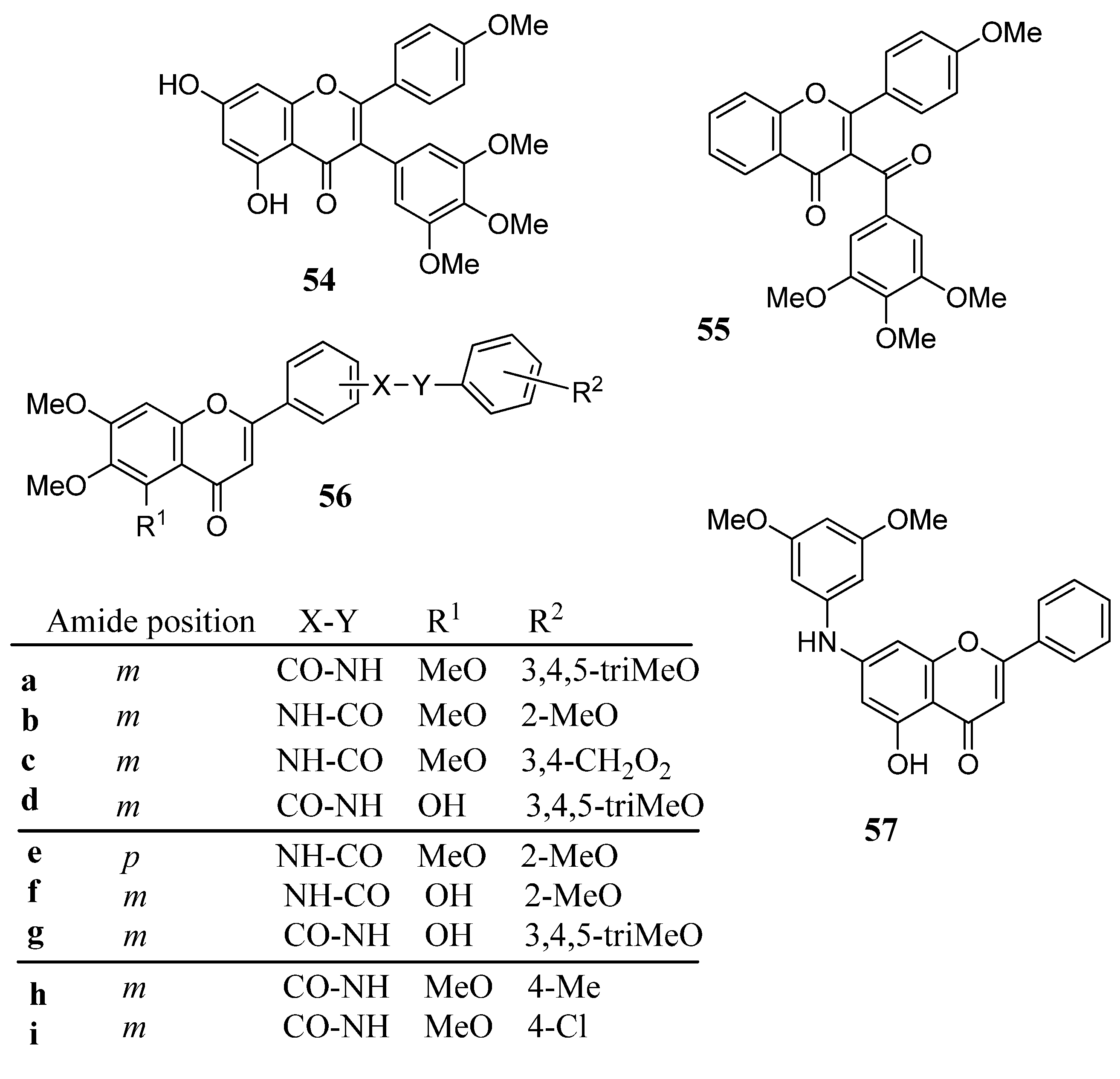
2.8. Aryl Derivatives of Flavones
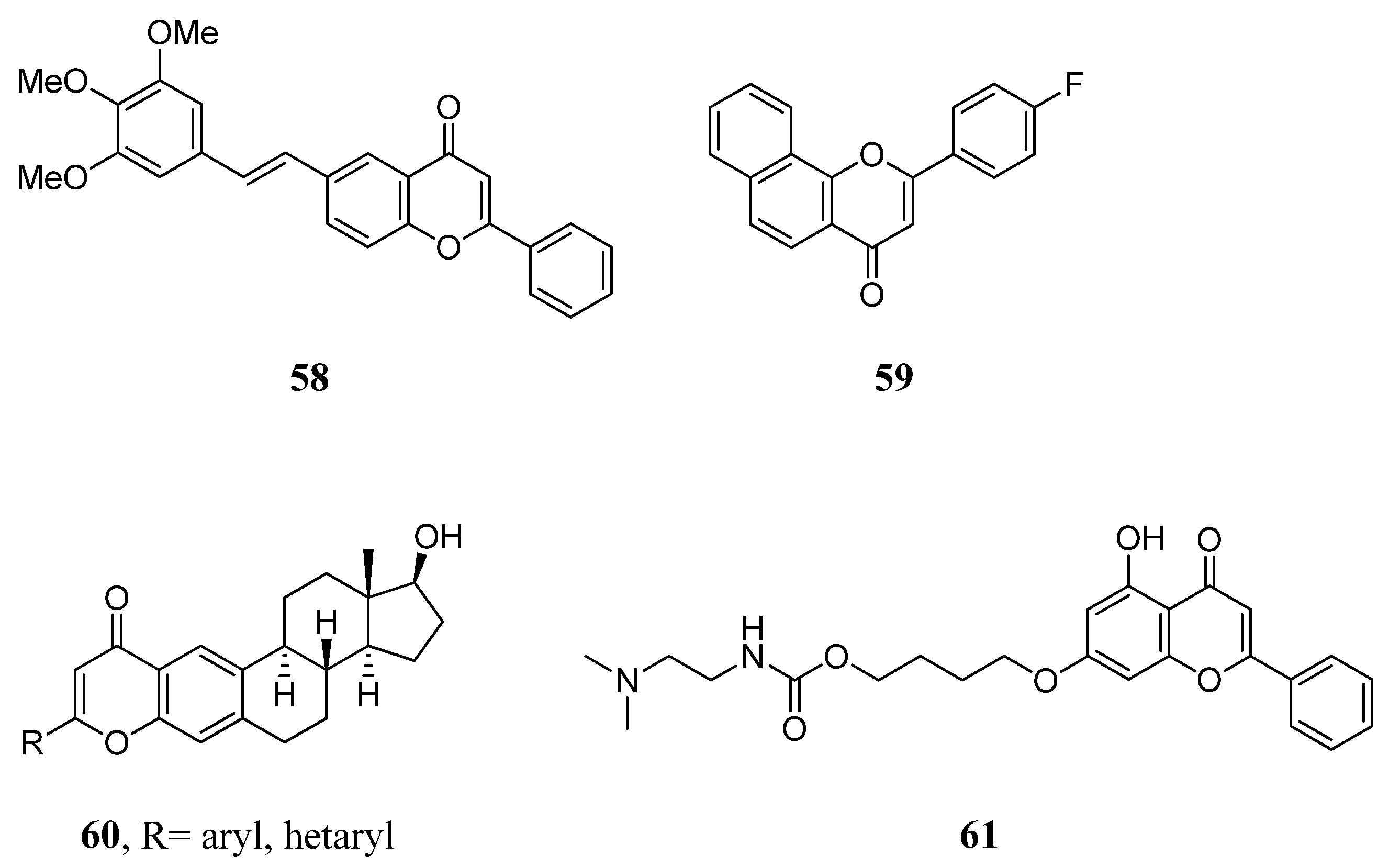
2.9. Miscellaneous
3. Scope and Limitations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beecher, G.R. Overview of dietary flavonoids: Nomenclature, occurrence and intake. J. Nutr. 2003, 133, 3248–3254. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising anticancer agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, S.H.; Nafees, S.; Zafaryab, M.; Khan, M.A.; Rizvi, M.M.A. Chrysin: A Promising Anticancer Agent its Current Trends and Future Perspectives. Eur. J. Exp. Biol. 2018, 8, 3–16. [Google Scholar] [CrossRef]
- Mei, Q.; Wang, C.; Yuan, W.; Zhang, G. Selective methylation of kaempferol via benzylation and deacetylation of kaempferol acetates. Beilstein J. Org. Chem. 2015, 11, 188–193. [Google Scholar] [CrossRef]
- Murakami, A.; Ashida, H.; Terao, J. Multitargeted cancer prevention by quercetin. Cancer Lett. 2008, 269, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Pawelczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Molecular consortia—Various structural and synthetic concepts for more effective therapeutics synthesis. Int. J. Mol. Sci. 2018, 19, 1104. [Google Scholar] [CrossRef]
- Spatafora, C.; Tringali, C. Natural-derived Polyphenols as Potential Anticancer Agents. Anti-Cancer Agents Med. Chem. 2012, 12, 902–918. [Google Scholar] [CrossRef]
- Singh, H.; Singh, J.V.; Kaur, N.; Sanduja, M.; Singh, G.; Singh, B.; Preet, M.; Sharma, S. Rational Approaches, Design Strategies, Structure Activity Relationship and Mechanistic Insights for Esterase Inhibitors. Mini-Rev. Med. Chem. 2018, 18, 837–894. [Google Scholar] [CrossRef]
- Mayer, S.; Keglevich, A.; Für Sepsey, C.; Bölcskei, H.; Ilkei, V.; Keglevich, P.; Hazai, L. Results in Chemistry of Natural Organic Compounds. Synthesis of New Anticancer Vinca Alkaloids and Flavone Alkaloids. Chemistry 2020, 2, 46. [Google Scholar] [CrossRef]
- Mayer, S.; Nagy, N.; Keglevich, P.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Hazai, L. Synthesis of Novel Vindoline-Chrysin Hybrids. Chem. Biodivers. 2021, 18, e202100725. [Google Scholar] [CrossRef]
- Pereira, D.; Pinto, M.; Correia-da-Silva, M.; Cidade, H. Recent Advances in Bioactive Flavonoid Hybrids Linked by 1,2,3-Triazole Ring Obtained by Click Chemistry. Molecules 2021, 27, 230. [Google Scholar] [CrossRef]
- Li, X.; Cai, Y.; Yang, F.; Meng, Q. Synthesis and molecular docking studies of chrysin derivatives as antibacterial agents. Med. Chem. Res. 2017, 26, 2225–2234. [Google Scholar] [CrossRef]
- Németh-Rieder, A.; Keglevich, P.; Hunyadi, A.; Latif, A.D.; Zupkó, I.; Hazai, L. Synthesis and In Vitro Anticancer Evaluation of Flavone-1,2,3-Triazole Hybrids. Molecules 2023, 28, 626. [Google Scholar] [CrossRef] [PubMed]
- Luan, T.; Quan, Z.; Fang, Y.; Yang, H. Design, Synthesis and Antiproliferative Activity of Chrysin Derivatives Bearing Triazole Moieties. Chin. J. Org. Chem. 2020, 40, 440–446. [Google Scholar] [CrossRef]
- Sowjanya, T.; Rao, Y.J.; Murthy, N.Y.S. Synthesis and Antiproliferative Activity of New 1,2,3-Triazole/Flavone Hybrid Heterocycles Against Human Cancer Cell Lines. Russ. J. Gen. Chem. 2017, 87, 1864–1871. [Google Scholar] [CrossRef]
- Kant, R.; Kumar, D.; Agarwal, D.; Gupta, R.D.; Tilak, R.; Awasthi, S.K.; Agarwal, A. Synthesis of newer 1,2,3-triazole linked chalcone and flavone hybrid compounds and evaluation of their antimicrobial and cytotoxic activities. Eur. J. Med. Chem. 2016, 113, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Mattarei, A.; Biasutto, L.; Rastrelli, F.; Garbisa, S.; Marotta, E.; Zoratti, M.; Paradisi, C. Regioselective O-derivatization of quercetin via ester intermediates. An improved synthesis of Rhamnetin and development of a new mitochondriatropic derivative. Molecules 2010, 15, 4722–4736. [Google Scholar] [CrossRef] [PubMed]
- Mattarei, A.; Sassi, N.; Durante, C.; Biasutto, L.; Sandoná, G.; Marotta, E.; Garbisa, S.; Gennaro, A.; Paradisi, C.; Zoratti, M. Redox properties and cytotoxicity of synthetic isomeric mitochondriatropic derivatives of the natural polyphenol quercetin. Eur. J. Org. Chem. 2011, 2011, 5577–5586. [Google Scholar] [CrossRef]
- Song, X.; Liu, Y.; He, J.; Zheng, X.; Lei, X.; Jiang, G.; Zjan, L. Synthesis of novel amino acid derivatives containing chrysin as anti-tumor agents against human gastric carcinoma MGC-803 cells. Med. Chem. Res. 2015, 24, 1789–1798. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Li, Y.; Liu, R.-F.; Xiao, J.; Zhou, B.-N.; Zhang, Q.-Z.; Song, J.-X. Synthesis, characterization and preliminary biological evaluation of chrysin amino acid derivatives that induce apoptosis and EGFR downregulation. J. Asian Nat. Prod. Res. 2021, 23, 39–54. [Google Scholar] [CrossRef]
- Kim, M.K.; Choo, H.; Chong, Y. Water-Soluble and Cleavable Quercetin−Amino Acid Conjugates as Safe Modulators for P-Glycoprotein-Based Multidrug Resistance. J. Med. Chem. 2014, 57, 7216–7233. [Google Scholar] [CrossRef]
- Pajtás, D.; Kónya, K.; Kiss-Szikszai, A.; Dzubák, B.; Pethő, Z.; Varga, Z.; Panyi, G.; Patonay, T. Optimization of the Synthesis of Flavone−Amino Acid and Flavone−Dipeptide Hybrids via Buchwald−Hartwig Reaction. J. Org. Chem. 2017, 82, 4578–4587. [Google Scholar] [CrossRef]
- Babu, K.S.; Babu, T.H.; Srinivas, P.V.; Kishore, K.H.; Murthy, U.S.N.; Rao, J.M. Synthesis and biological evaluation of novel C(7) modified chrysin analogues as antibacterial agents. Bioorg. Med. Chem. Lett. 2006, 16, 221–224. [Google Scholar] [CrossRef]
- Patel, R.V.; Mistry, B.M.; Syed, R.; Parekh, N.M.; Shin, H.-S. Sulfonylpiperazines based on a flavone as antioxidant and cytotoxic agents. Arch. Pharm. Chem. Life Sci. 2019, 352, e1900051. [Google Scholar] [CrossRef]
- Long, H.; Hu, X.; Wang, B.; Wang, Q.; Wang, R.; Liu, S.; Xiong, F.; Jiang, Z.; Zhang, X.-Q.; Ye, W.-C.; et al. Discovery of Novel Apigenin−Piperazine Hybrids as Potent and Selective Poly (ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors for the Treatment of Cancer. J. Med. Chem. 2021, 64, 12089–12108. [Google Scholar] [CrossRef]
- Raiguru, B.P.; Panda, J.; Mohapatra, S.; Nayak, S. Palladium-catalyzed facile synthesis of imidazo [1,2-a]pyridine-flavone hybrids and evaluation of their antiplasmodial activity. J. Mol. Struct. 2023, 1294, 136282. [Google Scholar] [CrossRef]
- Sivakumar, K.K.; Kumar, A.N.; Munirathinam, M.; Ponnilavarasan, I.; Preedeep, A.; Priya, A.S.M. Design, conventional and microwave assisted synthesis of molecular hybrids of thiazol-2-amine containing flavone/chalcone moieties possessing antimicrobial properties. Eur. J. Biomed. Pharm. Sci. 2018, 5, 246–256. [Google Scholar]
- Tangeti, V.S.; Vasundhara, D.; Satyanarayana, K.V.V.V.; Kumar, K.S.P. Synthesis and Antiproliferative Activity of Some Dihydro-1H-furo [2,3-c]pyrazole-Flavone Hybrids. Asian J. Chem. 2017, 29, 1525–1532. [Google Scholar] [CrossRef]
- Remya, R.S.; Ramalakshmi, N.; Nalini, C.N.; Amuthalakshmi, S. Design, synthesis, and in vitro evaluation of novel acridine derivatives as monoamine oxidase inhibitors. Rasayan J. Chem. 2022, 15, 2318–2325. [Google Scholar] [CrossRef]
- Liao, S.; Deng, H.; Huang, S.; Yang, J.; Wang, S.; Yin, B.; Zheng, T.; Zhang, D.; Liu, J.; Gao, G.; et al. Design, synthesis and evaluation of novel 5,6,7-trimethoxyflavone–6-chlorotacrine hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015, 25, 1541–1545. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Yoo, S.Y.; Lee, K.W.; Yoon, Y.M.; Ryu, H.W.; Jeong, Y.; Shin, J.-S.; Kang, S.-Y.; Kim, S.-Y.; Lee, H.-H.; et al. Repurposing mosloflavone/5,6,7-trimethoxyflavone-resveratrol hybrids: Discovery of novel p38-α MAPK inhibitors as potent interceptors of macrophage-dependent production of proinflammatory mediators. Eur. J. Med. Chem. 2019, 180, 253–267. [Google Scholar] [CrossRef]
- Singh, H.; Sharma, S.; Ojha, R.; Gupta, M.K.; Nepali, K.; Bedi, P.M.S. Synthesis and evaluation of naphthoflavones as a new class of non purine xanthine oxidase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4192–4197. [Google Scholar] [CrossRef]
- Molnár, B.; Gopisetty, M.K.; Nagy, F.I.; Adamecz, D.I.; Kása, Z.; Kiricsi, M.; Frank, É. Efficient access to domain-integrated estradiol-flavone hybrids via the corresponding chalcones and their in vitro anticancer potential. Steroids 2022, 187, 109099. [Google Scholar] [CrossRef]
- Lv, X.H.; Liu, H.; Ren, Z.L.; Wang, W.; Tang, F.; Cao, H.Q. Design, synthesis and biological evaluation of novel flavone Mannich base derivatives as potential antibacterial agents. Mol. Divers. 2019, 23, 299–306. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click Chemistry: 1,2,3-Triazoles as Pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Rao, Y.J.; Sowjanya, T.; Thirupathi, G.; Murthy, N.Y.S.; Kotapalli, S.S. Synthesis and biological evaluation of novel flavone/triazole/benzimidazole hybrids and flavone/isoxazole-annulated heterocycles as antiproliferative and antimycobacterial agents. Mol. Divers. 2018, 22, 803–814. [Google Scholar] [CrossRef]
- Qi, Y.; Ding, Z.; Yao, Y.; Ma, D.; Ren, F.; Yang, H.; Chen, A. Novel triazole analogs of apigenin-7-methyl ether exhibit potent antitumor activity against ovarian carcinoma cells via the induction of mitochondrial-mediated apoptosis. Exp. Ther. Med. 2019, 17, 1670–1676. [Google Scholar] [CrossRef]
- Thillainayagam, M.; Malathi, K.; Ramaiah, S. In-Silico molecular docking and simulation studies on novel chalcone and flavone hybrid derivatives with 1, 2, 3—Triazole linkage as vital inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. J. Biomol. Struct. Dyn. 2018, 36, 3993–4009. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.-W.; Qu, L.-B.; Chen, X.-L.; Qu, Z.-B.; Liu, X.-Q.; Ke, D.-D. An Efficient Synthesis of Mono and Bis-1,2,3-triazole AZT Derivatives via Copper(I)-catalyzed Cycloaddition. J. Chin. Chem. Soc. 2011, 58, 24–30. [Google Scholar] [CrossRef]
- Panova, N.A.; Shcherbakov, K.V.; Zhilina, E.F.; Burgart, Y.V.; Saloutin, V.I. Synthesis of Mono- and Polyazole Hybrids Based on Polyfluoroflavones. Molecules 2023, 28, 869. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Xiao, Y.; Fu, B.; Qin, Z. TPP-based mitocans: A potent strategy for anticancer drug design. RSC Med. Chem. 2020, 11, 858–875. [Google Scholar] [CrossRef]
- Gardner, Z.S.; Schumacher, T.J.; Ronayne, C.T.; Kumpati, G.P.; Williams, M.J.; Yoshimura, A.; Mereddy, V.R. Synthesis and biological evaluation of novel 2-alkoxycarbonylallylester phosphonium derivatives as potential anticancer agents. Bioorg. Med. Chem. Lett. 2021, 45, 128136. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod. 2017, 80, 2232–2239. [Google Scholar] [CrossRef]
- Biasutto, L.; Mattarei, A.; Paradisi, C. Synthesis and testing of novel isomeric mitochondriatropic derivatives of resveratrol and quercetin. In Mitochondrial Medicine, Volume II, Manipulating Mitochondrial Function; Methods in Molecular Biology, Volume 1265; Weissig, W., Edeas, M., Eds.; Springer: New York, NY, USA, 2015; Chapter 13; pp. 161–179. [Google Scholar] [CrossRef]
- Xing, L.; Lyu, J.-Y.; Yang, Y.; Cui, P.-F.; Gu, L.-Q.; Qiao, J.-B.; He, Y.-J.; Zhang, T.-Q.; Sun, M.; Lu, J.-J.; et al. pH-Responsive de-PEGylated nanoparticles based on triphenylphosphine-quercetin self-assemblies for mitochondria-targeted cancer therapy. Chem. Commun. 2017, 53, 8790–8793. [Google Scholar] [CrossRef]
- Veselovskaya, M.V.; Garazd, M.M.; Ogorodniichuk, A.S.; Garazd, Y.L.; Khilya, V.P. Synthesis of amino-acid derivatives of chrysin. Chem. Nat. Compd. 2008, 44, 704–711. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Hr, J.; Yu, W.; Xiao, J.; Guo, Y.; Zhu, X.; Liu, Y. Synthesis and biological evaluation of amino acid derivatives containing chrysin that induce apoptosis. Nat. Prod. Res. 2019, 35, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.-Y.; Tsai, M.-S.; Hsu, L.-C.; Lin, S.-W.; Liang, P.-H. Traversal of the Blood−Brain Barrier by Cleavable l-Lysine Conjugates of Apigenin. J. Agric. Food Chem. 2018, 66, 8124–8131. [Google Scholar] [CrossRef] [PubMed]
- Beutner, S.; Frixel, S.; Ernst, H.; Hoffmann, T.; Hernandez-Blanco, I.; Hundsdoerfer, C.; Kiesendahl, K.; Kock, S.; Martin, H.-D.; Mayer, B.; et al. Carotenylflavonoids, a novel group of potent, dual-functional antioxidants. Arkivoc 2007, 8, 279–295. [Google Scholar] [CrossRef]
- Hundsdörfer, C.; Stahl, W.; Müller, T.J.J.; De Spirt, S. UVA Photoprotective Properties of an Artificial Carotenylflavonoid Hybrid Molecule. Chem. Res. Toxicol. 2012, 25, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- DhunMati, K.; Nalin, C.N.; Ramalakshmi, N.; Niraimathi, V.; Amuthalakshmi, S. Synthesis, Molecular Docking and Invitro Evaluation of Nipecotic Acid-Flavone Hybrids as Anti Alzheimer Agents—A Multi Target Directed Ligand Approach. J. Pharm. Neg. Res. 2022, 13, 229–238. [Google Scholar]
- Zhang, Y.; Xu, Z.; Zhan, L.; Gao, Y.; Zheng, B.; Zhou, Y.; Sheng, Y.; Liang, G.; Song, Z. Design, synthesis and biological evaluation of novel chromone-maleimide hybrids as potent anti-inflammatory agents against LPS-induced acute lung injury. Bioorg. Chem. 2022, 128, 160049. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Verma, N.; Kumar, N.; Patel, A.; Roy, P.; Pruthi, V.; Ahmed, N. Design, Synthesis, Molecular Docking and Biological Studies of Novel Phytoestrogen-Tanaproget Hybrids. Synth. Commun. 2016, 46, 460–474. [Google Scholar] [CrossRef]
- Abu-Aisheh, M.N.; Mustafa, M.S.; Mubarak, M.S.; El-Abadelah, M.M.; Voelter, W. Synthesis of Some 1-(Flavone-7-yl)-4,5-dihydro-1,2,4-triazin-6(1H)-ones and Related Congeners. Lett. Org. Chem. 2012, 9, 465–473. [Google Scholar] [CrossRef]
- Kamal, A.; Ramu, R.; Khanna, G.B.R.; Saxena, A.K.; ShanMugavel, M.; Pandita, R.M. Synthesis and evaluation of new pyrrolo [2,1-c][1,4]benzodiazepine hybrids linked to a flavone moiety. Arkivoc 2005, 2005, 83–91. [Google Scholar] [CrossRef]
- Franke, K.; Porzel, A.; Schmidt, J. Flavone-coumarin hybrids from Gnidia socotrana. Phytochemistry 2002, 61, 873–878. [Google Scholar] [CrossRef]
- Pan, G.; Zhao, L.; Xiao, N.; Yang, K.; Ma, Y.; Zhao, X.; Fan, Z.; Zhang, Y.; Yao, Q.; Yu, P. Total synthesis of 8-(6″-umbelliferyl)-apigenin and its analogs as anti-diabetic reagents. Eur. J. Med. Chem. 2016, 122, 674–683. [Google Scholar] [CrossRef]
- Rao, H.S.P.; Tangeti, V.S. Synthesis of 3-Aroylcoumarin-Flavone Hybrids. Lett. Org. Chem. 2012, 9, 218–220. [Google Scholar] [CrossRef]
- Quintin, J.; Roullier, C.; Thoret, S.; Lewin, G. Synthesis and anti-tubulin evaluation of chromone-based analogues of combretastatins. Tetrahedron 2006, 62, 4038–4051. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Lee, K.-T.; Lee, Y.S. Flavone-based arylamides as potential anticancers: Design, synthesis and in vitro cell-based/cell-free evaluations. Eur. J. Med. Chem. 2020, 187, 111965. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.; Keglevich, P.; Ábrányi-Balogh, P.; Szigetvári, Á.; Dékány, M.; Szántay, C., Jr.; Hazai, L. Synthesis and in vitro evaluation of novel chrysin and 7-aminochrysin derivatives. Molecules 2020, 25, 888. [Google Scholar] [CrossRef] [PubMed]
- Leonte, D.; Ungureanu, D.; Zaharia, V. Flavones and Related Compounds: Synthesis and Biological Activity. Molecules 2023, 28, 6528. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Choi, E.; Yoon, Y.M.; Lee, K.W.; Yoo, S.Y.; Cho, M.C.; Yang, J.S.; Kim, H.I.; Hong, J.Y.; Shin, J.-S.; et al. Natural products hybrids: 3,5,4′-Trimethoxystilbene-5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur. J. Med. Chem. 2019, 161, 559–580. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Malhotra, R. Synthesis and antimicrobial activity of thiosemicarbazide induced hydrazone of 4-oxo-4H-chromene-3-carbaldehyde. AIP Conf. Proc. 2017, 1860, 020064. [Google Scholar] [CrossRef]
- Wei, M.; Xie, M.; Zhang, Z.; Wei, Y.; Zhang, J.; Pan, H.; Li, B.; Wang, J.; Song, Y.; Chong, C.; et al. Design and synthesis of novel flavone-based histone deacetylase inhibitors antagonizing activation of STAT3 in breast cancer. Eur. J. Med. Chem. 2020, 206, 112677. [Google Scholar] [CrossRef] [PubMed]
- Raiguru, B.P.; Mohapatra, S.; Nayak, S.; Sahal, D.; Yadav, M.; Acharya, B.N. Flavone-stilbene hybrids: Synthesis and evaluation as potential antimalarial agents. Eur. J. Med. Chem. Rep. 2022, 4, 100029. [Google Scholar] [CrossRef]
- Basha, S.J.; Mohan, P.; Yeggoni, D.P.; Babu, Z.R.; Kumar, P.B.; Rao, A.D.; Subramaynam, R.; Damu, A.G. New Flavone-Cyanoacetamide Hybrids with a Combination of Cholinergic, Antioxidant, Modulation of β-Amyloid Aggregation, and Neuroprotection Properties as Innovative Multifunctional Therapeutic Candidates for Alzheimer’s Disease and Unraveling Their Mechanism of Action with Acetylcholinesterase. Mol. Pharm. 2018, 15, 2206–2223. [Google Scholar] [CrossRef]
- Marc, M.A.; Kincses, A.; Rácz, B.; Nasim, M.J.; Sarfraz, M.; Lázaro-Milla, C.; Dominguez-Álvarez, E.; Jacob, C.; Spengler, G.; Almendros, P. Antimicrobial, Anticancer and Multidrug-Resistant Reversing Activity of Novel Oxygen-, Sulfur- and Selenoflavones and Bioisosteric Analogues. Pharmaceuticals 2020, 13, 453. [Google Scholar] [CrossRef]
- Kumar, D.; Singh, O.; Nepali, K.; Bedi, P.M.S.; Qayum, A.; Singh, S.; Jain, S.K. Naphthoflavones as antiproliferative agents: Design, synthesis and biological evaluation. Anti-Cancer Agents Med. Chem. 2016, 16, 881–890. [Google Scholar] [CrossRef]
- Samanta, R.; Narayan, R.; Antonchick, A.P. Rhodium(III)-Catalyzed Direct Oxidative Cross Coupling at the C5 Position of Chromones with Alkenes. Org. Lett. 2012, 23, 6108–6111. [Google Scholar] [CrossRef]
- Polbuppha, I.; Suthiphasipl, V.; Maneerat, T.; Charoensup, R.; Limtharakul, T.; Cheenpracha, S.; Pyne, S.G.; Laphookhieo, S. Desmoschinensisflavones A and B, two rare flavones having a hybrid benzyl benzoate esterflavone structural framework from Desmos chinensis Lour. RSC Adv. 2020, 10, 45076–45080. [Google Scholar] [CrossRef]
- Lu, C.; Huang, F.; Li, Z.; Ma, J.; Li, H.; Fang, L. Synthesis and Bioactivity of Quercetin Aspirinates. Bull. Korean Chem. Soc. 2014, 35, 518–520. [Google Scholar] [CrossRef]
- Jamali, Z.; Behbahani, G.R.R.; Zare, K.; Gheibi, N. Effect of chrysin omega—3 and 6 fatty acid esters on mushroom tyrosinase activity, stability, and structure. J. Food Biochem. 2018, 43, e12728. [Google Scholar] [CrossRef]
- Mayer, S.; Herr, D.M.; Nagy, N.; Donkó-Tóth, V.; Keglevich, P.; Weber, M.; Dékány, M.; Hazai, L. Synthesis and In Vitro Anticancer Evaluation of Chrysin Containing Hybrids and Other Chrysin Derivatives. Period. Polytech. Chem. Eng. 2023, 67, 316–336. [Google Scholar] [CrossRef]
- Jeon, K.-H.; Park, S.; Shin, J.-H.; Jung, A.-R.; Hwang, S.-Y.; Seo, S.H.; Jo, H.; Na, Y.; Kwon, Y. Synthesis and evaluation of 7-(3-aminopropyloxy)-substituted flavone analogue as a topoisomerase II catalytic inhibitor and its sensitizing effect to enzalutamide in castration-resistant prostate cancer cells. Eur. J. Med. Chem. 2023, 246, 114999. [Google Scholar] [CrossRef] [PubMed]
- Serafin, P.; Kleczkowska, P. Bombesins: A New Frontier in Hybrid Compound Development. Pharmaceutics 2023, 15, 2597. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.-H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef]




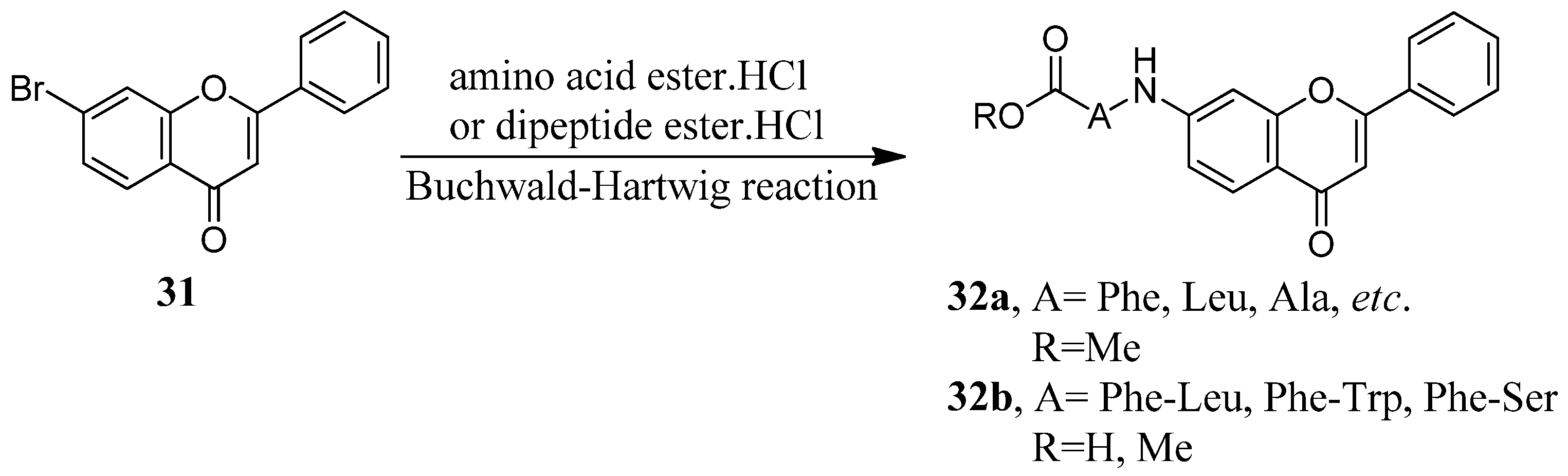




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
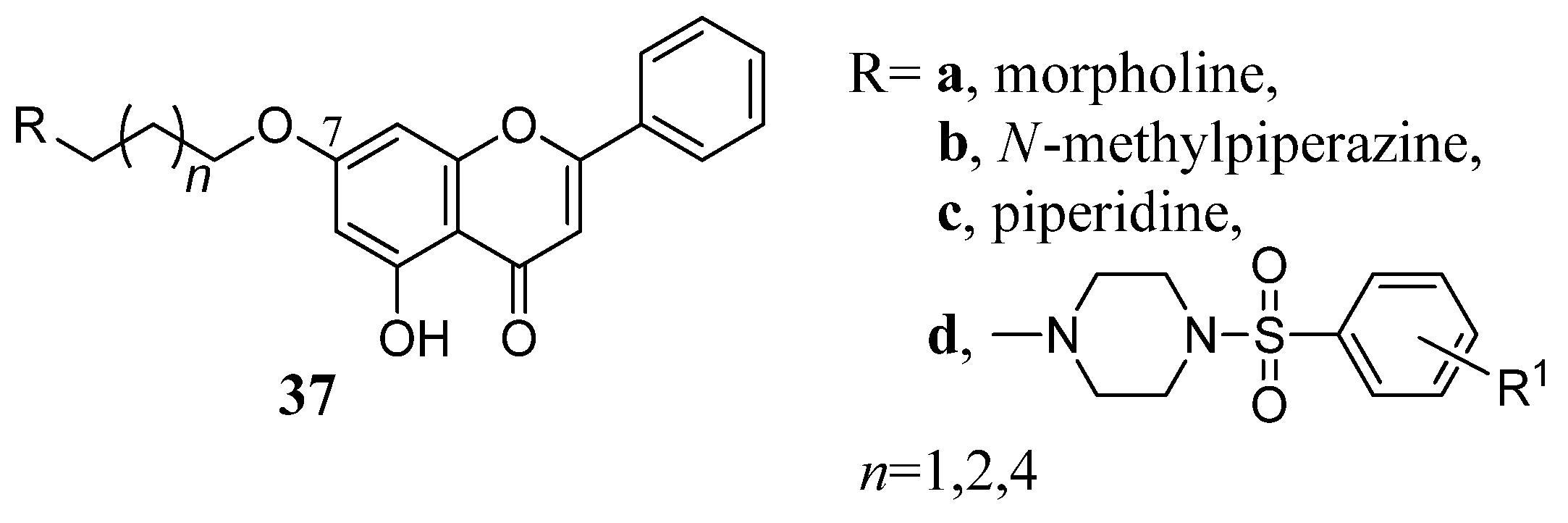{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavone | Pharmacophore | Biological Effect | Activity * | Control Activity | Section | Reference |
|---|---|---|---|---|---|---|
| chrysin | 1,2,3-triazoles | antibacterial | MIC = 6.25 µg/mL (E. coli) | penicillin, MIC = 50 µg/mL | 2.1. | [14] |
| chrysin | 1,2,3-triazoles | antiproliferative | IC50 = 0.733 µM (HeLa) | cisplatin, IC50 = 12.2 µM | 2.1. | [15,16] |
| 6-aminoflavone | 1,2,3-triazoles | antiproliferative | GI50 < 0.01 µM (MDA-MB-231) | paclitaxel, GI50 = 0.091 µM | 2.1. | [17] |
| luteolin | 1,2,3-triazoles | antiplasmodial | IC50 = 3.85 µM (P. falciparum 3D7) | artemisinin, IC50 = 1.12 µM | 2.1. | [18] |
| quercetin | triphenylphosphine | antioxidant | EC50 = 6.3 µM (DPPH assay) | quercetin, EC50 = 6.0 µM | 2.2. | [19,20] |
| chrysin | amino acids | antiproliferative | IC50 = 3.78 µM (MGC-803) | cisplatin, IC50 = 4.40 µM | 2.3. | [21,22] |
| quercetin | amino acids | MDR modulator | IC50 = 0.14 µM (MES-SA/Dx5) | doxorubicin, IC50 = 8.20 µM | 2.3. | [23] |
| 2-phenylchromen- 4-one | amino acids | antiproliferative | IC50 = 9.2 µM (CCRF-CEM) | n.d. | 2.3. | [24] |
| chrysin | vindoline | antiproliferative | GI50 = 1.1 µM (LOX IMVI) | n.d. | 2.4. | [12] |
| chrysin | morpholine | antibacterial | MIC = 6.25 µg/mL (B. sphaericus) | streptomycin, MIC = 12.5 µg/mL | 2.5. | [25] |
| chrysin | piperazines | antitumor | IC50 = 4.67 µM (HeLa) | gefitinib, IC50 = 17.9 µM | 2.5. | [26] |
| apigenin | piperazines | antitumor | IC50 = 0.0147 µM (PARP-1 inhibition) | olaparib, IC50 = 0.0051 µM | 2.5. | [27] |
| 2-phenylchromen- 4-one | imidazo[1,2-a] pyridine | antiplasmodial | IC50 = 1.98 µM (P. falciparum K1) | chloroquine, IC50 = 0.25 µM | 2.6. | [28] |
| 2-phenylchromen- 4-one | 2-amino-thiazole | antibacterial | MIC = 12.5 µg/mL (B. lintus) | ciprofloxacin, MIC < 1.5 µg/mL | 2.6. | [29] |
| 7-hydroxyflavone | dihydro-1H-furo[2,3-c]pyrazole | antitumor | IC50 = 2.5 µM (Hep2) | doxorubicin, IC50 = 10 µM | 2.6. | [30] |
| 7-hydroxyflavone | acridine | monoamine oxidase inhibitor | IC50 = 3.24 µM (MAO-B inhibition) | tacrine, IC50 = 7.85 µM | 2.6. | [31] |
| 5,6,7-trimethoxy- flavone | 6-chlorotacrine | anti-Alzheimer’s disease | IC50 = 0.0128 µM (AChE inhibition) | 6-chlorotacrine, IC50 = 0.0785 µM | 2.6. | [32] |
| chrysin | apigenin | antiproliferative | IC50 = 0.179 µM (PARP-1 inhibition) | olaparib, IC50 = 0.0051 µM | 2.7. | [27] |
| mosloflavone | resveratrol | anti-inflammatory | IC50 = 0.31 µM (PGE2 inhibition) | n.d. | 2.8. | [33] |
| 2-phenylchromen- 4-one | naphthopyran | xanthine oxidase inhibitor | IC50 = 0.62 µM (XO inhibition) | apigenin, IC50 = 1.11 µM | 2.9. | [34] |
| 2-phenylchromen- 4-one | estradiol | antiproliferative | IC50 = 3.9 µM (HeLa) | cisplatin, IC50 > 330 µM | 2.9. | [35] |
| kaempferol | carbohydrate | antibacterial | MIC = 0.05 µg/mL (S. gallinarum) | ciprofloxacin, MIC = 0.25 µg/mL | 2.9. | [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hazai, L.; Zsoldos, B.; Halmai, M.; Keglevich, P. Flavone Hybrids and Derivatives as Bioactive Agents. Appl. Sci. 2024, 14, 1039. https://doi.org/10.3390/app14031039
Hazai L, Zsoldos B, Halmai M, Keglevich P. Flavone Hybrids and Derivatives as Bioactive Agents. Applied Sciences. 2024; 14(3):1039. https://doi.org/10.3390/app14031039
Chicago/Turabian StyleHazai, László, Bernadett Zsoldos, Mónika Halmai, and Péter Keglevich. 2024. "Flavone Hybrids and Derivatives as Bioactive Agents" Applied Sciences 14, no. 3: 1039. https://doi.org/10.3390/app14031039
APA StyleHazai, L., Zsoldos, B., Halmai, M., & Keglevich, P. (2024). Flavone Hybrids and Derivatives as Bioactive Agents. Applied Sciences, 14(3), 1039. https://doi.org/10.3390/app14031039