
Prolonged Endurance Exercise Adaptations Counteract Doxorubicin Chemotherapy-Induced Myotoxicity in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Doxorubicin Treatment
2.3. Endurance Exercise Training
2.4. Dual Energy X-ray Absorptiometry (DEXA)
2.5. Tissue Collection
2.6. Hematoxylin and Eosin (H&E) Staining
2.7. Modified Gomori Trichrome (MGT) Staining
2.8. Protein Extraction and Western Blot Analysis
2.9. Statistics
3. Results
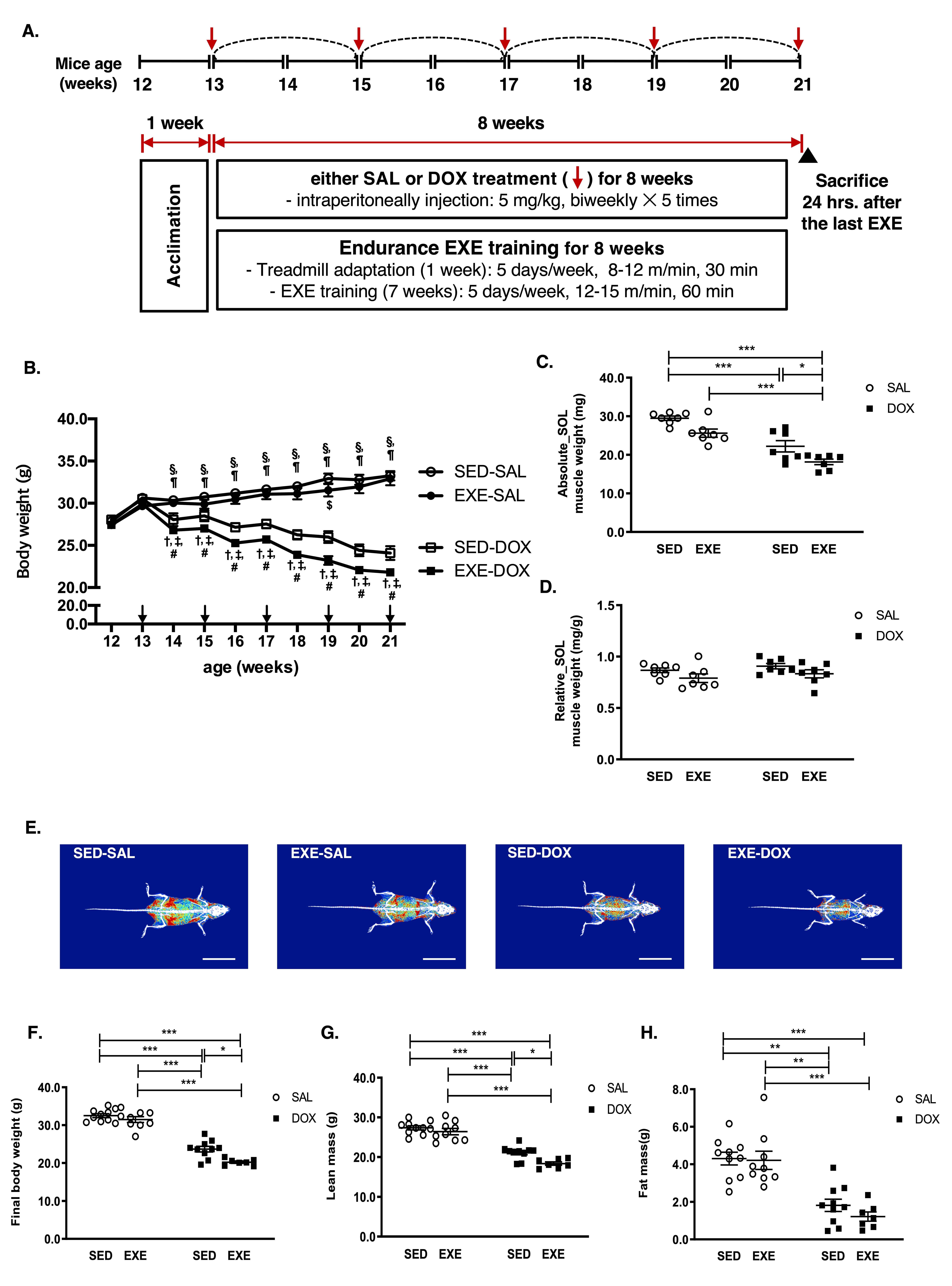
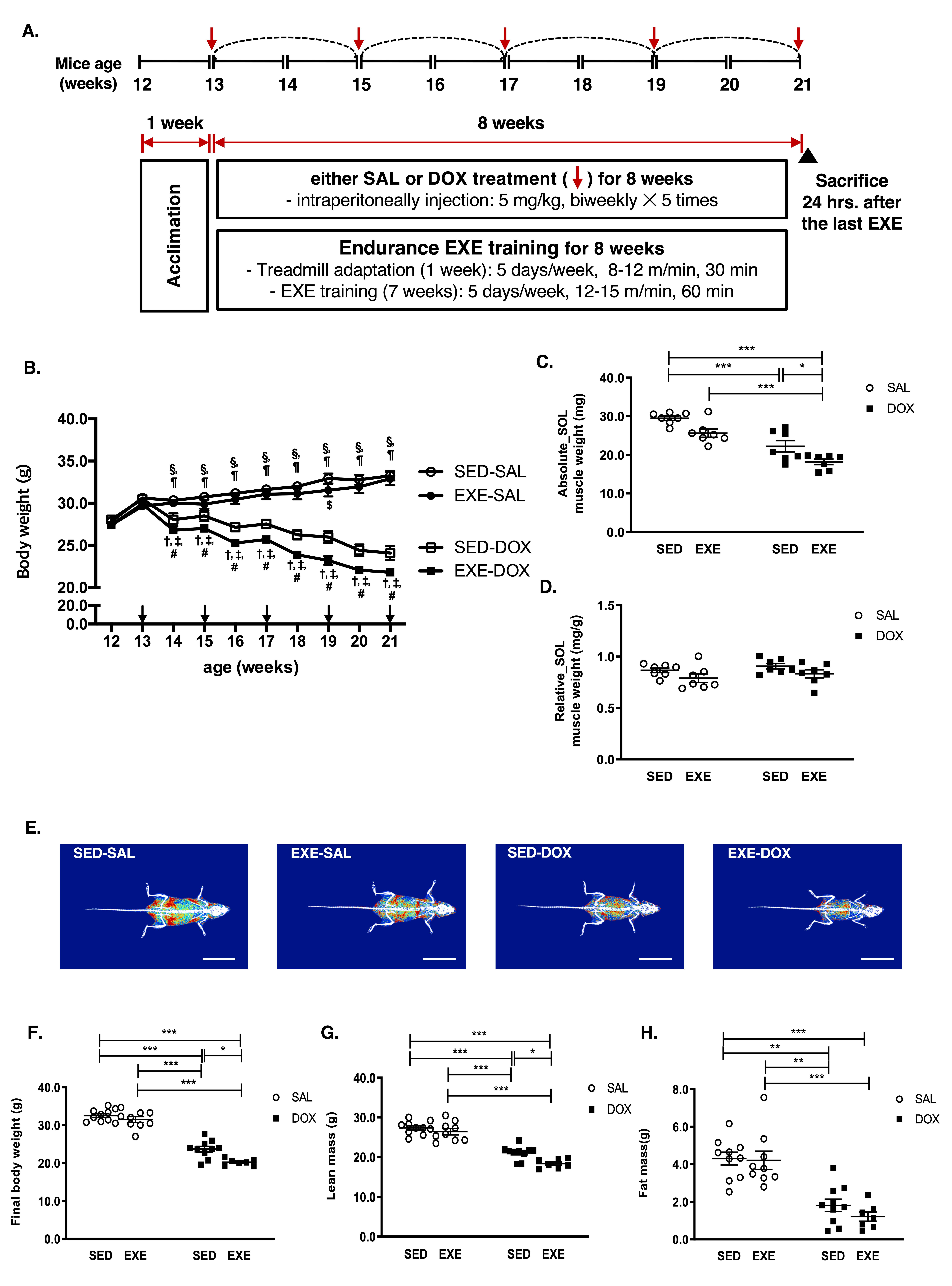
3.1. DOX Causes Loss of Body Weight, Lean Mass, Fat Mass, and Absolute SOL Muscle Mass
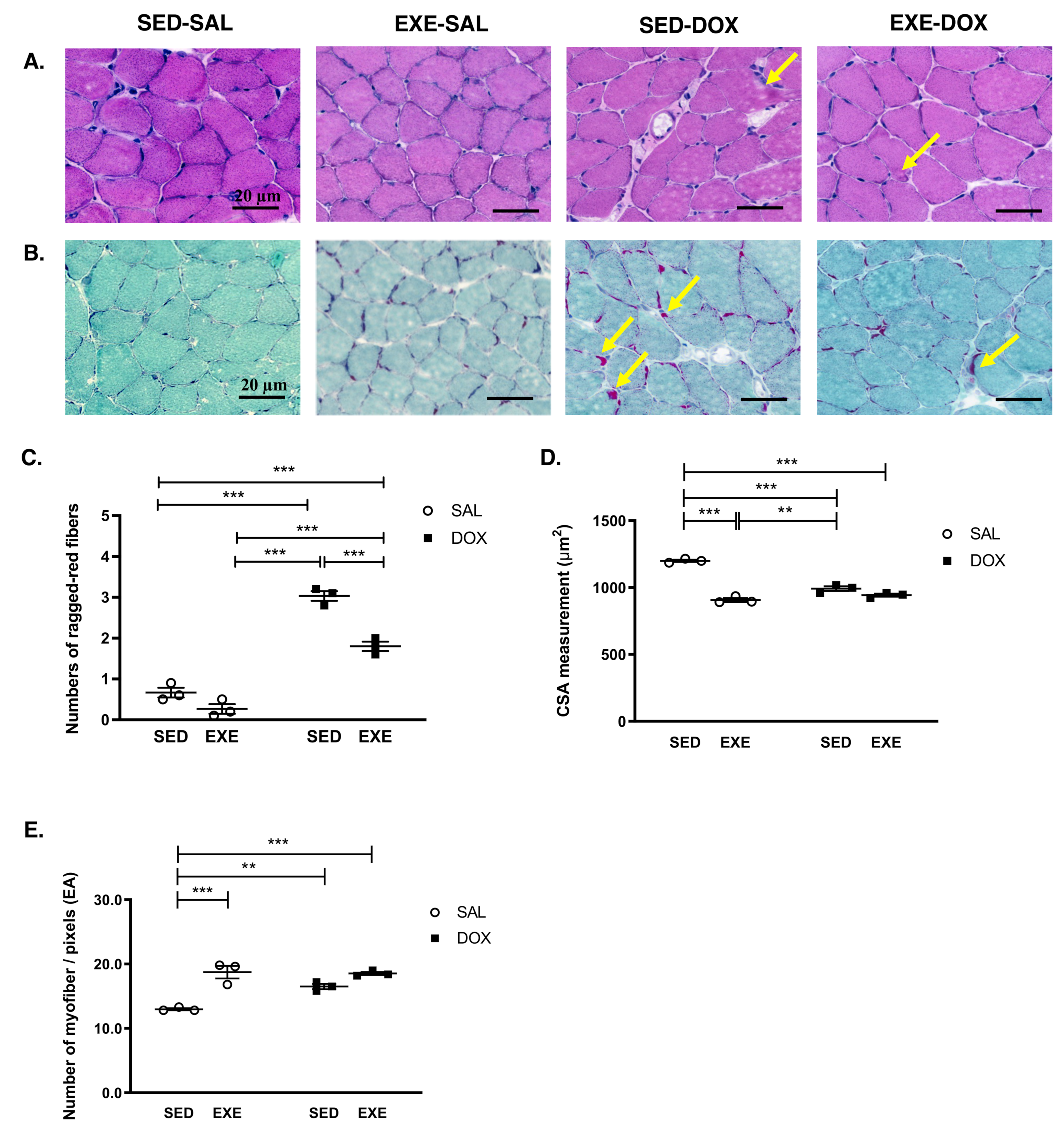
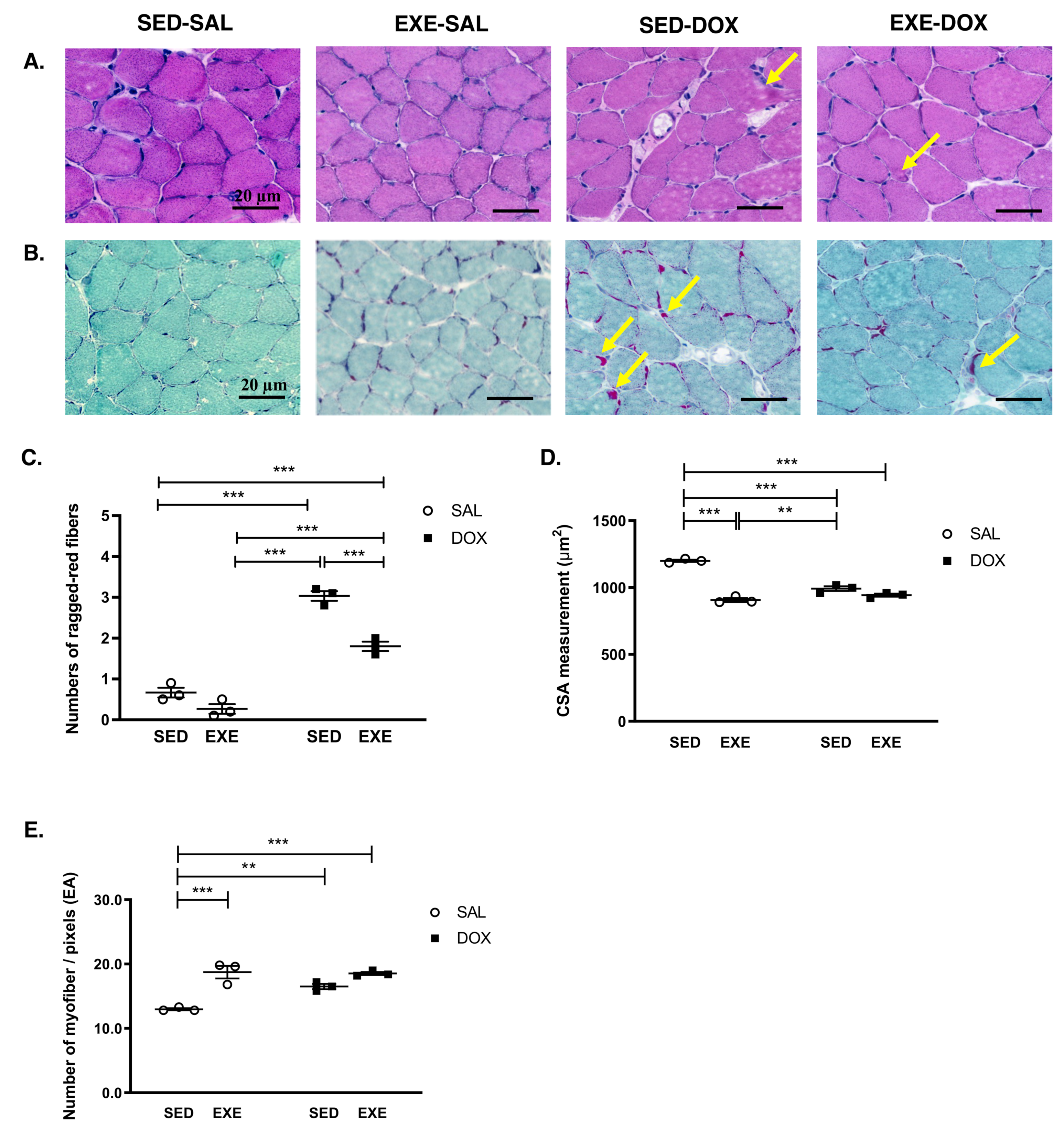
3.2. EXE Mitigates DOX-Induced Myotoxicity
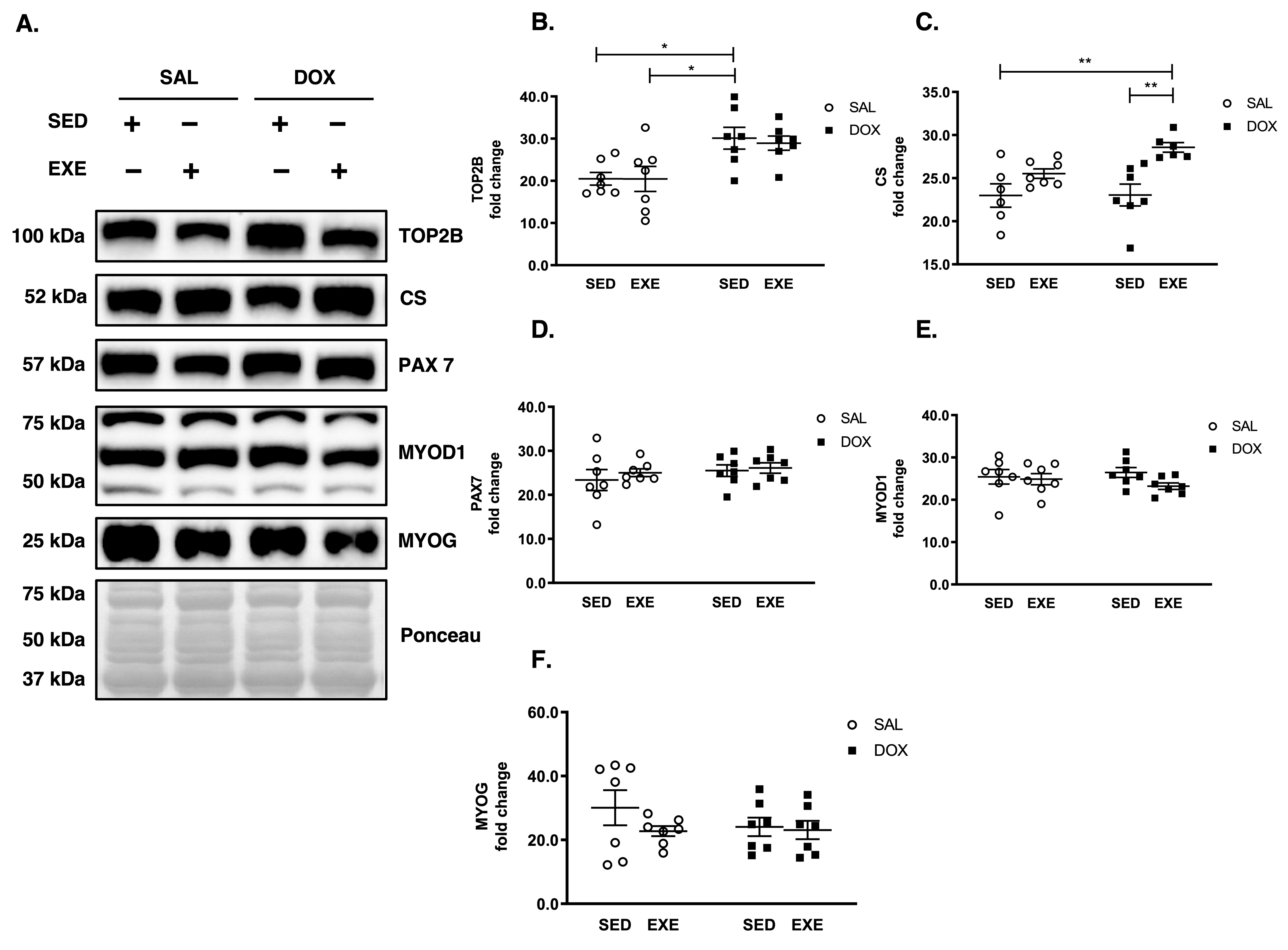
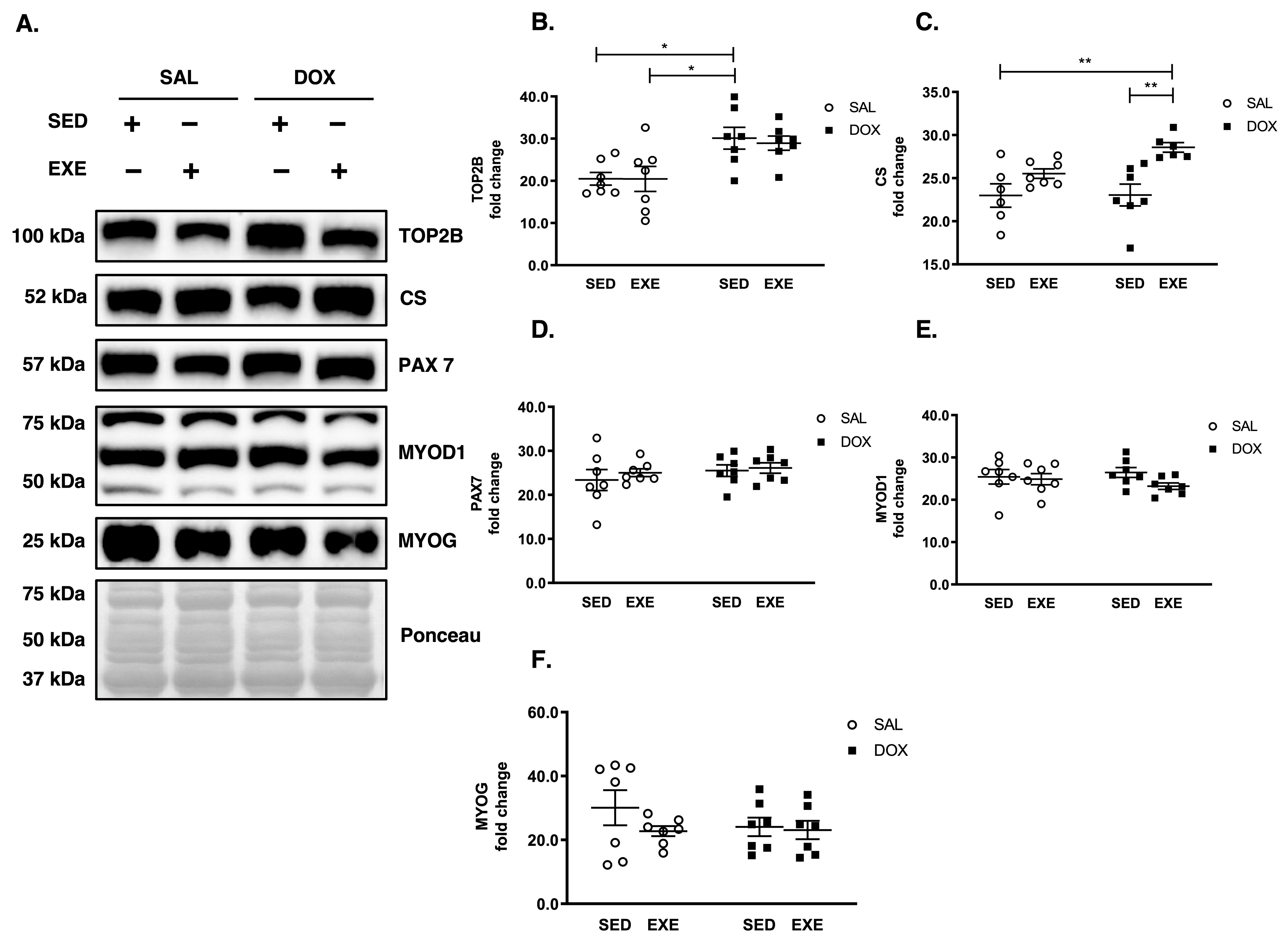
3.3. DOX Causes DNA Damage but Does Not Disrupt Satellite Cell Activation and Myogenesis
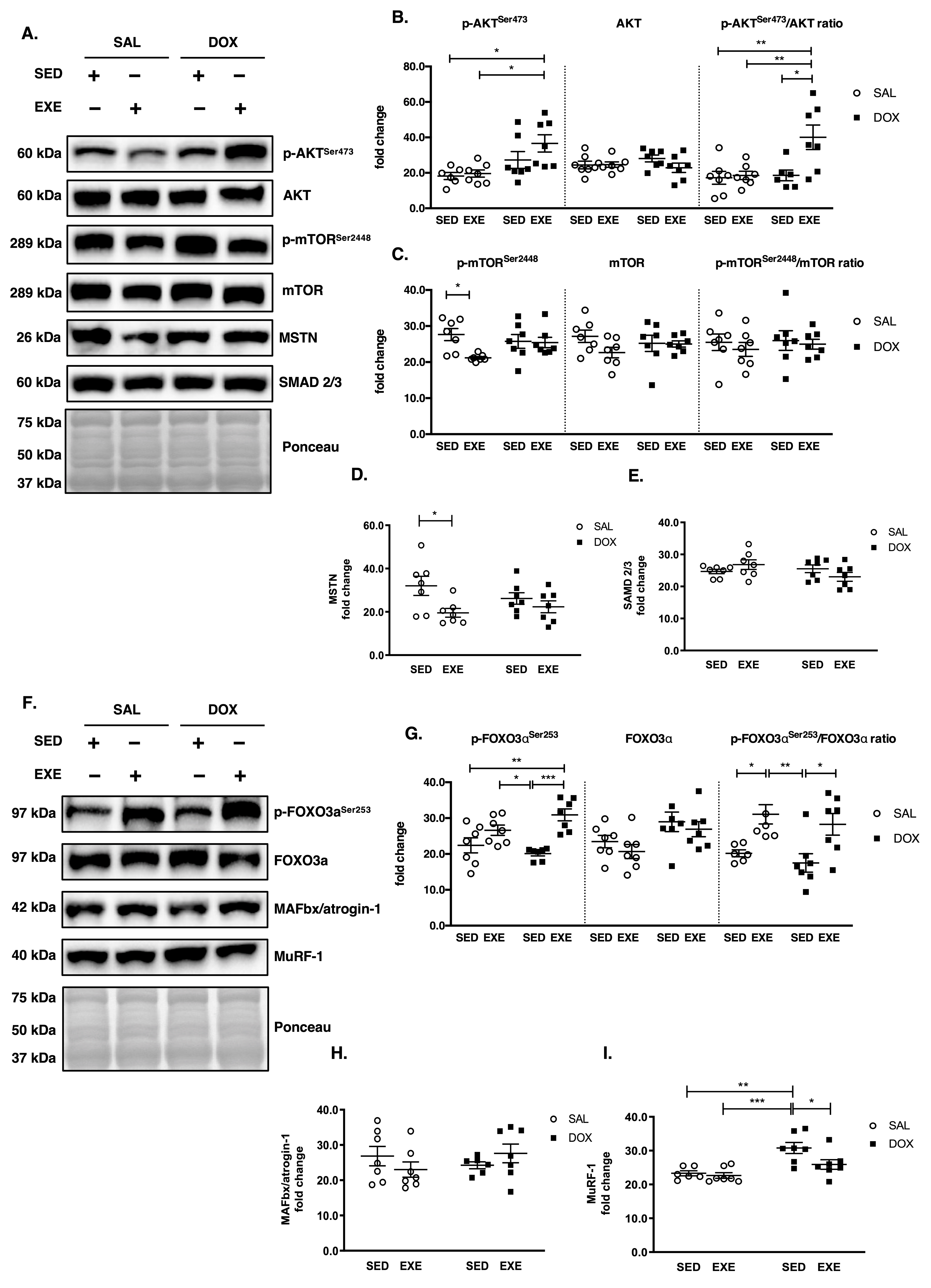
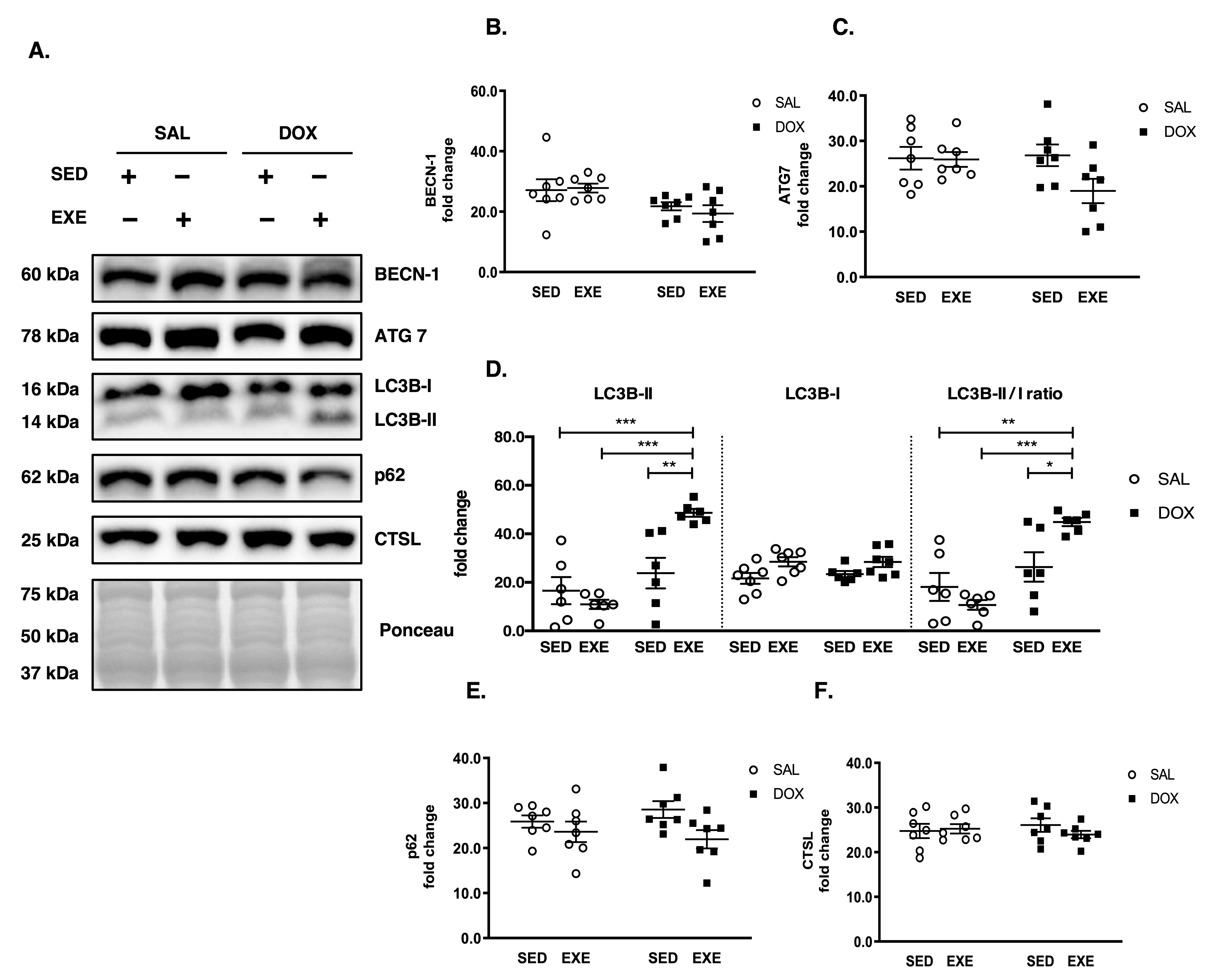
3.4. EXE Prohibits DOX-Induced Proteolytic Activation
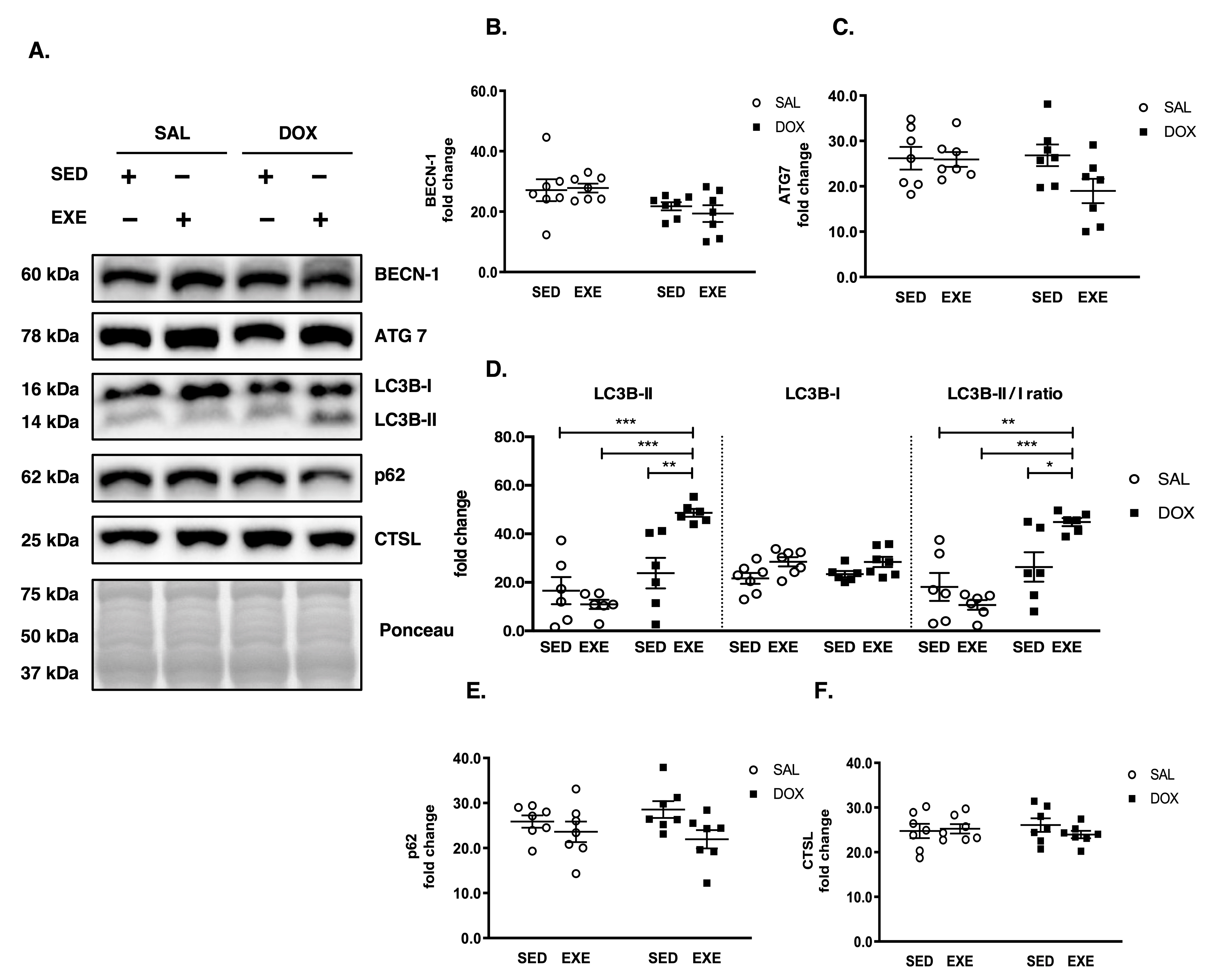
3.5. EXE Promotes Basal Autophagy
4. Discussion
5. Conclusions
Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheong, I.Y.; Yoo, J.S.; Chung, S.H.; Park, S.Y.; Song, H.J.; Lee, J.W.; Hwan, J.H. Functional loss in daily activity in ovarian cancer patients undergoing chemotherapy. Arch. Gynecol. Obstet. 2019, 299, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Mazurak, V.C.; Bhullar, A.S. Cancer cachexia is defined by an ongoing loss of skeletal muscle mass. Ann. Palliat. Med. 2019, 8, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Bredahl, E.C.; Pfannenstiel, K.B.; Quinn, C.J.; Hayward, R.; Hydock, D.S. Effects of Exercise on Doxorubicin-Induced Skeletal Muscle Dysfunction. Med. Sci. Sports Exerc. 2016, 48, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.M.; D’Lugos, A.C.; Mahmood, T.N.; Ormsby, J.C.; Salvo, L.; Dedmon, W.L.; Patel, S.H.; Katsma, M.S.; Mookadam, F.; Gonzales, R.J.; et al. Exercise Protects Skeletal Muscle during Chronic Doxorubicin Administration. Med. Sci. Sports Exerc. 2017, 49, 2394–2403. [Google Scholar] [CrossRef]
- Smuder, A.J. Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R662–R672. [Google Scholar] [CrossRef] [PubMed]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J. Appl. Physiol. 2011, 110, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharm. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Sarkar, S.; Scott, L.; Danelisen, I.; Trush, M.A.; Jia, Z.; Li, R.Y. Doxorubicin Redox Biology: Redox Cycling, Topoisomerase Inhibition, and Oxidative Stress. React. Oxyg. Species 2016, 1, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Mohan, N.; Endo, Y.; Shen, Y.; Wu, W.J. Type IIB DNA topoisomerase is downregulated by trastuzumab and doxorubicin to synergize cardiotoxicity. Oncotarget 2018, 9, 6095–6108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schuler, S.C.; Huttner, S.S.; von Eyss, B.; von Maltzahn, J. Adult stem cells at work: Regenerating skeletal muscle. Cell Mol. Life Sci. 2019, 76, 2559–2570. [Google Scholar] [CrossRef] [Green Version]
- Quinn, C.J.; Hydock, D.S. Effects of endurance exercise and doxorubicin on skeletal muscle myogenic regulatory factor expression. Muscles Ligaments Tendons J. 2017, 7, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Han, J.; Meng, Q.; Xi, Q.; Zhuang, Q.; Jiang, Y.; Han, Y.; Zhang, B.; Fang, J.; Wu, G. Muscle-specific E3 ubiquitin ligases are involved in muscle atrophy of cancer cachexia: An in vitro and in vivo study. Oncol. Rep. 2015, 33, 2261–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guigni, B.A.; Fix, D.K.; Bivona, J.J., 3rd; Palmer, B.M.; Carson, J.A.; Toth, M.J. Electrical stimulation prevents doxorubicin-induced atrophy and mitochondrial loss in cultured myotubes. Am. J. Physiol. Cell Physiol. 2019, 317, C1213–C1228. [Google Scholar] [CrossRef] [PubMed]
- Doerr, V.; Montalvo, R.N.; Kwon, O.S.; Talbert, E.E.; Hain, B.A.; Houston, F.E.; Smuder, A.J. Prevention of Doxorubicin-Induced Autophagy Attenuates Oxidative Stress and Skeletal Muscle Dysfunction. Antioxidants 2020, 9, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuiyan, M.S.; Pattison, J.S.; Osinska, H.; James, J.; Gulick, J.; McLendon, P.M.; Hill, J.A.; Sadoshima, J.; Robbins, J. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Investig. 2013, 123, 5284–5297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kwon, I.; Jang, Y.; Cosio-Lima, L.; Barrington, P. Endurance Exercise Attenuates Doxorubicin-induced Cardiotoxicity. Med. Sci. Sports Exerc. 2020, 52, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I. Protective effects of endurance exercise on skeletal muscle remodeling against doxorubicin-induced myotoxicity in mice. Phys. Act. Nutr. 2020, 24, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Ertunc, M.; Sara, Y.; Korkusuz, P.; Onur, R. Differential contractile impairment of fast- and slow-twitch skeletal muscles in a rat model of doxorubicin-induced congestive heart failure. Pharmacology 2009, 84, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Hydock, D.S.; Lien, C.Y.; Jensen, B.T.; Schneider, C.M.; Hayward, R. Characterization of the effect of in vivo doxorubicin treatment on skeletal muscle function in the rat. Anticancer Res. 2011, 31, 2023–2028. [Google Scholar] [PubMed]
- Romero-Calvo, I.; Ocón, B.; Martínez-Moya, P.; Suárez, M.D.; Zarzuelo, A.; Martínez-Augustin, O.; de Medina, F.S. Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal. Biochem. 2010, 401, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Stain-Free total protein staining is a superior loading control to β-actin for Western blots. Anal. Biochem. 2013, 440, 186–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilliam, L.A.; Moylan, J.S.; Patterson, E.W.; Smith, J.D.; Wilson, A.S.; Rabbani, Z.; Reid, M.B. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2012, 302, C195–C202. [Google Scholar] [CrossRef] [Green Version]
- Hayward, R.; Hydock, D.; Gibson, N.; Greufe, S.; Bredahl, E.; Parry, T. Tissue retention of doxorubicin and its effects on cardiac, smooth, and skeletal muscle function. J. Physiol. Biochem. 2013, 69, 177–187. [Google Scholar] [CrossRef]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N.; et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef] [Green Version]
- De Lima, E.A.; de Sousa, L.G.O.; de S Teiseira, A.A.; Marshall, A.G.; Zanchi, N.E.; Neto, J.C.R. Aerobic exercise, but not metformin, prevents reduction of muscular performance by AMPk activation in mice on doxorubicin chemotherapy. J. Cell. Physiol. 2018, 233, 9652–9662. [Google Scholar] [CrossRef] [PubMed]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J. Appl. Physiol. 2011, 111, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Mackay, A.D.; Marchant, E.D.; Munk, D.J.; Watt, R.K.; Hansen, J.M.; Thomson, D.M.; Hancock, C.R. Multitissue analysis of exercise and metformin on doxorubicin-induced iron dysregulation. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E922–E930. [Google Scholar] [CrossRef]
- Huertas, A.M.; Morton, A.B.; Hinkey, J.M.; Ichinoseki-Sekine, N.; Smuder, A.J. Modification of Neuromuscular Junction Protein Expression by Exercise and Doxorubicin. Med. Sci. Sports Exerc. 2020, 52, 1477–1484. [Google Scholar] [CrossRef]
- Huang, S.C.; Wu, J.F.; Saoviengm, S.; Chien, W.H.; Hsu, M.F.; Li, X.F.; Lee, S.D.; Huang, C.Y.; Kuo, C.H. Doxorubicin inhibits muscle inflammation after eccentric exercise. J. Cachexia Sarcopenia Muscle 2017, 8, 277–284. [Google Scholar] [CrossRef] [PubMed]
- De Lima, A.E.; Teixeira, A.A.S., Jr.; Biondo, L.A.; Diniz, T.A.; Silveira, L.S.; Coletti, D.; Rius, S.B.; Neto, J.C.R. Exercise Reduces the Resumption of Tumor Growth and Proteolytic Pathways in the Skeletal Muscle of Mice Following Chemotherapy. Cancers 2020, 12, 3466. [Google Scholar] [CrossRef]
- Jones, L.W.; Eves, N.D.; Courneya, K.S.; Chiu, B.K.; Baracos, V.E.; Hanson, J.; Johnson, L.; Mackey, J.R. Effects of exercise training on antitumor efficacy of doxorubicin in MDA-MB-231 breast cancer xenografts. Clin. Cancer Res. 2005, 11, 6695–6698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courneya, K.S.; Segal, R.J.; McKenzie, D.C.; Dong, H.; Gelmon, K.; Friedenreich, C.M.; Yasui, Y.; Reid, R.D.; Crawford, J.J.; Mackey, J.R. Effects of exercise during adjuvant chemotherapy on breast cancer outcomes. Med. Sci. Sports Exerc. 2014, 46, 1744–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, Y.L.; Kerrigan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIbeta mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Souza, V.A.; Ghiarone, T.; Sansonio, A.; Silva, S.K.A.; Tomazini, F.; Arcoverde, L.; Fyfe, J.; Perri, E.; Saner, N.; Kuang, J.; et al. Exercise twice-a-day potentiates markers of mitochondrial biogenesis in men. FASEB J. 2020, 34, 1602–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.N.; Richards, B.J.; Slavin, M.; Hood, D.A. Exercise Is Muscle Mitochondrial Medicine. Exerc. Sport Sci. Rev. 2021, 49, 67–76. [Google Scholar] [CrossRef]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Wojewoda, M.; Kmiecik, K.; Majerczak, J.; Ventura-Clapier, R.; Fortin, D.; Onopiuk, M.; Rog, J.; Kaminski, K.; Chlopicki, S.; Zoladz, J.A. Skeletal Muscle Response to Endurance Training in IL-6-/- Mice. Int. J. Sports Med. 2015, 36, 1163–1169. [Google Scholar] [CrossRef]
- Kurabayashi, M.; Jeyaseelan, R.; Kedes, L. Antineoplastic agent doxorubicin inhibits myogenic differentiation of C2 myoblasts. J. Biol. Chem. 1993, 268, 5524–5529. [Google Scholar] [CrossRef]
- Kavazis, A.N.; Smuder, A.J.; Powers, S.K. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl. Physiol. 2014, 117, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Hiensch, A.E.; Bolam, K.A.; Mijwel, S.; Jeneson, J.A.L.; Huitema, A.D.R.; Kranenburg, O.; van der wall, E.; Rundqvist, H.; Wengstrom, Y.; May, A.M. Doxorubicin-induced skeletal muscle atrophy: Elucidating the underlying molecular pathways. Acta Physiol. 2020, 229, e13400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumati, P.; Bonaldo, P. Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cells 2012, 1, 325–345. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Huber, R. Substrate access and processing by the 20S proteasome core particle. Int. J. Biochem. Cell Biol. 2003, 35, 606–616. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R. Inhibitors of the eukaryotic 20S proteasome core particle: A structural approach. Biochim. Biophys. Acta 2004, 1695, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Masiero, E.; Sandri, M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy 2010, 6, 307–309. [Google Scholar] [CrossRef] [Green Version]
- Kwon, I.; Lee, Y.; Cosio-Lima, L.M.; Cho, J.Y.; Yeom, D.C. Effects of long-term resistance exercise training on autophagy in rat skeletal muscle of chloroquine-induced sporadic inclusion body myositis. J. Exerc. Nutr. Biochem. 2015, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- He, C.C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.J.; Zou, Z.J.; An, Z.Y.; Loh, J.; Fisher, J.; Sun, Q.H.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, I.; Go, G.-W.; Lee, Y.; Kim, J.-H. Prolonged Endurance Exercise Adaptations Counteract Doxorubicin Chemotherapy-Induced Myotoxicity in Mice. Appl. Sci. 2022, 12, 3652. https://doi.org/10.3390/app12073652
Kwon I, Go G-W, Lee Y, Kim J-H. Prolonged Endurance Exercise Adaptations Counteract Doxorubicin Chemotherapy-Induced Myotoxicity in Mice. Applied Sciences. 2022; 12(7):3652. https://doi.org/10.3390/app12073652
Chicago/Turabian StyleKwon, Insu, Gwang-Woong Go, Youngil Lee, and Jong-Hee Kim. 2022. "Prolonged Endurance Exercise Adaptations Counteract Doxorubicin Chemotherapy-Induced Myotoxicity in Mice" Applied Sciences 12, no. 7: 3652. https://doi.org/10.3390/app12073652
APA StyleKwon, I., Go, G.-W., Lee, Y., & Kim, J.-H. (2022). Prolonged Endurance Exercise Adaptations Counteract Doxorubicin Chemotherapy-Induced Myotoxicity in Mice. Applied Sciences, 12(7), 3652. https://doi.org/10.3390/app12073652