
Grain Boundary—A Route to Enhance Electrocatalytic Activity for Hydrogen Evolution Reaction



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Compactional and Experimental Methods
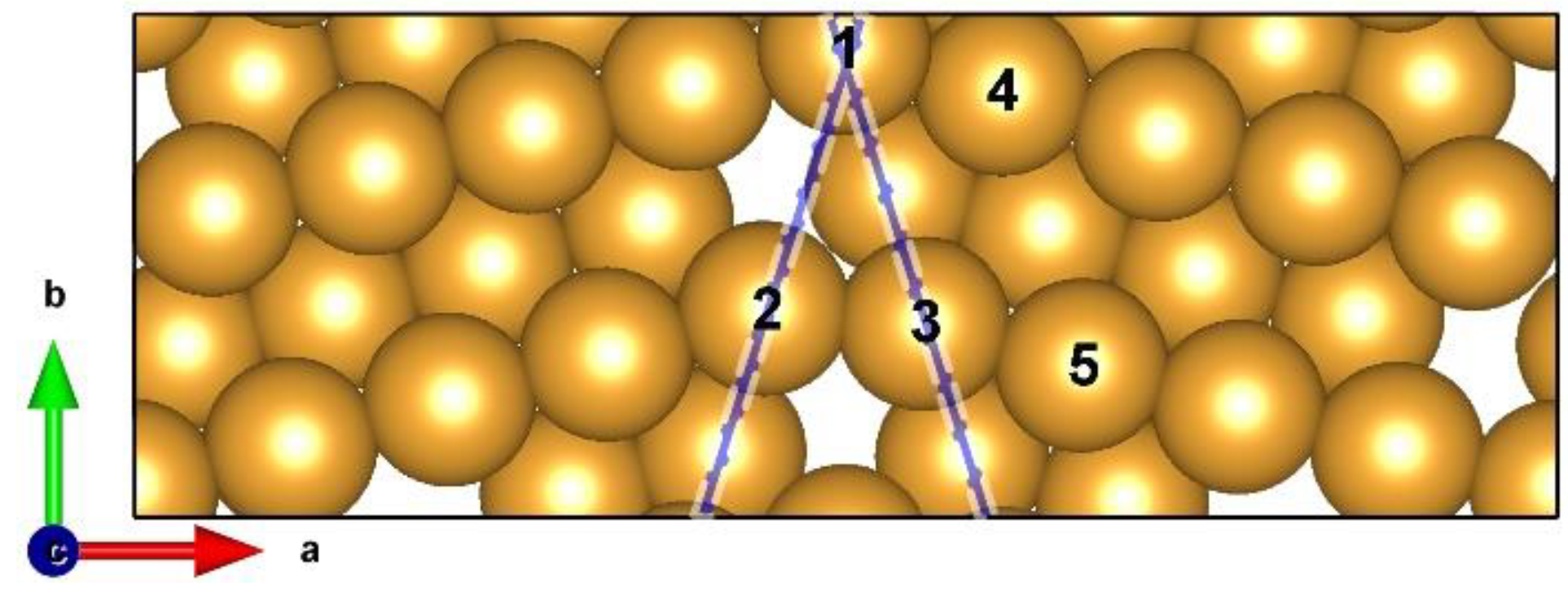
2.1. Computational Model and Method
2.2. Preparation of Au/CFP Electrodes
2.3. Electrochemical Characterization
3. Results and Discussion
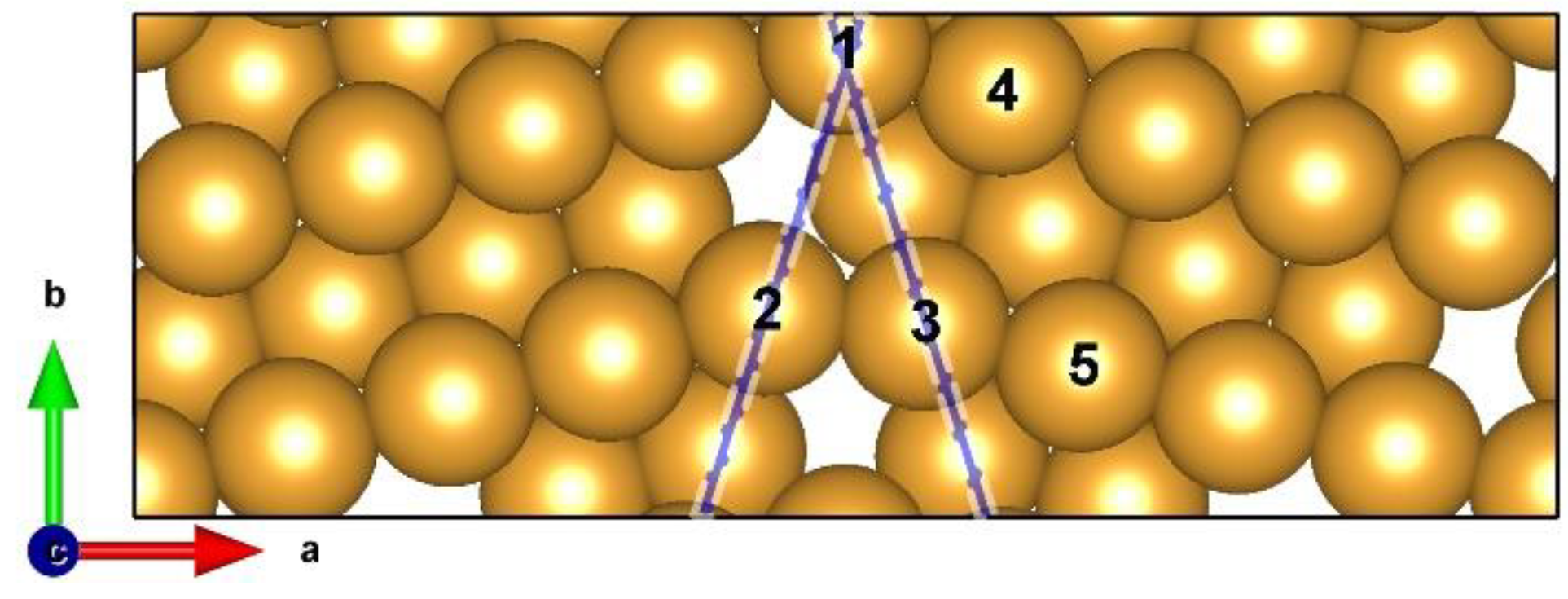
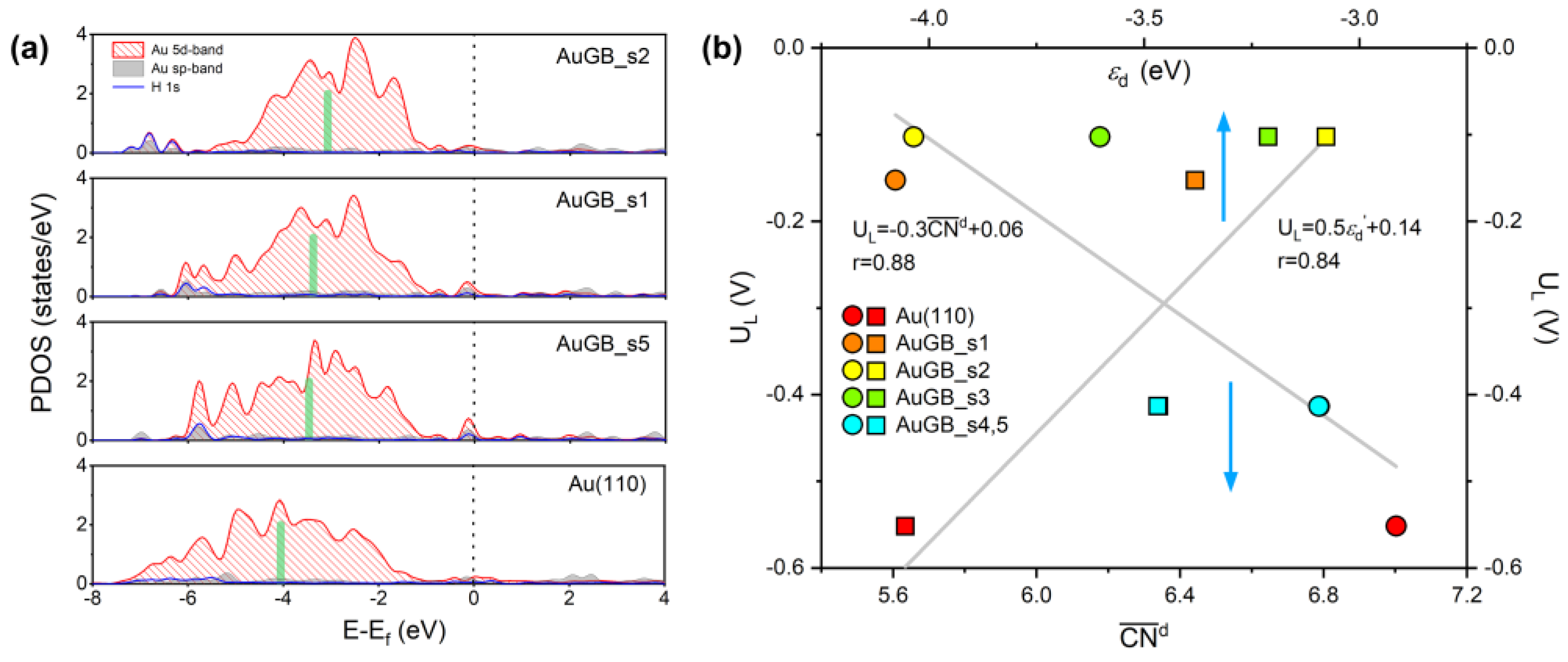
3.1. Theoretical Prediction of GB–Assisted HER
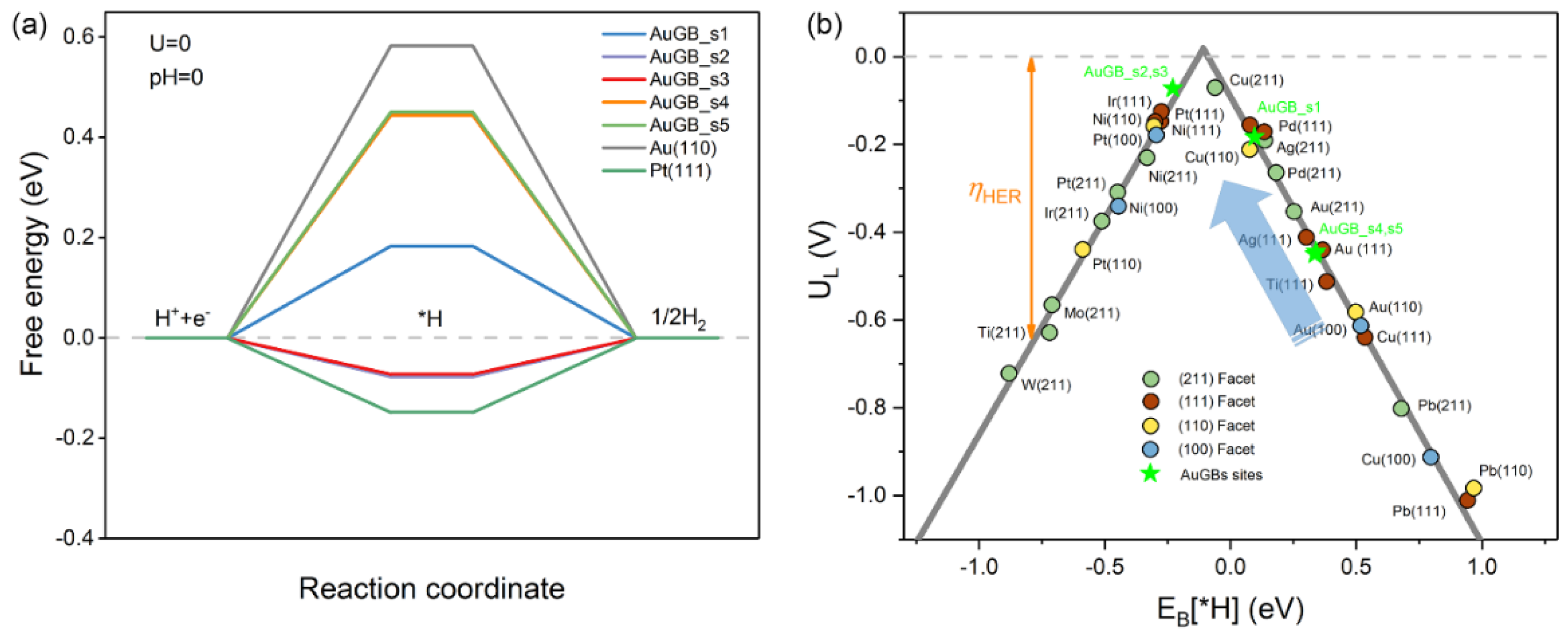
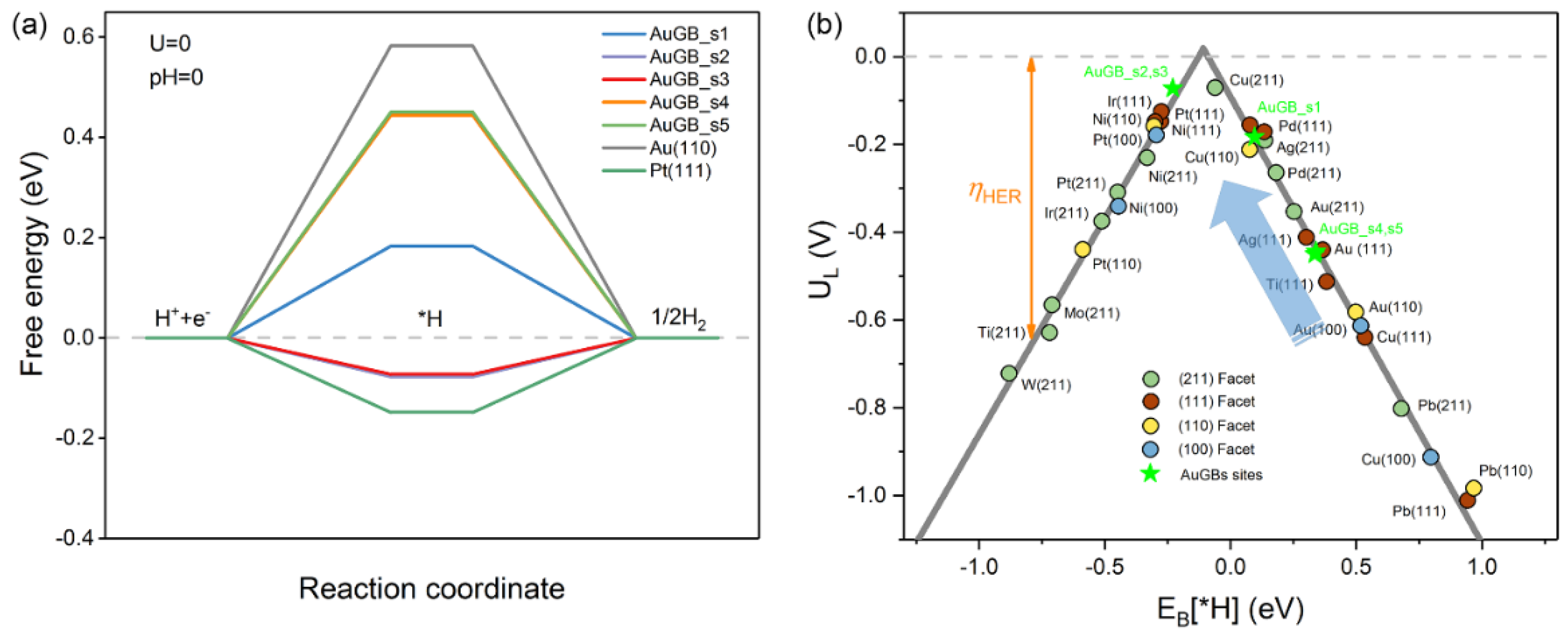
3.1.1. Gibbs Free Energy
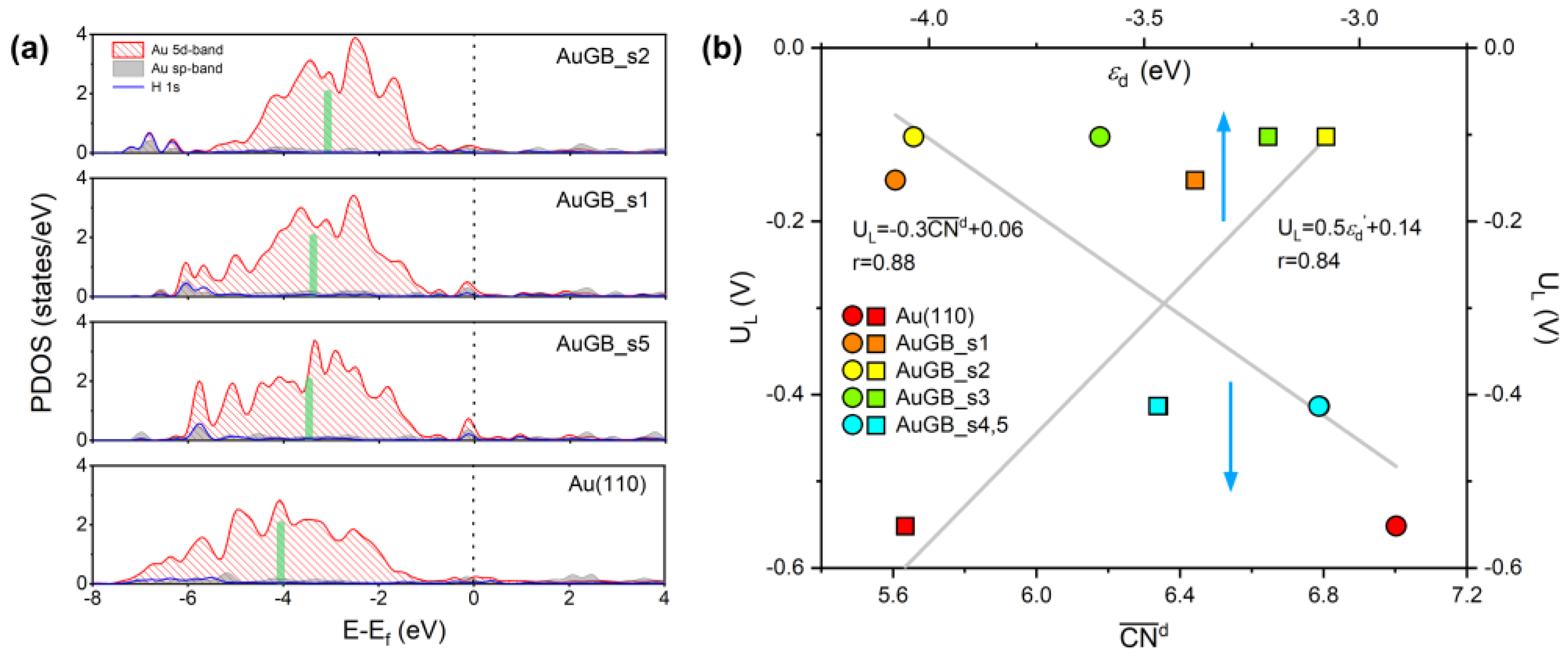
3.1.2. Limiting Potentials
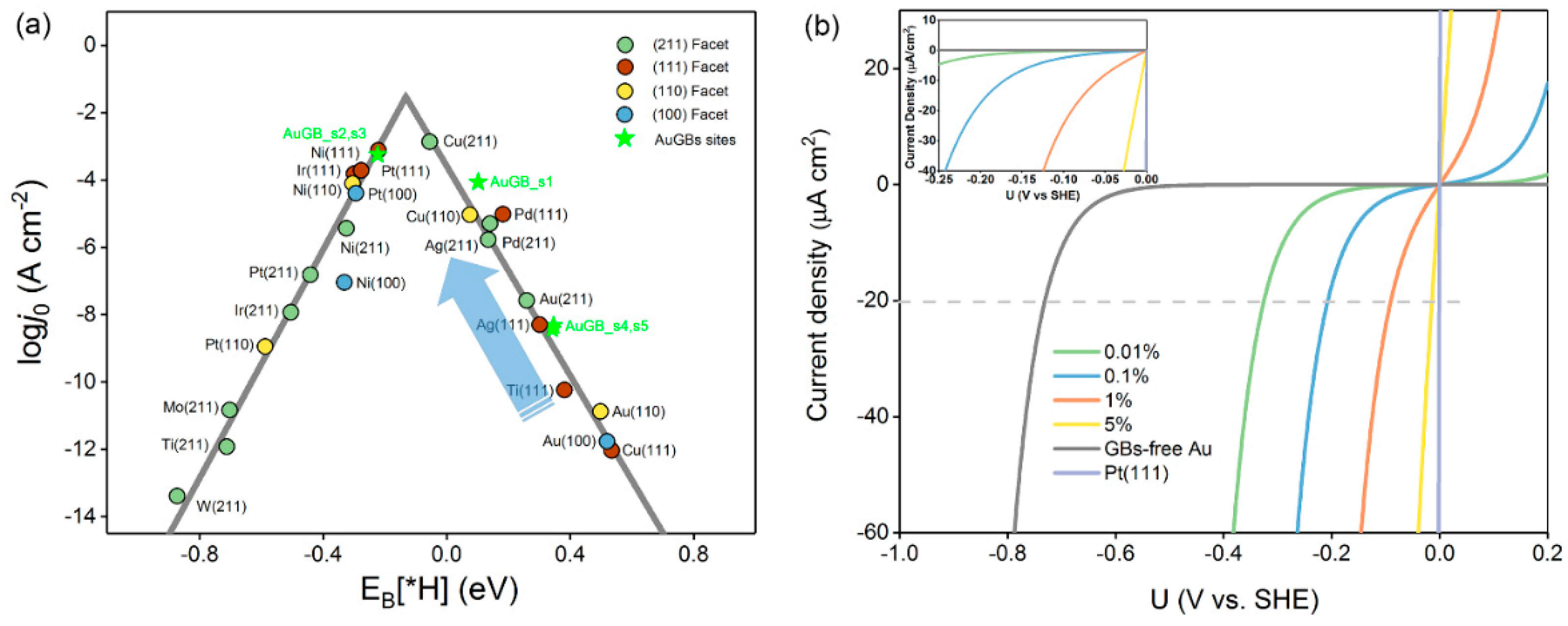
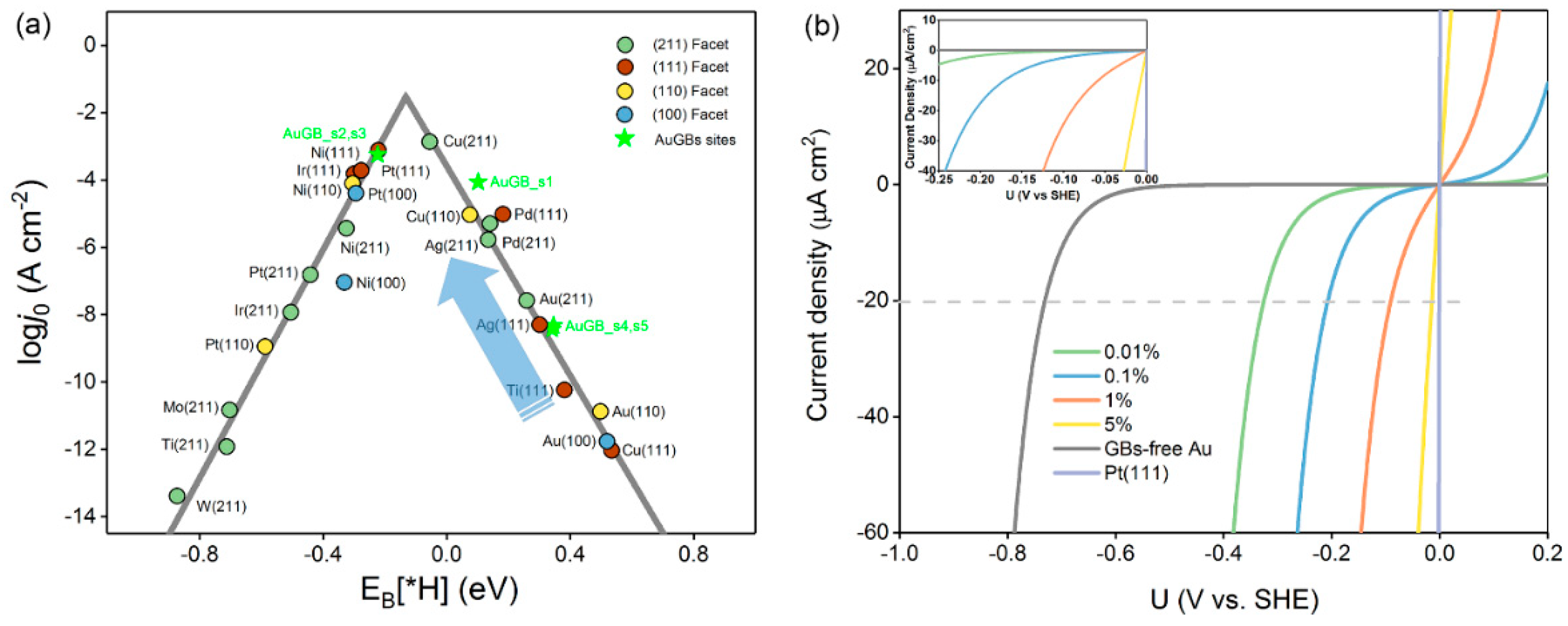
3.1.3. Exchange Current Density
3.1.4. GB–Mediated Current Density
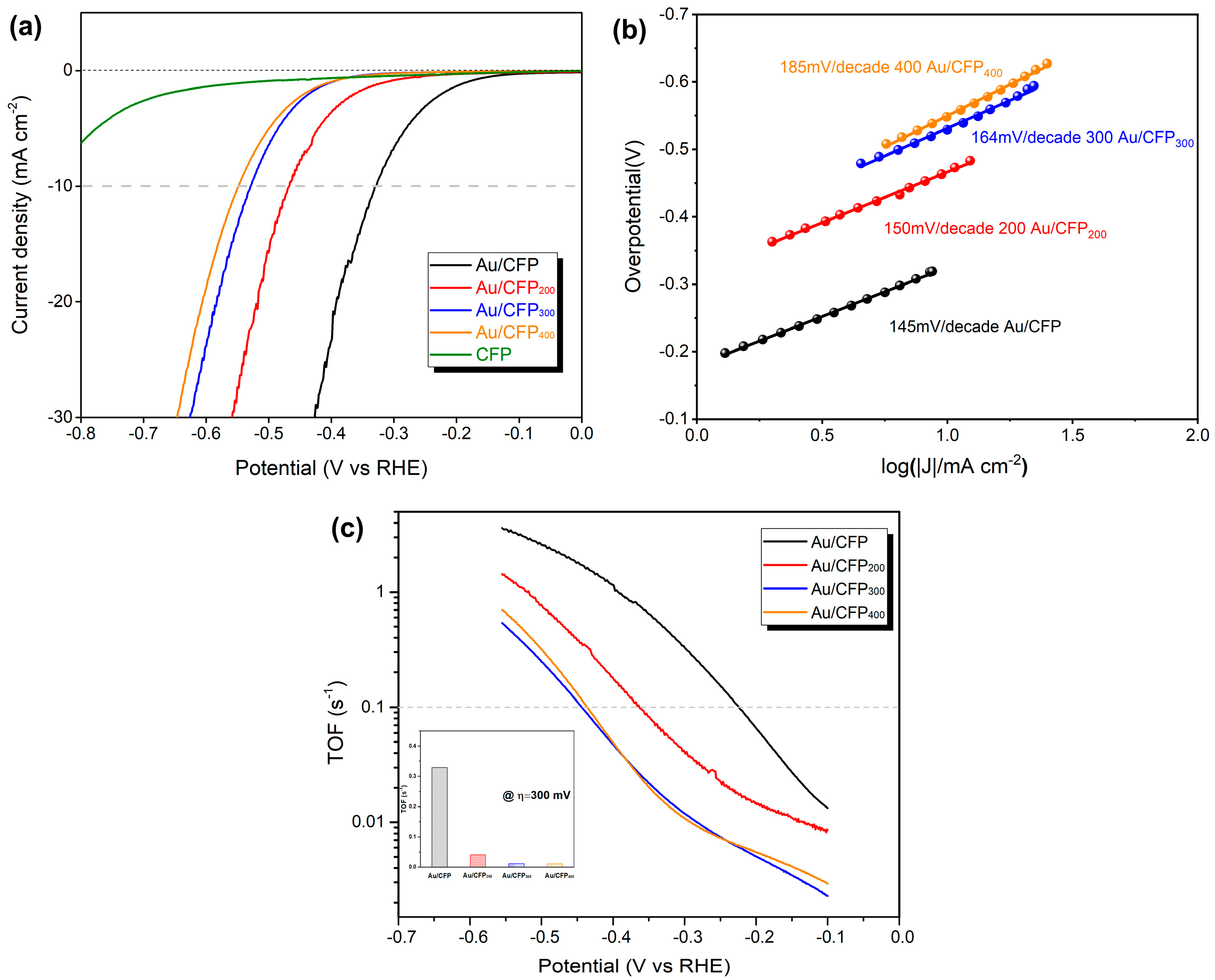
3.2. Experimental Verification of GB–Assisted HER Activity
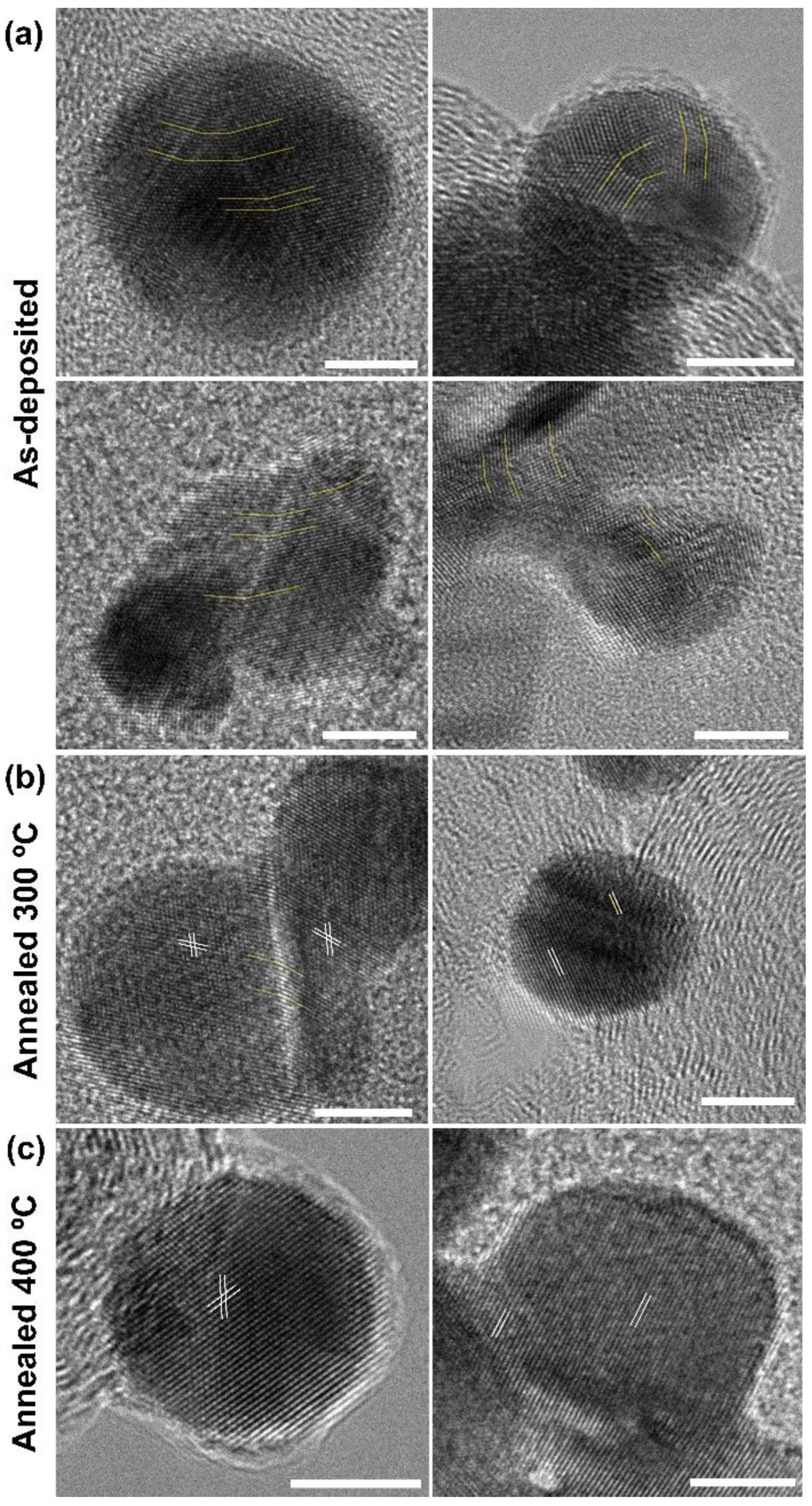
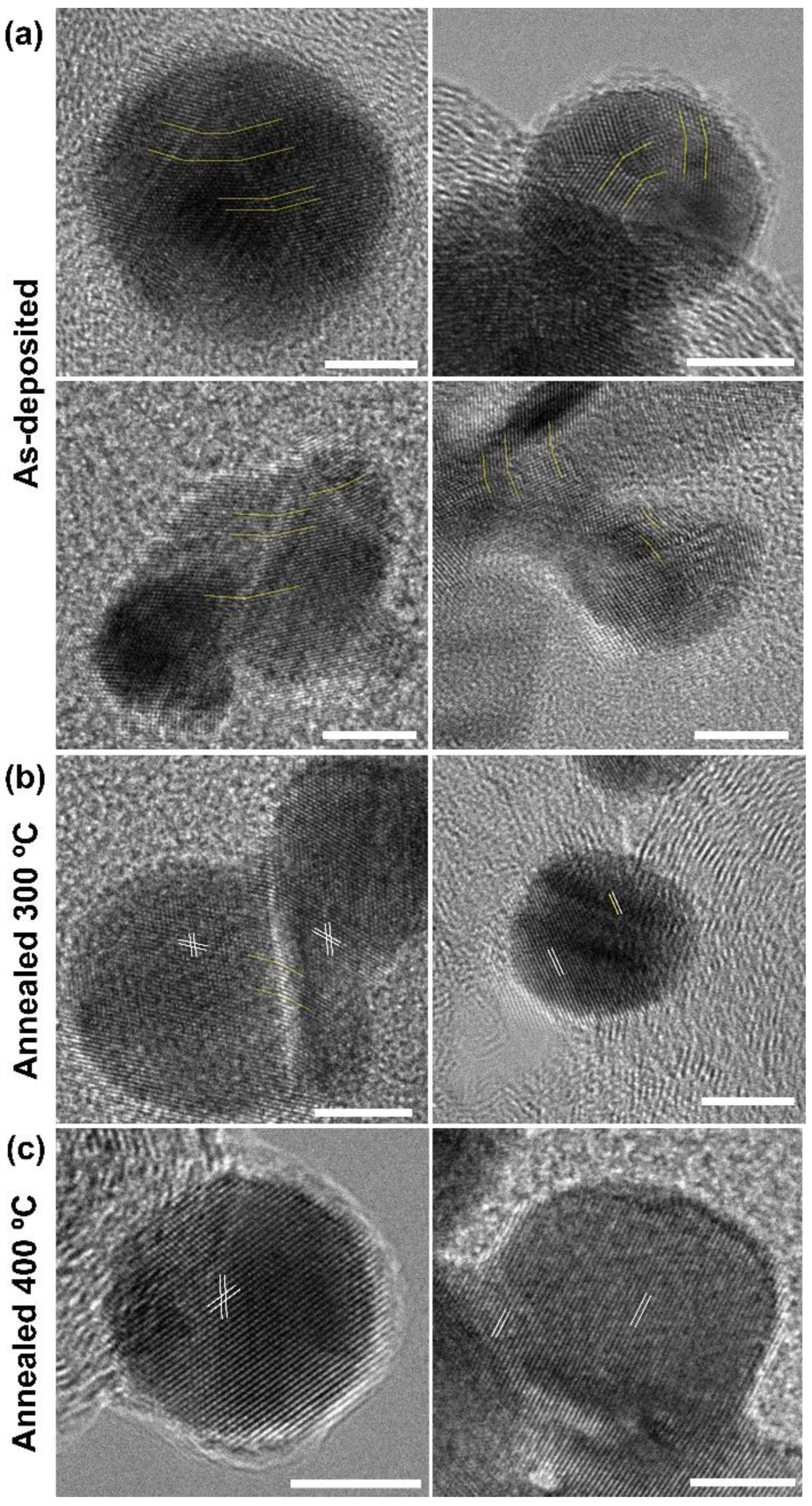
3.2.1. Exposed Surface Characterization
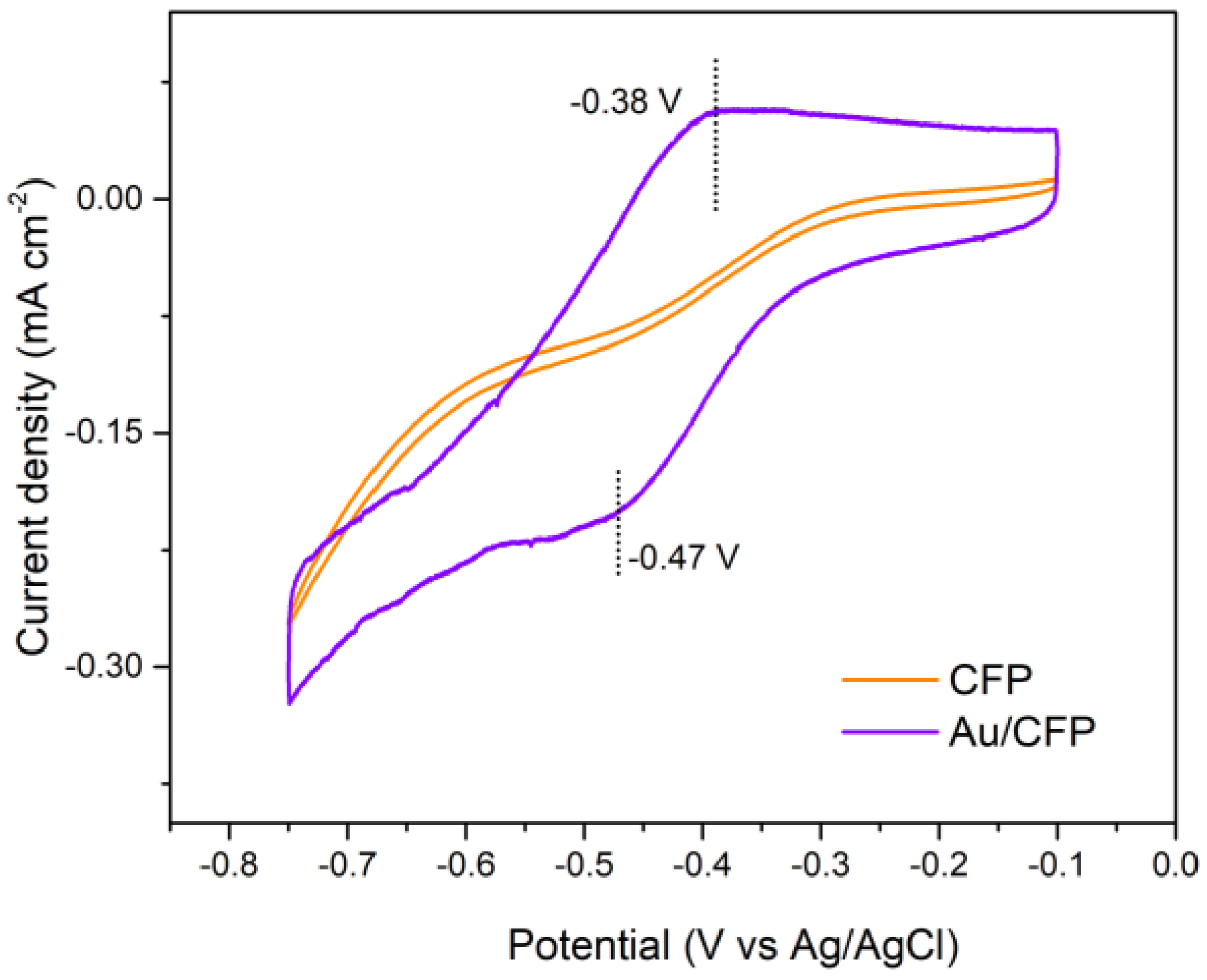
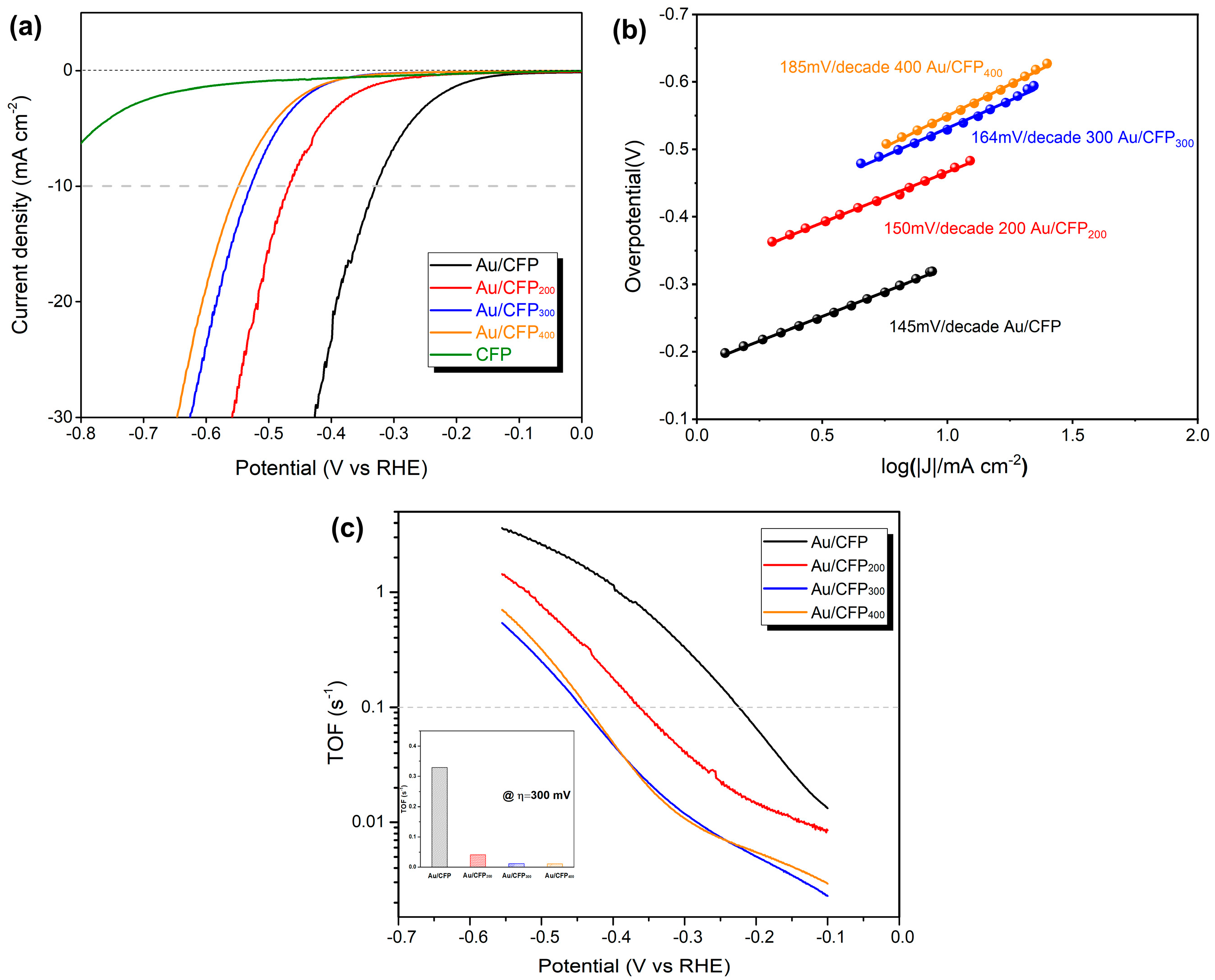
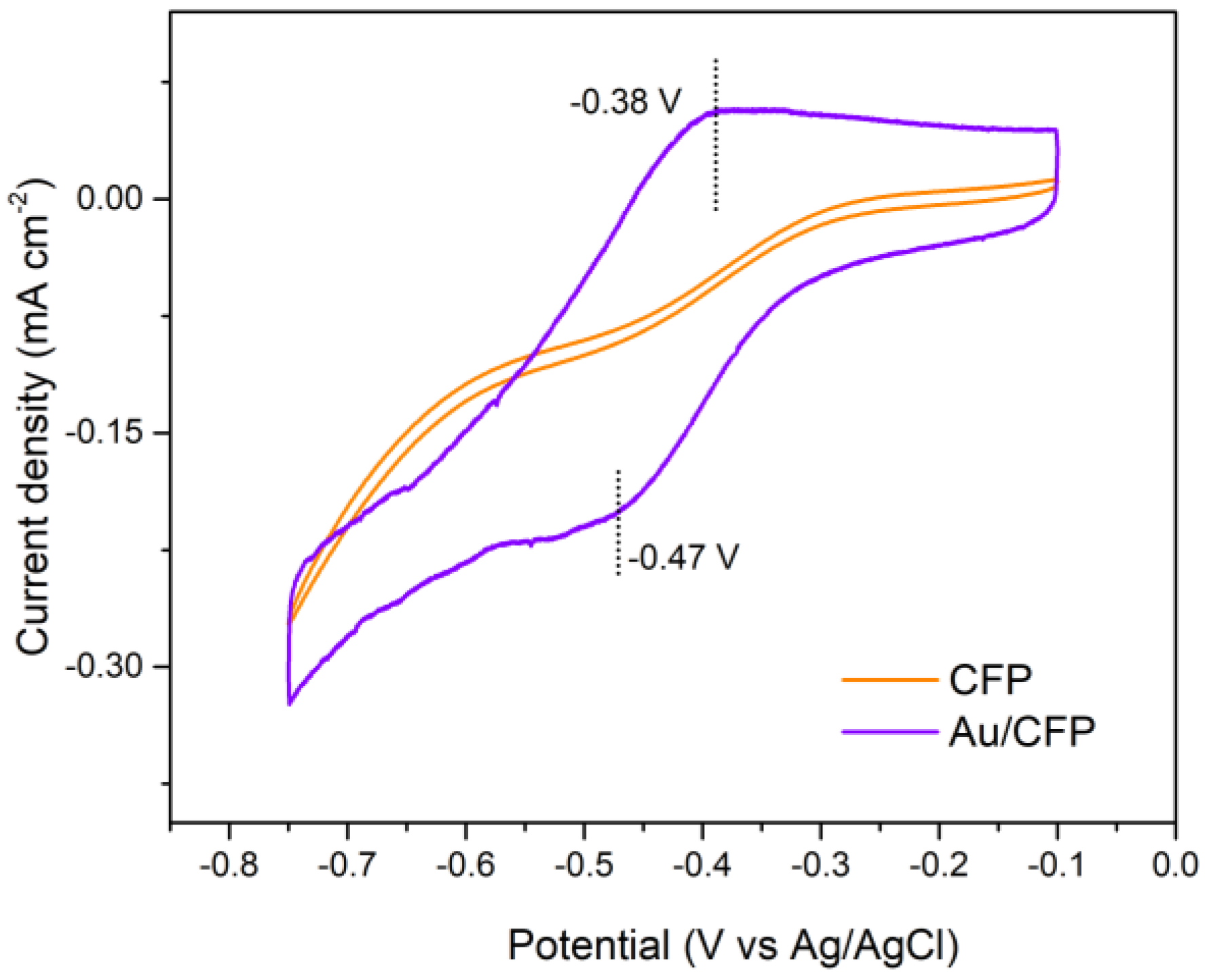
3.2.2. Electrochemical HER Characterization
3.3. Origin of GB–Enhanced HER
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobson, M.Z.; Colella, W.G.; Golden, D.M. Cleaning the air and improving health with hydrogen fuel-cell vehicles. Science 2005, 308, 1901–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Jiao, Y.; Jaroniec, M.; Qiao, S.Z. Advancing the Electrochemistry of the Hydrogen-Evolution Reaction through Combining Experiment and Theory. Angew. Chem. Int. Ed. 2015, 54, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Conway, B.E.; Tilak, B.V. Interfacial processes involving electrocatalytic evolution and oxidation of H2 and the role of chemisorbed H. Electrochim. Acta 2002, 47, 3571–3594. [Google Scholar] [CrossRef]
- Li, H.; Tsai, C.; Koh, A.L.; Cai, L.; Contryman, A.W.; Fragapane, A.H.; Zhao, J.; Han, H.S.; Manoharan, H.C.; Abild–Pedersen, F.; et al. Activating andoptimizing MoS2 basal planes for hydrogen evolution through the formation ofstrained sulphur vacancies. Nat. Mater. 2016, 15, 48–53. [Google Scholar] [CrossRef]
- Voiry, D.; Yamaguchi, H.; Li, J.; Silva, R.; Alves, D.C.B.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 2013, 12, 850–855. [Google Scholar] [CrossRef]
- Jaramillo, T.F.; Jørgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendorff, I. Identification of Active Edge Sites for Electrochemical H2 Evolution from MoS2 Nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef] [Green Version]
- Conway, B.E.; Bockris, J.O.M. Electrolytic Hydrogen Evolution Kinetics and Its Relation to the Electronic and Adsorptive Properties of the Metal. J. Chem. Phys. 1957, 26, 532–541. [Google Scholar] [CrossRef]
- Parsons, R. The rate of electrolytic hydrogen evolution and the heat of adsorption of hydrogen. Trans. Faraday Soc. 1958, 54, 1053–1063. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U.J. Trends in the exchange current for hydrogen evolution. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Xi, Z.; Chen, Z.; Guo, S.; Yu, Y.; Zhu, W.; Li, Q.; Zhang, X.; Pan, M.; Lu, G.; et al. A New Core/Shell NiAu/Au Nanoparticle Catalyst with Pt-like Activity for Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2015, 137, 5859–5862. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Guo, J.; Cui, X.; Yang, L.; Yang, Y.; He, H.; Xiao, P.; Zhang, Y. Three-Dimensional Dendritic Structures of NiCoMo as Efficient Electrocatalysts for the Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2017, 9, 22420–22431. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, D.; Qiao, Q.; Yu, Y.; Peterson, D.; Zafar, A.; Kumar, R.; Curtarolo, S.; Hunte, F.; Shannon, S.; et al. All the Catalytic Active Sites of MoS2 for Hydrogen Evolution. J. Am. Chem. Soc. 2016, 138, 16632–16638. [Google Scholar] [CrossRef]
- Vidruk, R.; Landau, M.V.; Herskowitz, M.; Talianker, M.; Frage, N.; Ezersky, V.; Froumin, N.J. Grain boundary control in nanocrystalline MgO as a novel means for significantly enhancingsurface basicity and catalytic activity. J. Catal. 2009, 263, 196–204. [Google Scholar] [CrossRef]
- Gavrilov, A.N.; Savinova, E.R.; Simonov, P.A.; Zaikovskii, V.I.; Cherepanova, S.V.; Tsirlina, G.A.; Parmon, V.N. On the influence of the metal loading on the structure of carbon–supported PtRu catalysts and their electrocatalytic activities in CO and methanol electrooxidation. Phys. Chem. Chem. Phys. 2007, 9, 5476–5489. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Ciston, J.; Kanan, M.W. Electroreduction of carbon monoxide to liquid fuel on oxide—derived nanocrystalline copper. Nature 2014, 508, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Kim, W.J.; Lim, H.-K.; Lee, E.K.; Kim, H. Tuned Chemical Bonding Ability of Au at Grain Boundaries for Enhanced Electrochemical CO2 Reduction. ACS Catal. 2016, 6, 4443–4448. [Google Scholar] [CrossRef]
- Dong, C.; Fu, J.; Liu, H.; Ling, T.; Yang, J.; Qiao, S.Z.; Du, X.-W. Tuning the selectivity and activity of Au catalysts for carbon dioxide electroreduction via grain boundary engineering: A DFT study. Mater. Chem. A 2017, 5, 7184–7190. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Nørskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented—Wave Method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gennero Chialvo, M.R.; Chialvo, A.C. Existence of Two Sets of Kinetic Parameters in the Correlation of the Hydrogen Electrode Reaction. J. Electrochem. Soc. 2000, 47, 1619–1622. [Google Scholar] [CrossRef]
- de Gennero Chialvo, M.R.; Chialvo, A.C. Hydrogen diffusion effects on the kinetics of the hydrogen electrode reaction. Part, I. Theoretical aspects. Phys. Chem. Chem. Phys. 2004, 6, 4009–4017. [Google Scholar] [CrossRef]
- Quaino, P.M.; de Gennero Chialvo, M.R.; Chialvo, A.C. Hydrogen diffusion effects on the kinetics of the hydrogen electrode reaction. Part, II. Evaluation of kinetic parameters. Phys. Chem. Chem. Phys. 2004, 6, 4450–4455. [Google Scholar] [CrossRef]
- Parsons, R. Catalysis in Electrochemistry; Santos, E., Schmickler, W., Eds.; Wiley: Hoboken, NJ, USA, 2011; Chapter 1; pp. 1–15. [Google Scholar]
- Zheng, Y.; Jiao, Y.; Zhu, Y.; Li, L.H.; Han, Y.; Chen, Y.; Du, A.; Jaroniec, M.; Qiao, S.Z. Hydrogen evolution by a metal-free electrocatalyst. Nat. Commun. 2014, 5, 3783. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, A. Underpotential deposition of lead on single crystal faces of gold: Part, I. The influence of crystallographic orientation of the substrate. J. Electroanal. Chem. 1984, 165, 167. [Google Scholar] [CrossRef]
- Hamelin, A.; Lipkowski, J. Underpotential deposition of lead on gold single crystal faces: Part II. General discussion. J. Electroanal. Chem. 1984, 171, 317. [Google Scholar] [CrossRef]
- Hernaández, J.; Solla–Gulloán, J.; Herrero, E. Gold nanoparticles synthesized in a water-in-oil microemulsion: Electrochemical characterization and effect of the surface structure on the oxygen reduction reaction. J. Electroanal. Chem. 2004, 574, 185. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia–Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Wu, D.; Xi, C.; Dong, C.; Liu, H.; Du, X.-W. Bond-energy-integrated coordination number: An accurate descriptor for transition—metal catalysts. J. Phys. Chem. C 2019, 123, 28248–28254. [Google Scholar] [CrossRef]
- Xi, C.; Zou, C.; Wang, M.; Wu, D.; Liu, H.; Dong, C.; Du, X.-W. A bond-energy-integrated-based descriptor for high-throughput screening of transition metal catalysts. J. Phys. Chem. C 2020, 124, 5241–5247. [Google Scholar] [CrossRef]
- Shustorovich, E.; Sellers, H. The UBI-QEP method: A practical theoretical approach to understanding chemistry on transition metal surfaces. Surf. Sci. Rep. 1998, 31, 1–119. [Google Scholar] [CrossRef]
- Kleis, J.; Greeley, J.; Romero, N.A.; Morozov, V.A.; Falsig, H.; Larsen, A.H.; Lu, J.; Mortensen, J.J.; Dułak, M.; Thygesen, K.S.; et al. Finite Size Effects in Chemical Bonding: From Small Clusters to Solids. Catal. Lett. 2011, 141, 1067–1071. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, R.; Fu, J.; Wang, Z.; Dong, C. Grain Boundary—A Route to Enhance Electrocatalytic Activity for Hydrogen Evolution Reaction. Appl. Sci. 2022, 12, 4290. https://doi.org/10.3390/app12094290
Jiang R, Fu J, Wang Z, Dong C. Grain Boundary—A Route to Enhance Electrocatalytic Activity for Hydrogen Evolution Reaction. Applied Sciences. 2022; 12(9):4290. https://doi.org/10.3390/app12094290
Chicago/Turabian StyleJiang, Ran, Jianyu Fu, Zhaoyang Wang, and Cunku Dong. 2022. "Grain Boundary—A Route to Enhance Electrocatalytic Activity for Hydrogen Evolution Reaction" Applied Sciences 12, no. 9: 4290. https://doi.org/10.3390/app12094290
APA StyleJiang, R., Fu, J., Wang, Z., & Dong, C. (2022). Grain Boundary—A Route to Enhance Electrocatalytic Activity for Hydrogen Evolution Reaction. Applied Sciences, 12(9), 4290. https://doi.org/10.3390/app12094290