
Rapid Detection of Direct Compound Toxicity and Trailing Detection of Indirect Cell Metabolite Toxicity in a 96-Well Fluidic Culture Device for Cell-Based Screening Environments: Tactics in Six Sigma Quality Control Charts


Abstract
:1. Introduction
1.1. A Fluidic Platform for Dual-Toxicity Screening
1.2. The Organ of Investigation Is Liver with Cell Culture Being Hepatoctyes
1.3. Exposure Compounds and Cell Health Readouts
2. Materials and Methods
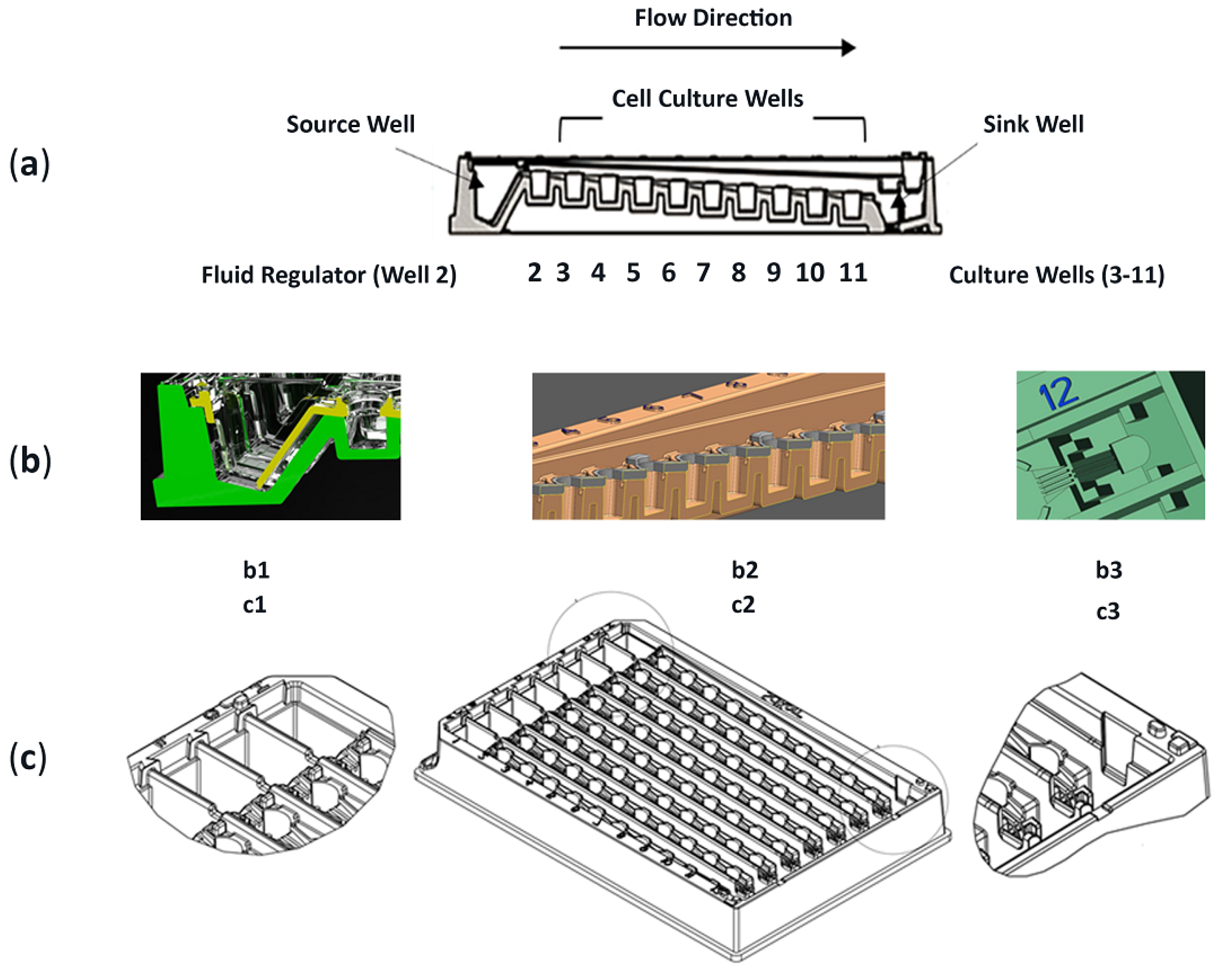
2.1. Device, Design, Material, Construction, Culture Surface, and Attributes
2.1.1. Device
Structure and Lab Compatibility
2.1.2. Material and Cell Compatibility
Mass Production for Repeatability
Culture Surface and Treatment
2.1.3. Assembly of µChannels
2.2. Device Preparation for Cell Seeding (Static) and µFluidic Operation (Flow)
2.2.1. Prepare Platform for Cell Seeding Using No Flow Conditions (Static)
2.2.2. Connecting Culture Wells and Initiating Fluidic Conditions (Flow)
2.3. Cell Seeding Conditions in µFluidic Device
2.3.1. Human HepaRG Cell Line
2.3.2. Rat Hepatocytes Freshly Isolated
2.4. Drug Treatment during Cell Culture
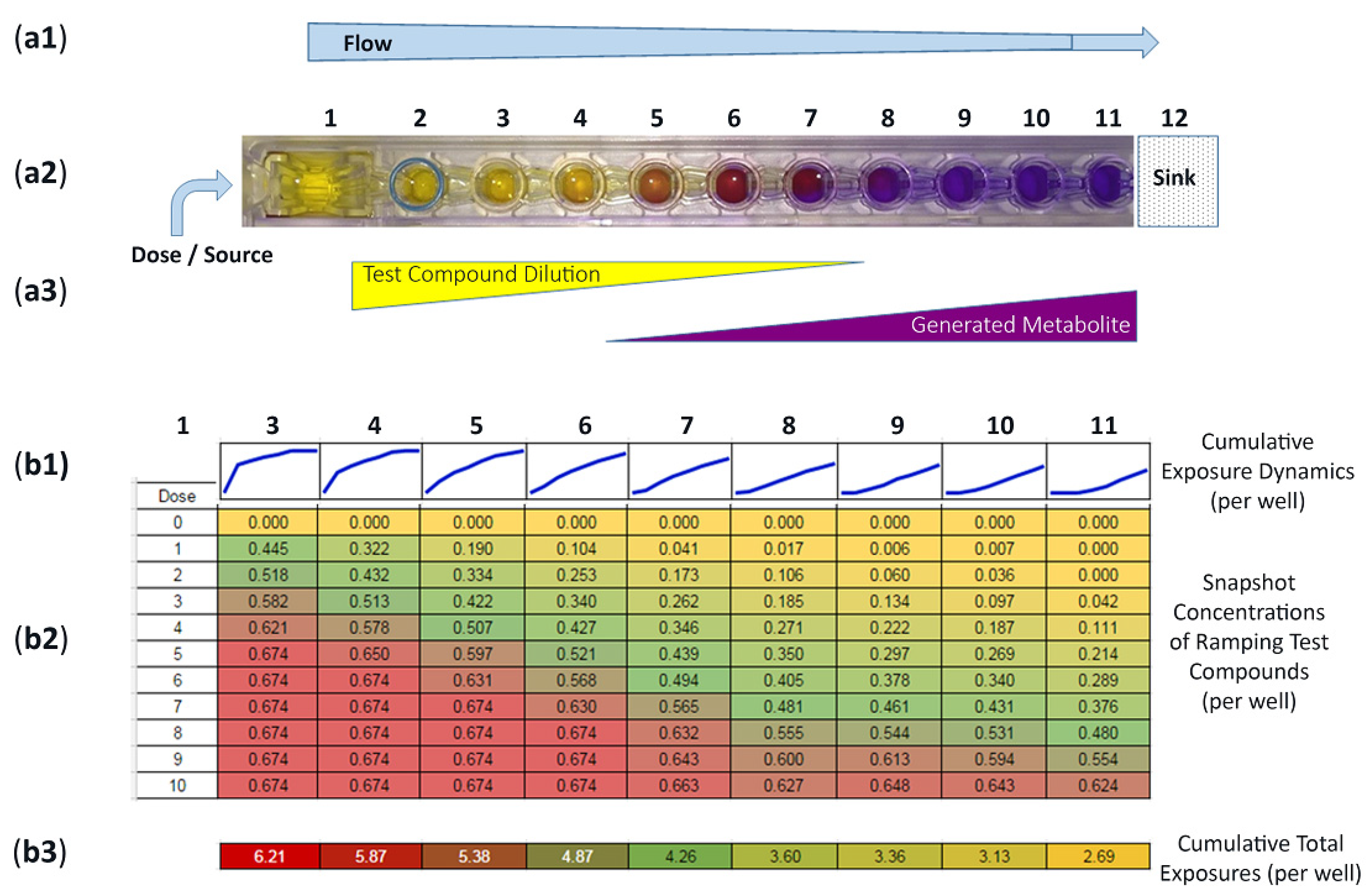
2.5. Suitability of FITC Surrogate Drug to Ascertain Compound Concentrations
2.6. Quantification of APAP Drug Metabolites
2.7. Assays to Evaluate Modulations in Cell Health
2.7.1. Live and Dead Assays (Imaging)
2.7.2. LDH Assay
2.7.3. CYP3A4 Luminescence Assay
2.7.4. Glutathione (GSH) Level
2.8. Statistical Analysis and Tactics for Lean Six Sigma Control Charts
2.8.1. Continuous Data for µFluidic Culture
2.8.2. Discrete Data
2.9. Standard Operating Procedure (SOP) for One Drug in One Fluidic Culture System
3. Results
3.1. Operational Insights for Compound Dosing, Quantified Concentration Gradients, Flow Distribution Patterns, and Transforming Well-to-Well Exposures
3.2. Incentive for Using FITC Surrogate to Track Actual Compound Distributions across Device Wells 3–11
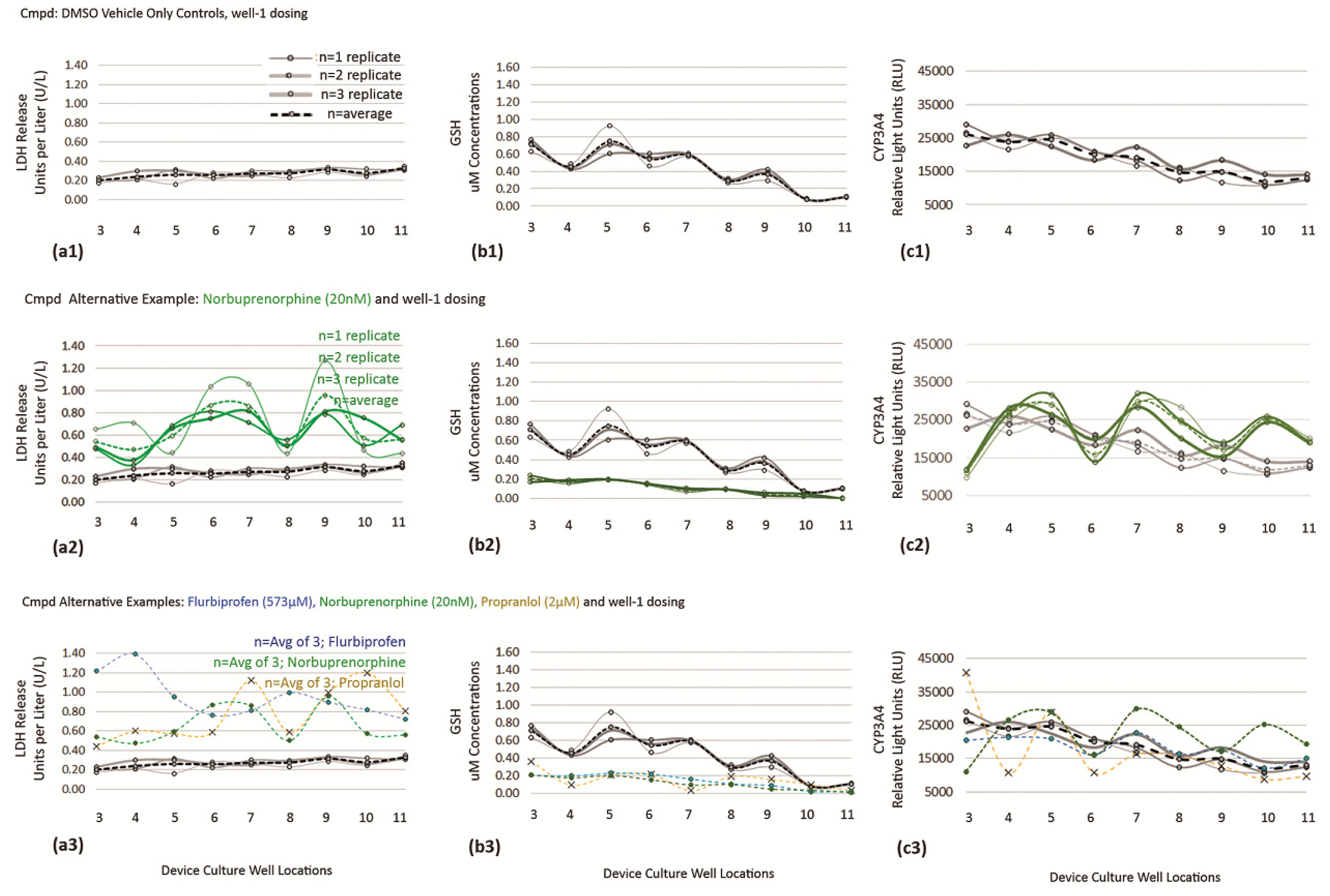
3.3. Validation of Tempered DMSO Compound as an Effective Control Vehicle Using High-Content Cell Imaging across the Sequential Assembly-Line of µFluidic Culture Sites
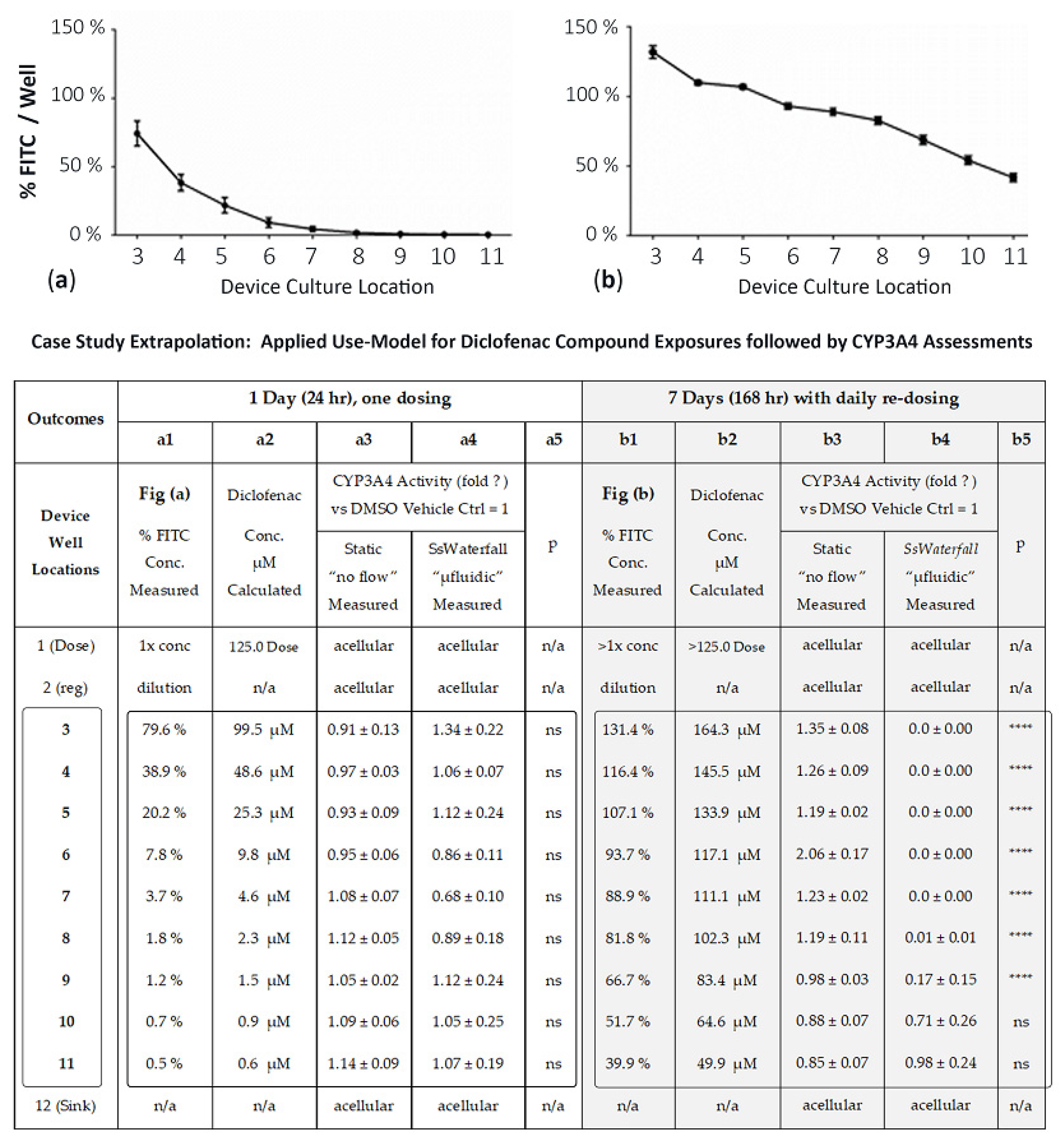
3.4. Use-Model, Drug Exposure, and CYP3A4 Comparisons between µFluidic Systems and Static Cultures
3.4.1. DMSO Vehicle as Baseline Control for Cell Functions
3.4.2. Extrapolation of Drug Exposures in µFluidic System, Diclofenac (Days 1 and 7)
3.5. Trending Effects on Cell Toxicity from DMSO Vehicle Control, APAP, NEF, and Indirect Cell Byproducts in µFluidic Device (Dual Toxicity Trends for Compoud Screening)
3.5.1. Data Generation
3.5.2. CV Values for VOC and Fluid-Flow Tracing across the Multiwell Channels
3.5.3. Data Reconfigure for Trending Applications Using X Control Charts Aligned with Statistical Process Control
3.6. Metabolite Generation and Dissemination in Multiwell µFluidic Culture System
3.6.1. Targeting APAP Metabolites
3.6.2. Distribution of APAP Metabolites
3.7. Metabolite Variance across Multiwell Fluidic System to Ascertain Sites of Generated Cell Byproducts
3.8. Cell Function Variance across Multiwell Fluidic System to Assess Bio-Activity Trends Affiliated with Direct Drug Exposure and Predictive Recognition for Indirect Cell-Byproduct Stimuli (Dual Assessment Screens)
3.8.1. Trending Relevance
LDH ASSAY—Functional Indicator (Studies 1–4)
CYP3A4 ASSAY—Functional Indicator (Studies 5–8)
GLUTATHIONE (GSH) ASSAY—Functional Indicator (Studies 9–12)
3.8.2. Vetting Trends in Cell Modulation by Drug or Cell Byproduct Stimuli (Studies 1–12)
4. Discussion
4.1. Drug Development Is a Very Long and Expensive Process
4.2. Natural 3D Organs with Instrinsic Fluid Flows
4.2.1. µFluidic Contemporary Technologies for In Vitro Cell Simulations
4.2.2. Unmasking the Cell-Byproduct Mechanism Using 1-Way In Vitro µFluidics
4.2.3. Trending Variations to Observe Different Types of Cell Toxicity
4.3. DILI Is One Reason That New Compounds Are Removed from Late-Stage Clinical Trials
4.4. Perpetually Adapating Cell Responses for Complementary Knowledge
4.4.1. Focused Modeling with Enhanced Tissue Engineering
4.4.2. LDH Release Is a Biomarker to Evaluate Liver Damage
4.4.3. CYP3A4 Is the Most Critical Drug-Metabolizing Enzyme Expressed in Liver Cells
4.4.4. Glutathione Is Produced in All Mammalian Cells
4.4.5. Observing Cell Behaviors
4.5. Emerging Advantages with Continued Optimization
4.6. Future Research Directions
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Terminology to Define Modulation Traits of Cell Data for Process Shift, Special Cause, and Unusual Outcome
Appendix A.2. Modulation of Cell Byproducts/Metabolites
- Hepatocyte → hepatocyte → hepatocyte → (channel multiwells 3–9 as presented in the current text);
- Liver zone-1 (wells 3–5) → liver zone-2 (wells 6–8) → liver zone-3 (wells 9–11); correlated to cell maturity;
- Intestinal (wells 3–4) → liver (wells 5–9) → target cancer (wells 10–11); correlated to multiorgan systems, etc.
Appendix B
Trending Effects of Vehicle DMSO Used for Normalization across Wells
References
- Choi, J.; Lee, E.K.; Choo, J.; Yuh, J.; Hong, J.W. Micro 3D cell culture systems for cellular behavior studies: Culture matrices, devices, substrates, and in-situ sensing methods. Biotechnol. J. 2015, 10, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.G.; Ferreira, I.C.C.; Marangoni, K.; Bastoz, V.A.F.; Goulart, V.A. Advances in Cell Culture: More than a Century after Cultivating Cells. J. Biotechnol. Biomater. 2016, 6, 1000221. [Google Scholar] [CrossRef]
- Coluccio, M.L.; Perozziello, G.; Malara, N.; Parrotta, E.; Zhang, P.; Gentile, F.; Limongi, T.; Raj, P.M.; Cuda, G.; Candeloro, P.; et al. Microfluidic platforms for cell cultures and investigations. Microelectron. Eng. 2019, 208, 14–28. [Google Scholar] [CrossRef]
- Beebe, D.; Ingber, D.; den Toonder, J. Organs on Chips. Lab A Chip 2013, 13, 3447–3448. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Dauth, S.; Hassell, B.; Herland, A.; Jain, A.; Jang, K.-J.; Karalis, K.; Kim, H.J.; MacQueen, L.; Mahmoodian, R.; et al. Engineered In Vitro Disease Models. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 195–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Ingber, D. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Bhise, N.S.; Ribas, J.; Manoharan, V.; Zhang, Y.S.; Polini, A.; Massa, S.; Dokmeci, M.R.; Khademhosseini, A. Organ-on-a-chip platforms for studying drug delivery systems. J. Control. Release 2014, 190, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Huh, D.; Torisawa, Y.-S.; Hamilton, G.A.; Kim, H.J.; Ingber, D.E. Microengineered physiological biomimicry: Organs-on-Chips. Lab A Chip 2012, 12, 2156–2164. [Google Scholar] [CrossRef]
- Van der Meer, A.D.; van der Berg, A. Organs-on-chips: Breaking the in vitro impasse. Integr. Biol. 2012, 4, 461–470. [Google Scholar] [CrossRef]
- Fiorina, G.; Chiu, D.T. Disposable microfluidic devices: Fabrication, function, and application. Biotechniques 2005, 38, 429–446. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.; Ali, Z. Fabrication Methods for Microfluidic Devices: An Overview. Micromachines 2021, 12, 319. [Google Scholar] [CrossRef] [PubMed]
- Convery, N.; Gadegaard, N. 30 years of microfluidics. Micro Nano Eng. 2019, 2, 76–91. [Google Scholar] [CrossRef]
- Van Meer, B.; de Vries, H.; Firth, K.; van Weerd, J.; Tertoolen, L.; Karperien, H.; Jonkheijm, P.; Denning, C.; Ijzerman, A.; Mummery, C. Small molecule absorption by PDMS in the context of drug response bioassays. Biochem. Biophys. Res. Commun. 2016, 482, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokaltun, A.; Kang, Y.B.A.; Yarmush, L.M.; Usta, O.B.; Asatekin, A. Simple Surface Modification of Poly(dimethylsiloxane) via Surface Segregating Smart Polymers for Bio-microfluidics. Sci. Rep. 2019, 9, 7377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Midwoud, P.M.; Janse, A.; Merema, M.T.; Groothuis, G.M.M.; Verpoorte, E. Comparison of Biocompatibility and Adsorption Properties of Different Plastics for Advanced Mi-crofluidic Cell and Tissue Culture Models. Anal. Chem. 2012, 84, 3938–3944. [Google Scholar] [CrossRef]
- Sinha, N.; Subedi, N.; Wimmers, F.; Soennichsen, M.; Tel, J. A Pipette-Tip Based Method for Seeding Cells to Droplet Microfluidic Platforms. J. Vis. Exp. 2019, 144, e57848. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.J.; Bhuiyan, N.H.; Shim, J.S. Fully integrated rapid microfluidic device translated from conventional 96-well ELISA kit. Sci. Rep. 2021, 11, 1986. [Google Scholar] [CrossRef]
- Berlanda, S.; Breitfeld, M.; Dietsche, C.L.; Dittrich, P.S. Recent Advances in Microfluidic Technology for Bioanalysis and Diagnostics. Anal. Chem. 2021, 93, 311–331. [Google Scholar] [CrossRef]
- Andria, B.; Bracco, A.; Cirino, G.; Chamuleau, R.A.F.M. Liver Cell Culture Devices. Cell Med. 2010, 1, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A. The Flaws and Human Harms of Animal Experimentation. Camb. Q. Healthc. Ethics 2015, 24, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Shanks, N.; Greek, R.; Greek, J. Are animal models predictive for humans? Philos Ethics Humanit. Med. 2009, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leenaars, C.H.C.; Kouwenaar, C.; Stafleu, F.R.; Bleich, A.; Ritskes-Hoitinga, M.; De Vries, R.B.M.; Meijboom, F.L.B. Animal to human translation: A systematic scoping review of reported concordance rates. J. Transl. Med. 2019, 17, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, B.A.; Sloan, D.J.; Jensen, T.C.; Ramaiahgari, S.C.; End, P.; Resh, G.E.; McClelland, R.E. A Unidirectional 96-Well Fluidic Culture Platform for Upstream Cell Dosing with Subsequent Downstream Nonlinear and Ascending Exposure Gradients for Real-Time and Cell-Based Toxicity Screening Environments. Appl. Vitr. Toxicol. 2021, 7, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T. Food for Thought Look Back in Anger—What Clinical Studies Tell Us About Preclinical Work. ALTEX 2013, 30, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Sacks, L.V.; Shamsuddin, H.H.; Yasinskaya, Y.I.; Bouri, K.; Lanthier, M.L.; Sherman, R.E. Scientific and Regulatory Reasons for Delay and Denial of FDA Approval of Initial Applications for New Drugs, 2000–2012. JAMA J. Am. Med. Assoc. 2014, 311, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Laverty, H.; Benson, C.; Cartwright, E.J.; Cross, M.J.; Garland, C.; Hammond, T.; Holloway, C.; McMahon, N.; Milligan, J.; Park, B.K.; et al. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br. J. Pharmacol. 2001, 163, 675–693. [Google Scholar] [CrossRef]
- Real, M.; Barnhill, M.S.; Higley, C.; Rosenberg, J.; Lewis, J.H. Drug-Induced Liver Injury: Highlights of the Recent Literature. Drug Saf. 2018, 42, 365–387. [Google Scholar] [CrossRef]
- McNaughton, R.; Huet, G.; Shakir, S. An investigation into drug products withdrawn from the EU market between 2002 and 2011 for safety reasons and the evidence used to support the decision-making. BMJ Open 2014, 4, e004221. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.L.; Baker, T.K. The future of drug safety testing: Expanding the view and narrowing the focus. Drug Discov. Today 2009, 14, 162–167. [Google Scholar] [CrossRef]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Correction to: Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2019, 17, 56. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Kaplowitz, N. Mechanisms of Drug-induced Liver Injury. Clin. Liver Dis. 2013, 17, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaemmaghami, A.; Hancock, M.J.; Harrington, H.; Kaji, H.; Khademhossein, A. Biomimetric tissues on a chip for drug discovery. Drug Discov. Today 2012, 17, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiati, S.; Kompella, U.B.; Damiati, S.A.; Kodzius, R. Microfluidics Devices for Drug Delivery Systems and Drug Screening. Genes 2018, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewing, S.; Boess, F.; Moisan, A.; Bertinetti-Lapatki, C.; Minz, T.; Hedtjaern, M.; Tessier, Y.; Schuler, F.; Singer, T.; Roth, A.B. Establishment of a Predictive In Vitro Assay for Assessment of the Hepatotoxic Potential of Oligonucleotide Drugs. PLoS ONE 2016, 11, e0159431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamola, P.J.; Maratou, K.; Wilson, P.A.; Rush, K.; Mullaney, T.; McKevitt, T.; Evans, P.; Ridings, J.; Chowdhury, P.; Roulois, A.; et al. Strategies for In Vivo Screening and Mitigation of Hepatotoxicity Associated with Antisense Drugs. Mol. Ther. Nucleic Acids 2017, 8, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Timm, M.; Saaby, L.; Moesby, L.; Hansen, E.W. Considerations regarding use of solvents in in vitro cell based assays. Cytotechnology 2013, 65, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, Y.; Sasaki, Y.; Terasaki, N.; Kawataki, T.; Katekawa, K.; Iwase, Y.; Shimizu, T.; Sanoh, S.; Ohta, S. Comparison of Drug Metabois and Its Related Hepatotoxic Effect in HepaRG, Cryopreserved Human Hepatocytes, and HepG2 Cell Cultures. Biol. Pharm. Bull. 2018, 41, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Geng, X.-C.; Wang, J.-F.; Miao, Y.-F.; Lu, Y.-L.; Li, B. The HepaRG cell line, a superior in vitro model to L-02, HepG2 and hiHeps cell lines for assessing drug-induced liver injury. Cell Biol. Toxicol. 2016, 32, 37–59. [Google Scholar] [CrossRef]
- Proctor, W.R.; Foster, A.J.; Vogt, J.; Summers, C.; Middleton, B.; Pilling, M.; Shienson, D.; Kijanska, M.; Ströbel, S.; Kelm, J.M.; et al. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Arch. Toxicol. 2017, 91, 2849–2863. [Google Scholar] [CrossRef]
- Garside, H.; Marcoe, K.F.; Chesnut-Speelman, J.; Foster, A.J.; Muthas, D.; Kenna, J.G.; Warrior, U.; Bowes, J.; Baumgartner, J. Evaluation of the use of imaging parameters for the detection of compound-induced hepatoxicity in 384-well cultures of HepG2 cells and cryopreserved primary human hepatocytes. Toxicol. In Vitro 2014, 28, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Byrne, B.; McDermott, O.; Noonan, J. Applying Lean Six Sigma Methodology to a Pharmaceutical Manufacturing Facility: A Case Study. Processes 2021, 9, 550. [Google Scholar] [CrossRef]
- EPA, U.S. Lean Thinking and Methods—Six Sigma. Available online: https://www.epa.gov/sustainability/lean-thinking-and-methods-six-sigma (accessed on 8 June 2021).
- Wheeler, D.J.; Chambers, D.S. Understanding Statistical Process Control, 2nd ed.; SPC Press: Knoxville, TN, USA, 1992; 406p. [Google Scholar]
- Nelson, L.S. The Shewhart Control Chart—Tests for Special Causes. J. Qual. Technol. 1984, 16, 237–239. [Google Scholar] [CrossRef]
- Mazaleuskaya, L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 2016, 25, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, A.; Chaker, J.; Léger, T.; Al-Salhi, R.; Dalgaard, M.D.; Styrishave, B.; Bury, D.; Koch, H.M.; Jegou, B.; Kristensen, D.M. Acetaminophen metabolism revisited using non-targeted analysis: Implications for human biomonitoring. Environ. Int. 2021, 149, 106388. [Google Scholar] [CrossRef] [PubMed]
- Vliegenthart, A.; Kimmitt, R.; Seymour, J.; Homer, N.; Clarke, J.; Eddleston, M.; Gray, A.; Wood, D.M.; Dargan, P.; Cooper, J.; et al. Circulating acetaminophen metabolites are toxicokinetic biomarkers of acute liver injury. Clin. Pharmacol. Ther. 2016, 101, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassouna, M.; Mohamed, M. Application of Lean Six Sigma Methodologies and In-Vitro Dissolution Studies for Simulta-neous Determination of Cefdinir and Sodium Benzoate by RP-HPLC and UPLC Methods in their Dosage Forms. Biomed. Sci. Technol. Res. 2019, 16, 12288–12300. [Google Scholar]
- Ghassabian, S.; Rawling, T.; Zhou, F.; Doddareddy, M.R.; Tattam, B.N.; Hibbs, D.E.; Edwards, R.J.; Cui, P.H.; Murray, M. Role of human CYP3A4 in the biotransformation of sorafenib to its major oxidized metabolites. Biochem. Pharmacol. 2012, 84, 215–223. [Google Scholar] [CrossRef]
- Kaplowitz, N.; Aw, T.Y.; Ookhtens, M. The regulation of hepatic glutathione. Annu. Rev. Pharmacol. Toxicol. 1985, 25, 715–744. [Google Scholar] [CrossRef]
- Sullivan, T. A Tough Road: Cost to Develop One New Drug Is $2.6 Billion; Approval Rate for Drugs Entering Clinical De-velopment is Less Than 12%. Available online: https://www.policymed.com/2014/12/a-tough-road-cost-to-develop-one-new-drug-is-26-billion-approval-rate-for-drugs-entering-clinical-de.html (accessed on 21 March 2019).
- Prasad, V.; Mailankody, S. Research and Development Spending to Bring a Single Cancer Drug to Market and Revenues After Approval. JAMA Intern. Med. 2017, 177, 1569–1575. [Google Scholar] [CrossRef]
- Herper, M. The Cost of Developing Drugs Is Insane. That Paper That Says Otherwise Is Insanely Bad. Available online: https://www.forbes.com/sites/matthewherper/2017/10/16/the-cost-of-developing-drugs-is-insane-a-paper-that-argued-otherwise-was-insanely-bad/?sh=6688f78f2d45 (accessed on 16 October 2017).
- Herper, M. The Cost of Creating A New Drug Now $5 Billion, Pushing Big Pharma To Change. Available online: https://www.forbes.com/sites/matthewherper/2013/08/11/how-the-staggering-cost-of-inventing-new-drugs-is-shaping-the-future-of-medicine/?sh=421de82e13c3 (accessed on 11 August 2013).
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the Toxicity of Pharmaceuticals in Humans and in Animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef]
- Stokes, W. Animals and the 3Rs in toxicology research and testing: The way forward. Hum. Exp. Toxicol. 2015, 34, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- EPA Press Office, U.S. EPA Finalizes Guidance to Waive Toxicity Tests on Animal Skin; U.S. Environmental Protection Agency, Ed.; EPA—Chemical Safety and Pollution Prevention: Washington, DC, USA, 2021.
- Eipel, C.; Abshagen, K.; Vollmar, B. Regulation of hepatic blood flow: The hepatic arterial buffer response revisited. World J. Gastroenterol. 2010, 16, 6046–6057. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejczak-Silicka, M. History of Cell Culture. In New Insights into Cell Culture Technology; IntechOpen: London, UK, 2017. [Google Scholar]
- Gebhardt, R.; Hengstler, J.G.; Müller, D.; Glöckner, R.; Buenning, P.; Laube, B.; Schmelzer, E.; Ullrich, M.; Utesch, D.; Hewitt, N.; et al. New hepatocyte in vitro systems for drug metabolism: Metabolic capacity and recommendations for ap-plication in basic research and drug development, standard operation procedures. Drug Metab. Rev. 2003, 35, 145–213. [Google Scholar] [CrossRef] [PubMed]
- Serras, A.S.; Rodrigues, J.S.; Cipriano, M.; Rodrigues, A.V.; Oliveira, N.G.; Miranda, J.P. A Critical Perspective on 3D Liver Models for Drug Metabolism and Toxicology Studies. Front. Cell Dev. Biol 2021, 9, 203. [Google Scholar] [CrossRef]
- Underhill, G.H.; Khetani, S.R. Advances in Engineered Human Liver Platforms for Drug Metabolism Studies. Drug Metab. Dispos. 2018, 46, 1626–1637. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Valadez, A.V.; Zuo, P.; Nie, Z. Microfluidic 3D cell culture: Potential application for tissue-based bioassays. Bioanalysis 2012, 4, 1509–1525. [Google Scholar] [CrossRef] [Green Version]
- Vollertsen, A.R.; De Boer, D.; Dekker, S.; Wesselink, B.A.M.; Haverkate, R.; Rho, H.S.; Boom, R.J.; Skolimowski, M.; Blom, M.; Passier, R.; et al. Modular operation of microfluidic chips for highly parallelized cell culture and liquid dosing via a fluidic circuit board. Microsyst. Nanoeng. 2020, 6, 107. [Google Scholar] [CrossRef]
- Lasser, K.E.; Allen, P.D.; Woolhandler, S.J.; Himmelstein, D.U.; Wolfe, S.M.; Bor, D.H. Timing of New Black Box Warnings and Withdrawals for Prescription Medications. JAMA: J. Am. Med. Assoc. 2002, 287, 2215–2220. [Google Scholar] [CrossRef] [Green Version]
- Siramshetty, V.B.; Nickel, J.; Omieczynski, C.; Gohlke, B.-O.; Drwal, M.N.; Preissner, R. WITHDRAWN—A resource for withdrawn and discontinued drugs. Nucleic Acids Res. 2015, 44, D1080–D1086. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Tayara, H.; Chong, K. Prediction of Drug-Induced Liver Toxicity Using SVM and Optimal Descriptor Sets. Int. J. Mol. Sci. 2021, 22, 8073. [Google Scholar] [CrossRef]
- Gupta, R.; Schrooders, Y.; Hauser, D.; van Herwijnen, M.; Albrecht, W.; ter Braak, B.; Brecklinghaus, T.; Castell, J.V.; Elenschneider, L.; Escher, S.; et al. Comparing in vitro human liver models to in vivo human liver using RNA-Seq. Arch. Toxicol. 2020, 95, 573–589. [Google Scholar] [CrossRef]
- Kammerer, S.; Küpper, J.-H. Human hepatocyte systems for in vitro toxicology analysis. J. Cell. Biotechnol. 2018, 3, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Bale, S.; Borenstein, J. Microfluidic Cell Culture Platforms to Capture Hepatic Physiology and Complex Cellular Interactions. Drug Metab. Dispos. 2018, 46, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tan, Z.; Huang, S.; Huang, Z.; Ou-Yang, D.; Zhou, H. Isozyme-specific induction of low-dose aspirin on cytochrome P450 in healthy subjects. Clin. Pharmacol. Ther. 2003, 73, 264–271. [Google Scholar] [CrossRef]
- Von Moltke, L.; Greenblatt, D.J.; Granda, B.W.; Grassi, J.M.; Schmider, J.; Harmatz, J.S.; Shader, R.I. Nefazodone, meta-chlorophenylpiperazine, and their metabolites in vitro: Cytochromes mediating transformation, and P450-3A4 inhibitory actions. Psychopharmacology 1999, 145, 113–122. [Google Scholar] [CrossRef]
- Palleria, C.; Roberti, R.; Iannone, L.F.; Tallarico, M.; Barbieri, M.A.; Vero, A.; Manti, A.; De Sarro, G.; Spina, E.; Russo, E. Clinically relevant drug interactions between statins and antidepressants. J. Clin. Pharm. Ther. 2019, 45, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Lu, S. Glutathione synthesis. Biochem. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Dysregulation of glutathione synthesis in liver disease. Liver Res. 2020, 4, 64–73. [Google Scholar] [CrossRef]
- Rohr-Udilova, N.; Bauer, E.; Timelthaler, G.; Eferl, R.; Stolze, K.; Pinter, M.; Seif, M.; Hayden, H.; Reiberger, T.; Schulte-Hermann, R.; et al. Impact of glutathione peroxidase 4 on cell proliferation, angiogenesis and cytokine production in hepatocellular carcinoma. Oncotarget 2018, 9, 10054–10068. [Google Scholar] [CrossRef]
- Choi, S. Nefazodone (Serzone) withdrawn because of hepatotoxicity. Can. Med. Assoc. J. 2003, 169, 1187. [Google Scholar]
- LiverTox. Clinical and Research Information on Drug-Induced Liver Injury [Internet]; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012; About LIVERTOX. [Updated 2020 July 1].
- Domansky, K.; Inman, S.W.; Serdy, J.G.; Dash, A.; Lim, M.H.M.; Griffith, L.G. Perfused multiwell plate for 3D liver tissue engineering. Lab A Chip 2009, 10, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | 96-well geometric footprint (½ area). 8 replicate flow channels (A-H) with linked wells. |
| External Dimensions: | Lab equipment agnostic. Compatible with most plate readers and most imagers. |
| Disposable Unit: | 1× use. Amendable application models such as modular cell-system configurations. |
| Cell Alignment & Analysis | Biologic support and functions. |
| Accessibility: | Cell harvesting (cellular analysis.). Media aspiration (fluid analysis). |
| Cell Phenotype: | Single cell phenotype. Multi-cell phenotypes. Cell-cell signaling. |
| Fluorescence Detection: | Optically clear wells. Image through plate bottom. Plate shall not auto-fluoresce. |
| Format | (x, y) well locations mimic traditional positioning. Z-height varies across 4 mm vertical. |
| Growth Area per Well: | Surface area is 0.167 cm2. 2D, flat surfaces. Side walls have edges for manufacturing. |
| Each Channel Includes: | Dose well (1); fluid regulator well (1); culture wells (9); waste well (1). |
| Device Composition: | Dosing wells (8); fluid regulator wells (8); culture wells (72); waste wells (8). |
| Z-Height Offset: | Wells 2–11, surfaces have 0.5 mm z-height offset amid adjacent (serially linked) wells. |
| Channels for Fluid Flow | Paths connect wells. Capillarity involves both 3-wall and 4-wall designs; air venting. |
| Fluid Traverse: | Passageway transport initiates in well 1 and terminates at well 12. Unidirectional flow. |
| Dose Well (Well 1): | Max vol. 500 μL. Fluid flow passageway from bottom of well 1 into top of well 2. |
| Regulator Well (Well 2): | Max vol. 100 μL. Force equilibrator w/fluid hydrodynamic. Fulcrum amid flow paths. |
| Culture Wells (Wells 3–11): | Working vol. of 30–100 μL; ½ area of 96-well format; 4 mm well height. |
| Sink Well (Well 12): | Max vol. 350 μL. Evaporation wick (syphon) pulls fluid from well 1 to well 12 (1-way). |
| Device Fluid Volume: | Per channel working volume is 1850 μL (1.85 mL). Per device (8 channels), 14.8 mL. |
| Fluidic Mixing in Wells: | Rapid mixing. Involves surface tension, laminar flow, well shapes, and fluid dispersion. |
| Flow Forces: | Gravity driven, hydrodynamics, capillarity, wicking. |
| Materials in Fabrication | Polystyrene, optically clear wells, conventional surface tension, base system with lid. |
| Thickness of Platform: | 0.5 mm polymer thickness; parameter for injection molding and QA/QC non-warping. |
| Smoothness: | Well bottoms. Avoid bubbles in resin (injection mold flow rate). Contamination free. |
| Manufacture Units | Mass produced, 2 components, injection molding. Large quantity production capacity. |
| Manufacture Components: | Lid (1 per unit). Row cover (8 per unit). Wick (8 per unit). |
| Assembly of Units: | Ultrasound welding of molded parts. No glue. No adhesive. No bonding contaminants. |
| Surface Treatment: | Tissue culture plastic (TCP). Can be surface coated (e.g., proteins, non-binding, etc.). |
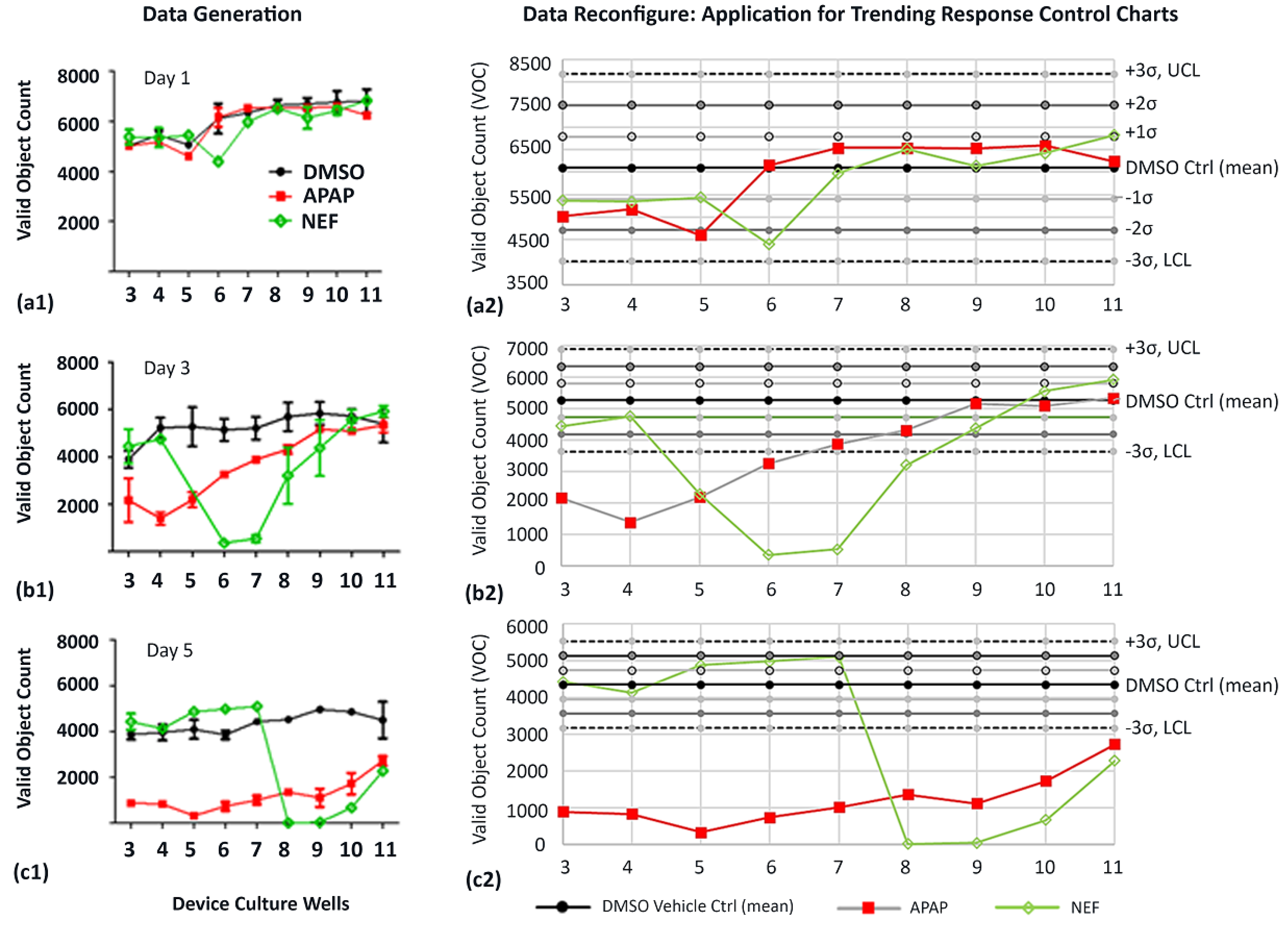
| Valid Obj Counts (Figure 7(a1,b1,c1)) | Well 3 | Well 7 | Well 11 |
|---|---|---|---|
| DMSO Vehicle Controls (Days 1, 3, 5) | (2.1%, 11.4%, 7.8%) | (2.6%, 13.0%, 0.64%) | (9.8%, 7.2%, 14.8%) |
| APAP (Days 1, 3, 5) | (2.8%, 20.1%, 12.8%) | (8.6%, 6.3%, 12.8%) | (1.9%, 8.2%, 5.8%) |
| NEF (Days 1, 3, 5) | (7.5%, 23.1%, 11.4%) | (3.3%, 5.8%, 0.04%) | (3.5%, 5.6%, 3.3%) |
| Tracing Fluid Flow Movements [23] (First Dose and Repeat Dosing) | Upstream (Wells 3–5) | Midstream (Wells 6–8) | Downstream (Wells 9–11) |
| ≤1 h after each dose into well 1 | not appraised | not appraised | not appraised |
| (acute hydrodynamic forces in action) | SOP waits 1 h before analysis. Fnc of user interface (manual pipette dosing). | ||
| 1 h > dosing ≤ 3.3 h | 5–8% | 5–6% | 4–6% |
| (transitional forces creating stability) | |||
| >3.3 h till re-dosing | 4–5% | 4–5% | 4–5% |
| (sustained equilibrium forces) | |||
| Day | DMSO Mean UCL/LCL | APAP Correlations Superimposed over DMSO Data | NEF Correlations Superimposed over DMSO Data |
|---|---|---|---|
| 1 | 6098.3 VOC 8173.8/4022.8 Figure 7(a1,a2) |
|
|
| Conjecture: APAP concentrations and exposure time not yet influenced cells | Conjecture: NEF concentrations and exposure time not yet influenced cells | ||
| 3 | 5265.7 VOC 6885.8/3645.6 Figure 7(b1,b2) “Time-delayed provocations” |
|
|
| Conjecture: APAP w/direct negative influence at highest conc in upstream wells. APAP conc dilutions are less detrimental in downstream wells | Conjecture: NEF w/o direct upstream influence. Generating cell-byproducts w/negative influence in midstream wells 5–8 but not yet reached downstream wells 9–11 | ||
| 5 | 4353.2 VOC 5534.9/3171.4 Figure 7(c1,c2) “Time-delayed provocations” |
|
|
| Conjecture: APAP w/direct negative influence at all ramped concentrations and time durations | Conjecture: NEF w/o upstream influence. Generating cell byproducts w/negative influence at downstream wells 8–11 |
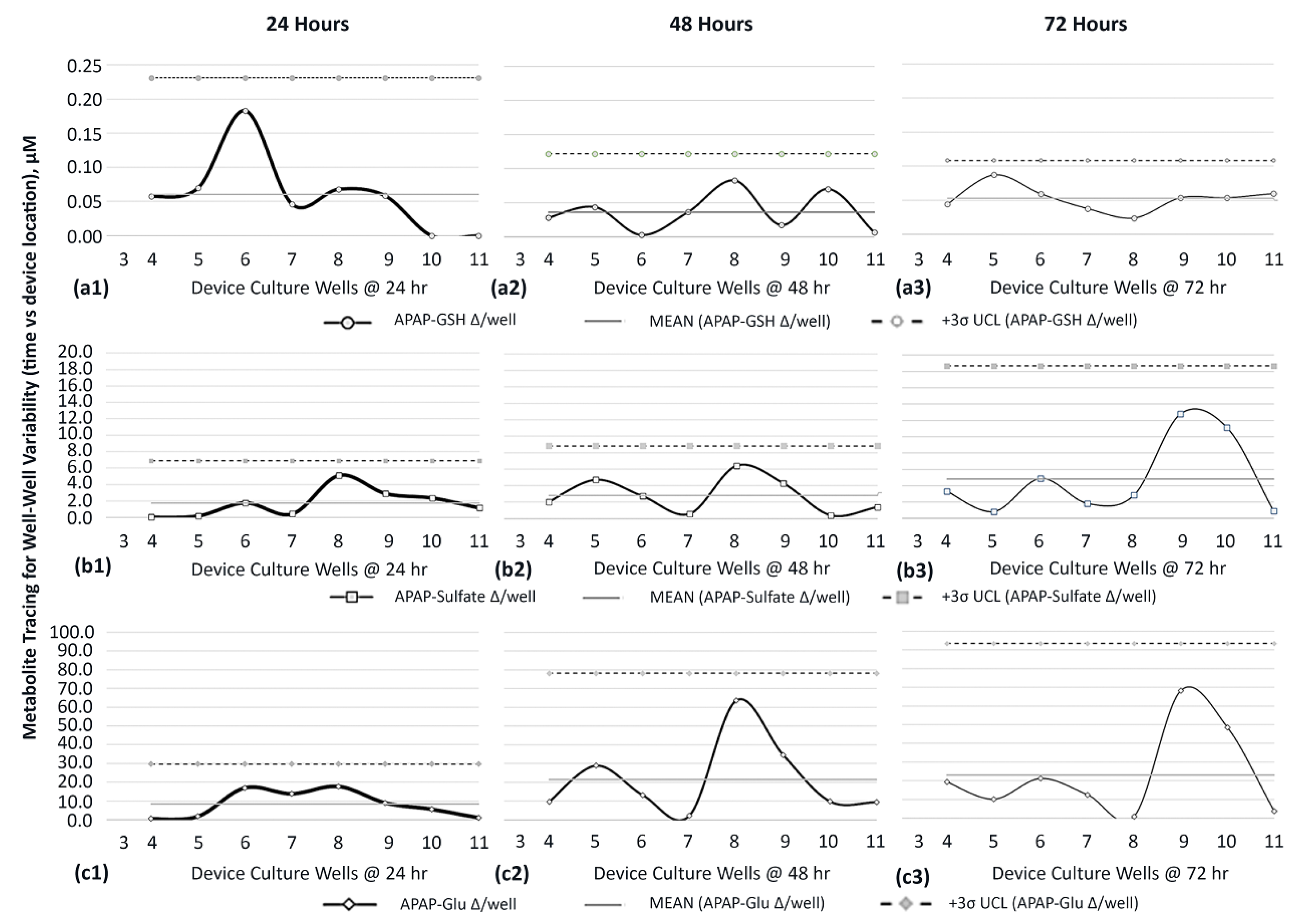
| APAP-GSH | Highest MR @ 24 h | Most well–well ∆ is 0.18 µM occurs at 24 h in midstream well 6 (Figure 9(a1)). |
| Mean MR @ 24 h | Uniform well–well ∆ is measured in wells 4, 7, 8, and 9 (0.05 µM) (Figure 9(a1)). | |
| Negligeable MR @ 24 h | No well–well ∆ found in downstream wells 10 and 11 (0.0 µM) (Figure 9(a1)). | |
| MR Compilations @ 24, 48, 72 h |
| |
| In Brief (Figure 9(a1–a3)): | 3σ UCL is maximum at 24 h and decreases over time to indicate that cell generated APAP-GSH has the highest variance during early APAP exposure periods in upstream sites (wells 3–5; 0.05 µM; Figure 9(a1)) and conditions midstream culture wells with cell byproducts (well 6; 0.18 µM). | |
| APAP-Sulfate | Highest MR @ 72 h | Most well–well ∆s are 12.79 µM and 11.11 µM occurring at 72 h in downstream wells 9 and 10 (Figure 9(b3)). |
| Mean MR @ 72 h | Uniform well–well ∆s are present in upstream wells 4–8 (0.84→4.9 µM) and downstream well 11 (0.9 µM) (Figure 9(b3)). | |
| Negligeable MR @ 24 h | Limited well–well ∆ found in upstream wells 4 and 5 (~0.0 µM) (Figure 9(b1)). | |
| MR Compilations @ 24, 48, 72 h |
| |
| In Brief (Figure 9(b1–b3)): | 3σ UCL is minimum at 24 h and increases over time to indicate that cell-generated APAP-GSH has the highest variance at later APAP exposure periods with both upstream sites and prolonged exposure times augmenting downstream device sites with conditioned media; Figure 9b3 wells 9 and 10. | |
| APAP-GLU | Highest MR @ 72 h | Most well–well variations are 68.1 µM & 48.6 µM occurring at 72 h in downstream wells 9 and 10 (Figure 9(c3)). |
| Mean MR @ 72 h | Uniform well–well ∆s are present in upstream wells 4–8 (0.91→21.24 µM) and downstream well 11 (3.9 µM) (Figure 9c3). | |
| Negligeable MR @ 24 h | Limited well–well ∆ found in upstream wells 4 and 5 (~0.0 µM) (Figure 9(c1)). | |
| MR Compilations @ 24, 48, 72 h |
| |
| In Brief (Figure 9(c1–c3)): | 3σ UCL is minimum at 24 h and increases over time to indicate that cell-generated APAP-Glu has high variance at both intermediate and late APAP exposure periods (Figure 9(c2) wells 8–9 and Figure 9(c3) wells 9–10). |
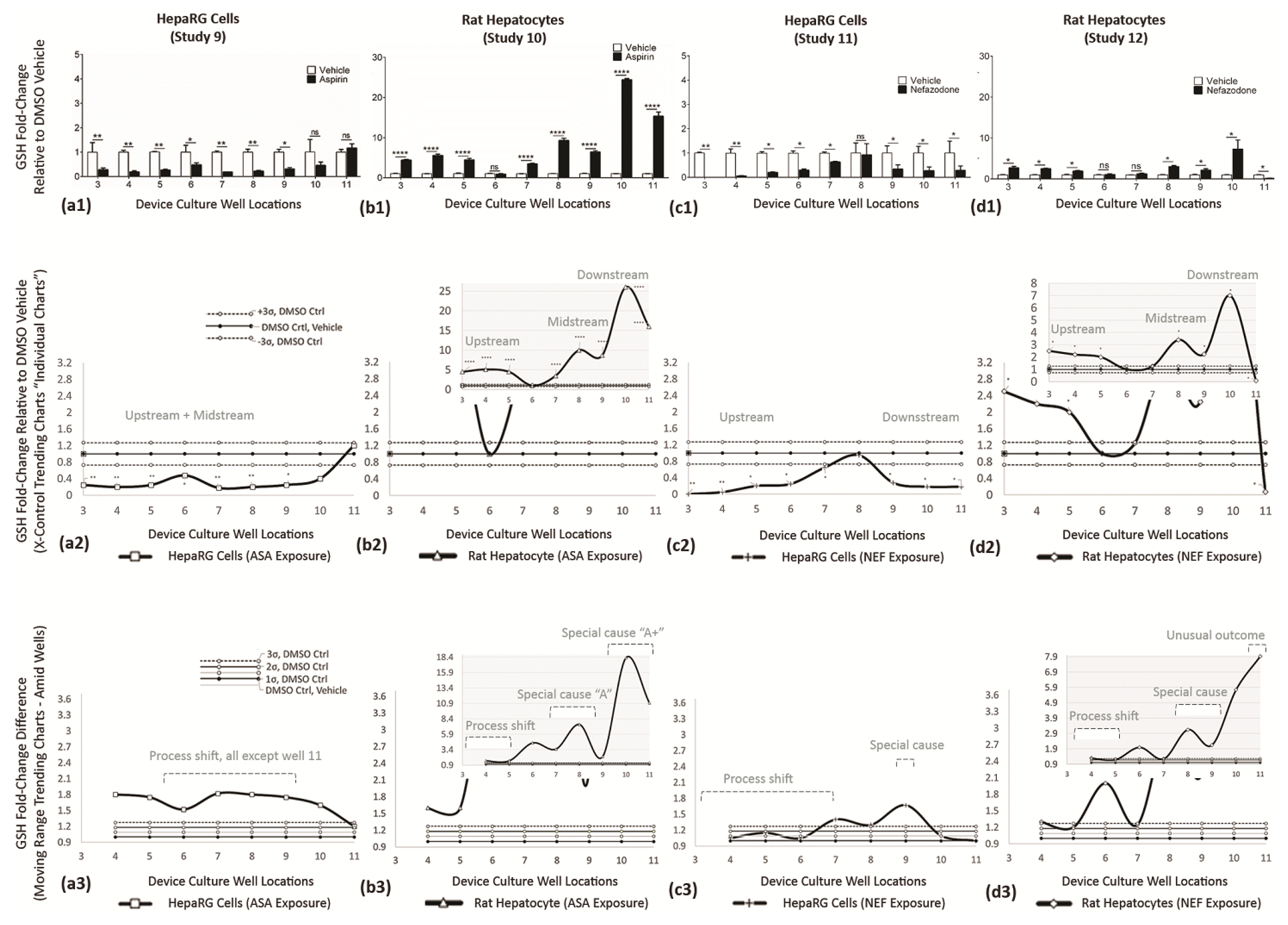
| Rows | Purpose | Description |
|---|---|---|
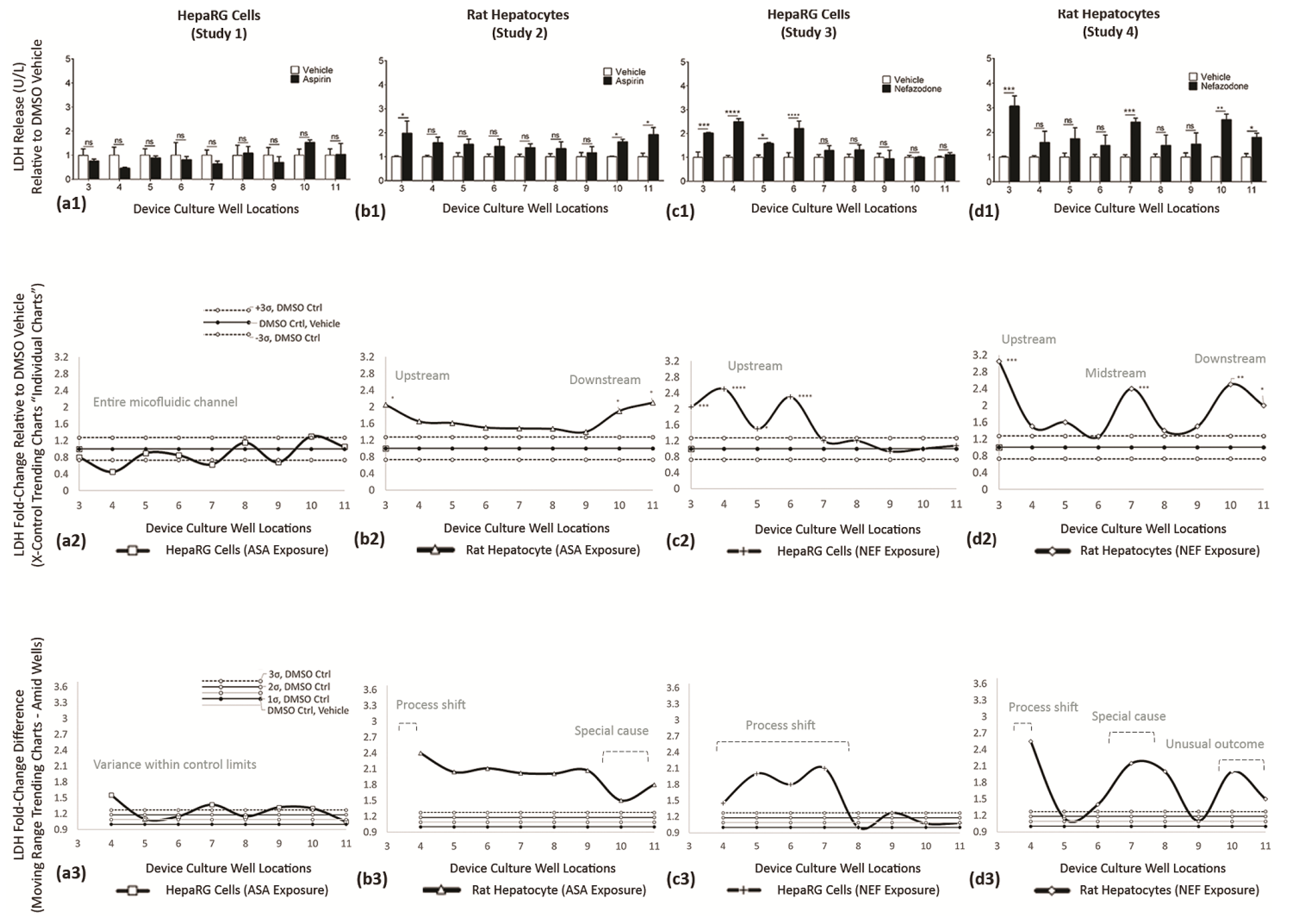
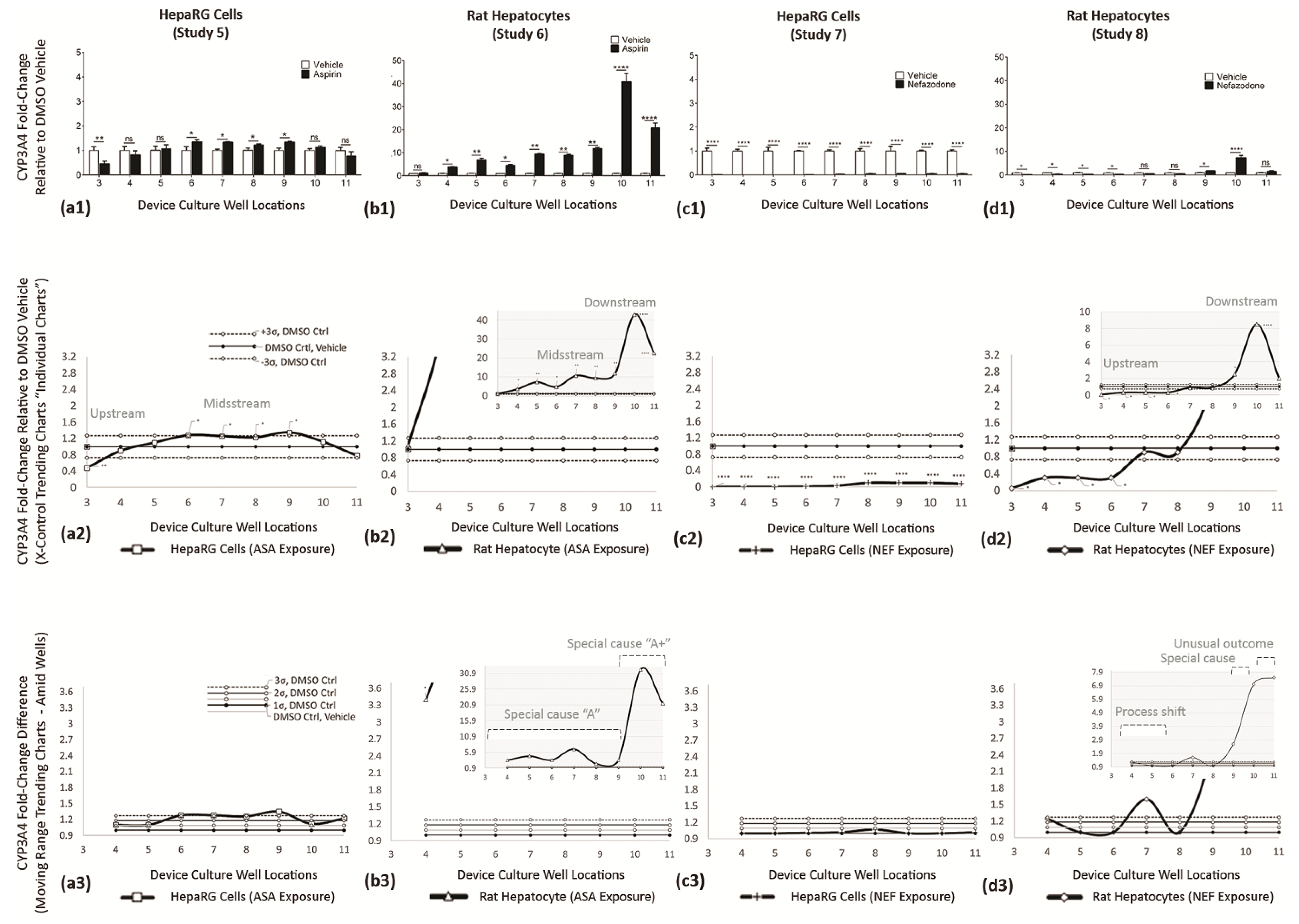
| a1→d1 | Reveal cell functions having discrete statistical analysis, i.e., Generation of Data. The vertical axis are fold-changes related to DMSO vehicle control. | DMSO vehicle studies (white bars) are normalized at 1, i.e., 100% normal function per well; the compound-of-interest data (black bars) are function fold-changes versus DMSO controls. |
| a2→d2 (system) | Re-expressions of data using X control charts, continuous systems (e.g., fluidics assembly line), to identify DMSO control averages (Mean) and ±3σ DMSO offsets (UCL and LCL). The vertical axis are fold-changes related to DMSO vehicle control. | Superimposed over DMSO controls are compound-of-interest results to reveal statistical shifts in cell bioactivity along upstream (wells 3–5), midstream (wells 6–8), and downstream (wells 9–11) cell stations. |
| a3→d3 (adjacent) | Display adjacent-well variations using MR control charts, i.e., Application to Diagnose Cell-Generated Stimuli. The vertical axis is fold-changed to probe well–well irregularities, magnitudes are absolute values, orientation with DMSO vehicle = 1 (line shifts @ 1). Standard deviations are +1σ, +2σ, +3σ. | Blending of system trends with adjacent-well analysis facilitate deductions for direct drug influence (i.e., process shift), indirect drug influence (i.e., special cause (cell generated secretions)), and indirect tertiary influence (i.e., unusual outcomes (secondary cell metabolite)). |
| Study 1 ASA on HepaRG cells | Trending Chart (system wide): | Discloses a cyclical culture response across the µfluidic channel with most datapoints inside ± 3σ DMSO boundaries, Figure 10(a2). |
| Variability Chart (adjacent well): | Shows well 4 is differentiable from wells 5–11 (1.6-fold vs. 0.9–1.35-fold), Figure 10(a3), yet the adjacent-well asymmetry is negligible as determined “ns” in Figure 10(a1). | |
| Blended Data (Figure 10(a1–a3)): | Infers ASA does not induce LDH release or incite hepatoxicity in HepaRG cells. | |
| Study 2 ASA on Rat Heps | Trending Chart (system wide): | Discloses an inverted bell curve response, Figure 10(b2), revealing rat hepatocytes have greater activity above HepaRG (1.35-to-2.2-fold vs. 0.5-to-1.3-fold). The rat hepatocytes up-, mid-, and downstream cell functions are tempered (i.e., fewer cyclical fluctuations) compared with HepaRG cells, Figure 10(b2) vs. Figure 10(a2). |
| Variability Chart (adjacent well): | Confirms irregular functions at upstream well 4 (i.e., 2.4-fold change) implying direct ASA exposure induces a process shift. Moreover, irregularity in downstream wells 10–11 infer indirect ASA exposures have special cause implications (e.g., cell generating impacts) as midstream wells have lower functions; Figure 10(b2,b3). | |
| Blended Data (Figure 10(b1–b3)): | Infers ASA-induced endogenous metabolites stimulate LDH release in downstream rat hepatocytes | |
| Study 3 NEF on HepaRG cells | Trending Chart (system wide): | Discloses upstream activity with fold changes 1.4 to 2.6 above DMSO controls, with cyclical data patterns; Figure 10(c2). |
| Variability Chart (adjacent well): | Confirms irregularities in upstream wells 4–5 (1.45 and 2.1-fold ∆) and midstream wells 6–7 (1.8 and 2.2-fold ∆); Figure 10(c3). | |
| Blended Data (Figure 10(c1–c3)): | Infers NEF directly induces an upstream process shift in HepaRG cells (wells 3–6); i.e., direct NEF influence. | |
| Study 4 ASA on Rat Heps | Trending Chart (system wide): | Discloses three cell-function peaks to at upstream well 3 (3.1-fold ∆), midstream well 7 (2.3-fold ∆), and downstream well 10 (2.4-fold ∆) that are interspersed between normal cell-function outcomes; Figure 10(d2). |
| Variability Chart (adjacent well): | Confirms irregularities in upstream well 4 (i.e., process shift = direct NEF influence), midstream wells 7–8 (i.e., special cause, cell secretions, or generated metabolite), and downstream wells 10–11 (i.e., unusual outcome or tertiary/secondary metabolite = indirect NEF influence); Figure 10(d3). | |
| Blended Data (Figure 10(d1–d3)): | Infers NEF directly, indirectly, and tertiary stimulate LDH release in rat heps. |
| Study 5 ASA on HepaRG cells | Trending Chart (system wide): | Discloses a bell curve response having inferior function at upstream well 3 (0.4-fold ∆), maximum functions in midstream wells 6–9 (1.3-fold ∆), and diminishing function at downstream well 11 (0.7-fold ∆); Figure 11(a2). |
| Variability Chart (adjacent well): | Confirms irregularities in upstream well 4 (process shift = direct ASA influence), and midstream wells 6–9 (special cause or generated metabolite = indirect ASA influence) given that well 5 has lower functions; Figure 11(a3). | |
| Blended Data (Figure 11(a1–a3)): | Infers ASA directly and indirectly stimulate CYP3A4 activity in HepaRG cells. | |
| Study 6 ASA on Rat Heps | Trending Chart (system wide): | Discloses an exponential growth curve response, Figure 11(b2), revealing rat hepatocyte CYP3A4 functions 7.5× to 33.1× are higher than HepaRG functions, ranges being 3.0-to-43.0 fold versus 0.4-to-1.3 fold, respectively. |
| Variability Chart (adjacent well): | Confirms irregularities having partitioned influences. Influence-1, well 3, being unchanged and replicates DMSO control functions (i.e., no direct ASA influence = no process shift). Influence-2, wells 4–9, have upregulated CYP3A4 at 1.5-to-7.0-fold ranges (i.e., special cause A (e.g., metabolite A = indirect ASA influence)). Influence-3, wells 10–11, have top-heavy upregulated CYP3A4 with 20-to-40-fold ranges (i.e., special cause A↑ (e.g., additional cell generating impacts = indirect ASA influence)); Figure 11(b3). | |
| Blended Data (Figure 11(b1–b3)): | Infers ASA endogenous metabolites stimulate CYP3A4 induction in rat hepatocytes, i.e., wells 4–11. | |
| Study 7 NEF on HepaRG cells | Trending Chart (system wide): | Discloses NEF outcomes with fold levels 0.9–1.0-fold below the DMSO vehicle control. A marginal rise is evident at wells 7–8; Figure 11(c2). |
| Variability Chart (adjacent well): | Confirms activity as minimal, no irregularity across the entire µchannel (i.e., a process shift as direct NEF exposure inhibits CYP3A4 functions); Figure 11(c3). | |
| Blended Data (Figure 11(c1–c3)): | Infers that NEF directly impedes CYP3A4 in HepaRG cells. | |
| Study 8 ASA on Rat Heps | Trending Chart (system wide): | Discloses three notable cell-function influencers; Influence-1 with upstream wells 3–6 at fold levels 0.6–1.0 below the DMSO vehicle control, Influence-2 shows wells 9–10 at fold levels 2.3-to-8.3-fold above DMSO control, Influence-3 displays downstream well 11 at 1.9-fold above the DMSO control; Figure 11(d2). |
| Variability Chart (adjacent well): | Confirms irregularities in upstream wells 4–6 (i.e., process shift = direct NEF influence), highly irregular in downstream wells 9–10 (i.e., special cause or generated metabolite = indirect NEF influence), and a decline in downstream well 11 (i.e., unusual outcome = tertiary indirect NEF influence); Figure 11(d3). | |
| Blended Data (Figure 11(d1–d3)): | Infers NEF has direct, indirect, and tertiary influences on CYP3A4 mutable responses in rat hepatocytes. |
| Study 9 ASA on HepaRG cells | Trending Chart (systemwide): | Discloses a delayed exponential growth curve with inferior functions upstream and midstream (0.2–0.6-fold ∆) and ramping at downstream well 11 (1.19-fold ∆). Only well 11 resides in ±3σ DMSO boundary limits; Figure 12(a2). |
| Variability Chart (adjacentwell): | Confirms functions remain similar across the entire µchannel (not well 11), inferring direct ASA exposure induces a process shift; Figure 12(a3). | |
| Blended Data (Figure 12(a1–a3)): | Infers ASA directly impedes GSH in HepaRG cells. | |
| Study 10 ASA on Rat Heps | Trending Chart (system wide): | Disclose an atypical-inverted bell curve response with upstream increases (4.5 to 5.8-fold), midstream DMSO vehicle levels at 1.0, a 2nd midstream increase (3.0 to 9.0-fold), and ramping downstream activity at 15.0 to 26.5-fold ∆’s; Figure 12b2. |
| Variability Chart (adjacent well): | Confirms functions are similar in upstream wells 3–5 (process shift = direct ASA influence), then upregulated in midstream wells 7–9 (3.4 to 7.0-fold) occurring after non-significant well 6, (indirect ASA influence = special cause A (e.g., metabolite A)), then top-heavy upregulation in wells 10–11 (10.9 to 18.4-fold), i.e., indirect ASA influence = special cause A↑ (e.g., additional cell generating impacts); Figure 12(b3). | |
| Blended Data (Figure 12(b1–b3)): | Infers ASA metabolites stimulate GSH induction in rat hepatocytes (wells 7–11). | |
| Study 11 NEF on HepaRG cells | Trending Chart (system wide): | Discloses a bell curve response having upstream wells below the DMSO LCL (−3σ), well 8 approaching DMSO control at 1, and downstream wells below the DMSO LCL (−3σ); Figure 12(c2). |
| Variability Chart (adjacent well): | Confirms functions are similar in upstream wells 4–7 (process shift = direct NEF influence) and similar in downstream wells 9–11 (special cause or generated metabolite = indirect NEF influence); Figure 12(c3). | |
| Blended Data (Figure 12(c1–c3)): | Infers that NEF directly and indirectly impedes GSH in HepaRG cells. | |
| Study 12 ASA on Rat Heps | Trending Chart (system wide): | Discloses an inverted bell curve response having segmented influencers; Influence-1 in upstream wells 3–5 at fold levels 2.0–2.5 above the DMSO vehicle control; Influence-2 in wells 8–10 at fold levels 2.0–7.0 above DMSO controls, Influence-3 in downstream well 11 displaying fold-level 0.1 below the DMSO control; Figure 12(d2). |
| Variability Chart (adjacent well): | Confirms functions are similar in upstream wells 3–5 (process shift = direct NEF influence), returns to DMSO levels at well 7, becomes upregulated in downstream wells 8–10 (special cause or generated metabolite = indirect NEF influence), then experiences an irregularity in downstream well 11 (infers an unusual outcome); Figure 12(d3). | |
| Blended Data (Figure 12(d1–d3)): | Infers NEF directly, indirectly, and tertiary stimulate GSH mutable responses in rat hepatocytes. |
| Study | Feature | {-----Upstream-----} Device Culture Wells {--Downstream--} | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Phenotype | Exposure Drug | Cell Function | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Ctrl | ALL | DMSO | ALL | DMSO vehicle control: 0.1% dimethyl sulfoxide (labeled as 0.1 v/v) Establishing baseline and standard cell culture limits = Normalized at 1 | ||||||||
| 1 | HepaRG | ASA | LDH | w/i control limits (i.e., not significant [ns] compared with the DMSO vehicle) | ||||||||
| 3 | HepaRG | NEF | LDH | Process Shift (↑fnc) | w/i Control Limits | |||||||
| 5 | HepaRG | ASA | CYP3A4 | Process Shift (↓fnc) | w/i Control Limits | Special Cause (* minimal ↑fnc) | w/i Control Limits | |||||
| 7 | HepaRG | NEF | CYP3A4 | Process Shift (↓fnc) | ||||||||
| 9 | HepaRG | ASA | GSH | Process Shift (↓fnc) | w/i Control Limits | |||||||
| 11 | HepaRG | NEF | GSH | Process Shift (↓fnc) | w/i Control Limits | Special Cause (*minimal ↓fnc) | ||||||
| 2 | Rat Hep | ASA | LDH | Process Shift (↑fnc) | w/i Control Limits | Special Cause (↑fnc) | ||||||
| 4 | Rat Hep | NEF | LDH | Process Shift (↑fnc) | w/i Control Limits | Special Cause (↑fnc) | w/i Control Limits | Unusual Outcome (↑fnc) | ||||
| 6 | Rat Hep | ASA | CYP3A4 | w/i Control Limits | Special Cause (A); (↑fnc) | Special Cause (A+); (↑fnc) | ||||||
| 8 | Rat Hep | NEF | CYP3A4 | Process Shift (↓fnc) | w/i Control Limits | Special Cause (↑fnc) | Unusual Outcome (↓fnc) | |||||
| 10 | Rat Hep | ASA | GSH | Process Shift (↑fnc) | w/i Control Limits | Special Cause (A); (↑fnc) | Special Cause (A↑); (↑fnc) | |||||
| 12 | Rat Hep | NEF | GSH | Process Shift (↑fnc) | w/i Control Limits | Special Cause (↑fnc) | Unusual Outcome (↓fnc) | |||||
| Company | Device | Transport | Microenvironment |
|---|---|---|---|
| BellBrook | IUVO | Cell movement through ECM | Micro conduits for invasion cell analysis across 3D environments |
| BioIVT | HepatoPac | Static co-cultures | Liver modules that are based culture patterning for segmented hepatocytes and stromal cells. |
| CNBio | Microphysiologic Platforms | Mixing chamber and recirculating flow | Scaffolds for 3D tissue formation and transwells for basal component re-circulation |
| Emulate | Organ on Chips | Molecular scaffolds in microchannels | Suite of organ-chip models with automated flow, instruments, consumables, and software; microfluidic channels lined with cells |
| Hurel | Hurelflow | Recirculating | Three devices on a biochip. Each device with two cell compartments aligning co-cultures with reactive metabolites |
| InSphero | Akura Platforms | Gravity driven and rocker | InSight microtissue models in 3D for co-culture liver cells. Microslides, robotics, and cell spheroid pipette transfers |
| Kirkstall | Quasi Vivo | Recirculating | Interconnected flows, peristaltic pump, chambers for cultures, tubes for media transports, and Lego-like building system. |
| Millipore | CellAsic ONIX2 | Flow gradients | Automated platform, manipulation of culture parameters, and continuous culture observation |
| Mimetas | Organoplate | Gravity driven | Perfusable vascular liver models with Phaseguide meniscus channels and barriers for chemical and compound gradients |
| TissUse | Human on Chip | Micropump chips and dynamic circulations | To simulate activity of multiple connected human organs; systemic. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lubamba, B.; Jensen, T.; McClelland, R. Rapid Detection of Direct Compound Toxicity and Trailing Detection of Indirect Cell Metabolite Toxicity in a 96-Well Fluidic Culture Device for Cell-Based Screening Environments: Tactics in Six Sigma Quality Control Charts. Appl. Sci. 2022, 12, 2786. https://doi.org/10.3390/app12062786
Lubamba B, Jensen T, McClelland R. Rapid Detection of Direct Compound Toxicity and Trailing Detection of Indirect Cell Metabolite Toxicity in a 96-Well Fluidic Culture Device for Cell-Based Screening Environments: Tactics in Six Sigma Quality Control Charts. Applied Sciences. 2022; 12(6):2786. https://doi.org/10.3390/app12062786
Chicago/Turabian StyleLubamba, Bob, Timothy Jensen, and Randall McClelland. 2022. "Rapid Detection of Direct Compound Toxicity and Trailing Detection of Indirect Cell Metabolite Toxicity in a 96-Well Fluidic Culture Device for Cell-Based Screening Environments: Tactics in Six Sigma Quality Control Charts" Applied Sciences 12, no. 6: 2786. https://doi.org/10.3390/app12062786
APA StyleLubamba, B., Jensen, T., & McClelland, R. (2022). Rapid Detection of Direct Compound Toxicity and Trailing Detection of Indirect Cell Metabolite Toxicity in a 96-Well Fluidic Culture Device for Cell-Based Screening Environments: Tactics in Six Sigma Quality Control Charts. Applied Sciences, 12(6), 2786. https://doi.org/10.3390/app12062786