
Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part I: Emerging Platforms and Perspectives



Abstract
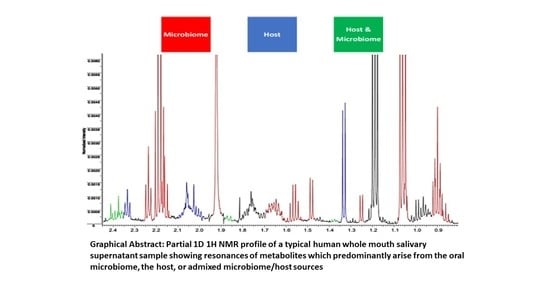
:
1. Introduction to NMR-Based Salivary/Oral Fluid Metabolomics
2. Sources and Composition of Human Saliva: Host and Microbial Contributions
Nutrient Availability for the Oral Microbiome
3. Advantages and Disadvantages of Saliva as a Bioanalytical Medium for the Seeking, Identification, and Determination of Biomarkers
3.1. Ease of Collection and Metabolite Concentration Considerations
3.2. Value of NMR Analysis Techniques for the Metabolomics Analysis of Human Saliva
4. Types of Salivary Metabolomics Experiments
5. Brief Early History of Salivary NMR Analysis and Associated Metabolomics Technology Investigations
6. Recommended Protocols for the Collection, Storage, and Preparation of Human Saliva Specimens for NMR Analysis: Precautions against Artefacts, and Artefactual Metabolite Generation or Consumption
6.1. Saliva Sample Collection
6.2. Abstention from Oral Activities Prior to Saliva Sample Donation: How Long Is Really Necessary?
6.3. Post-Collection Maintenance of Salivary Biomolecular Profiles: Preparation, Transport, and Storage of Saliva Samples for NMR Analysis
7. NMR Pulse Sequences: Recent Advances and Future Prospects
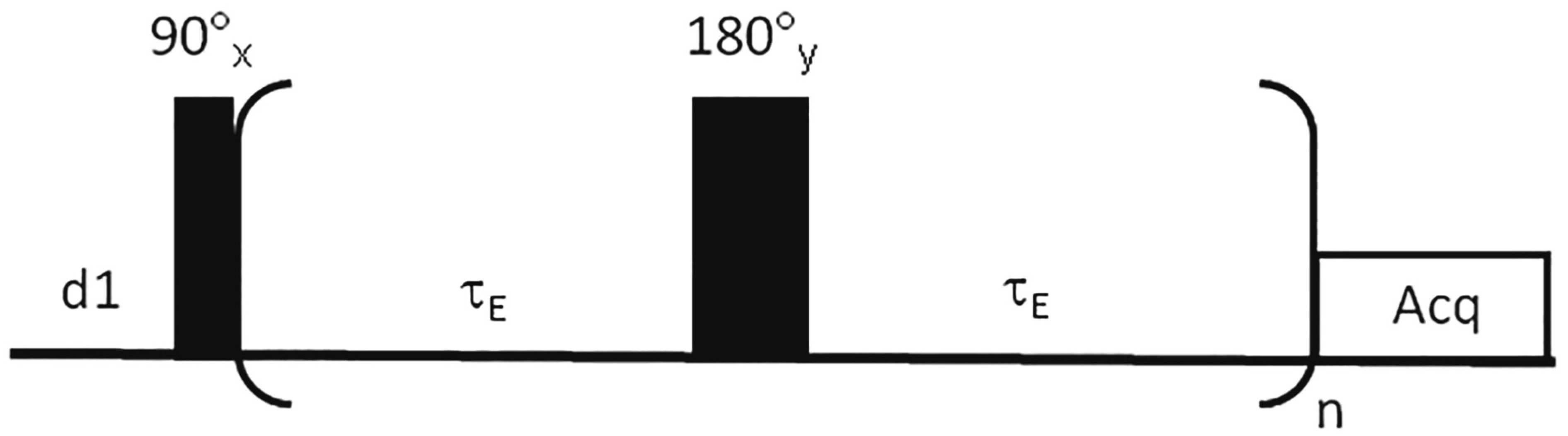
7.1. Value Offered by the Application of Spin-Echo (CPMG) Pulse Sequences to Human Saliva Sample Analysis
7.2. ‘State-of-the Art’ Advances beyond the CPMG Pulse Sequence
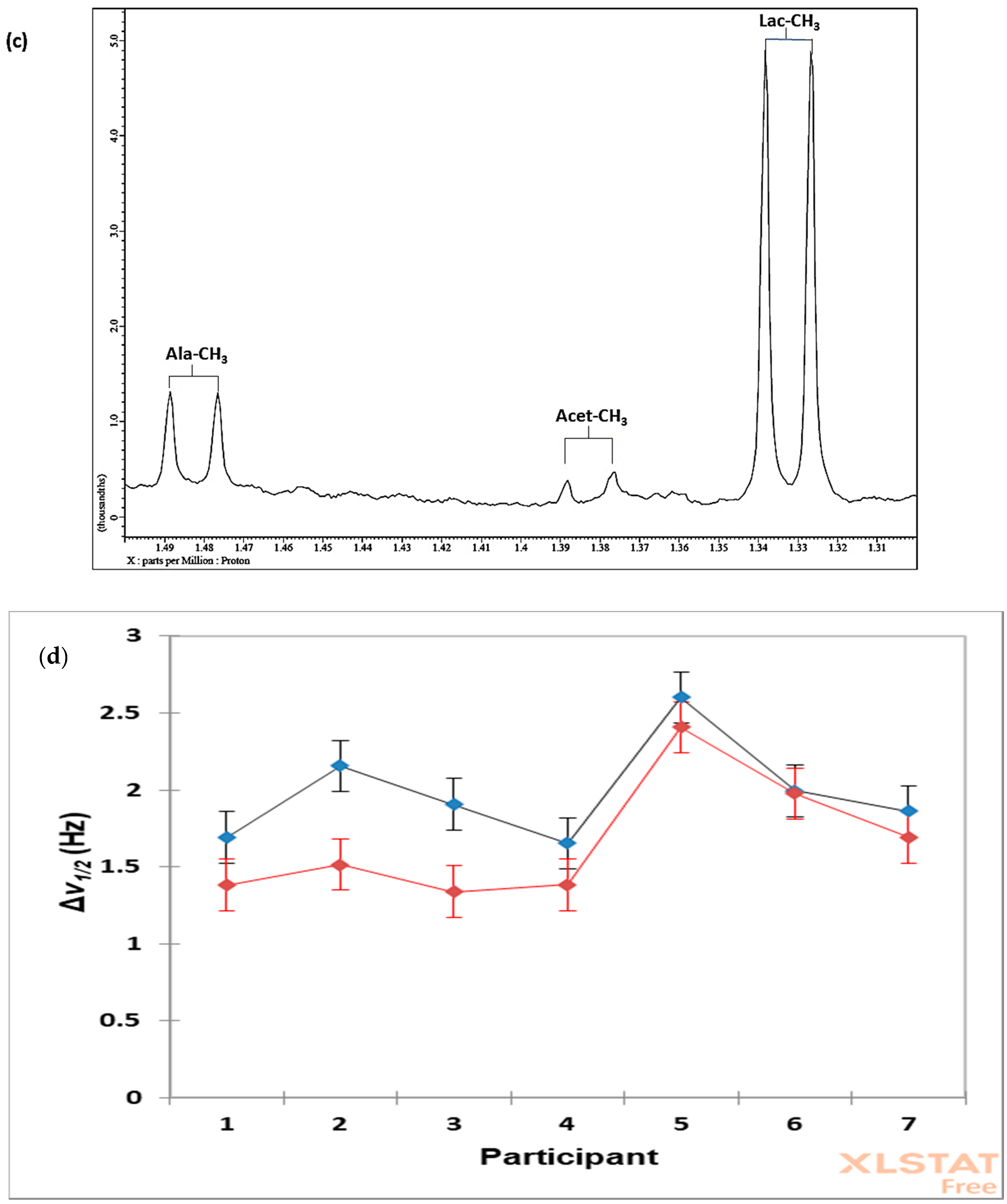
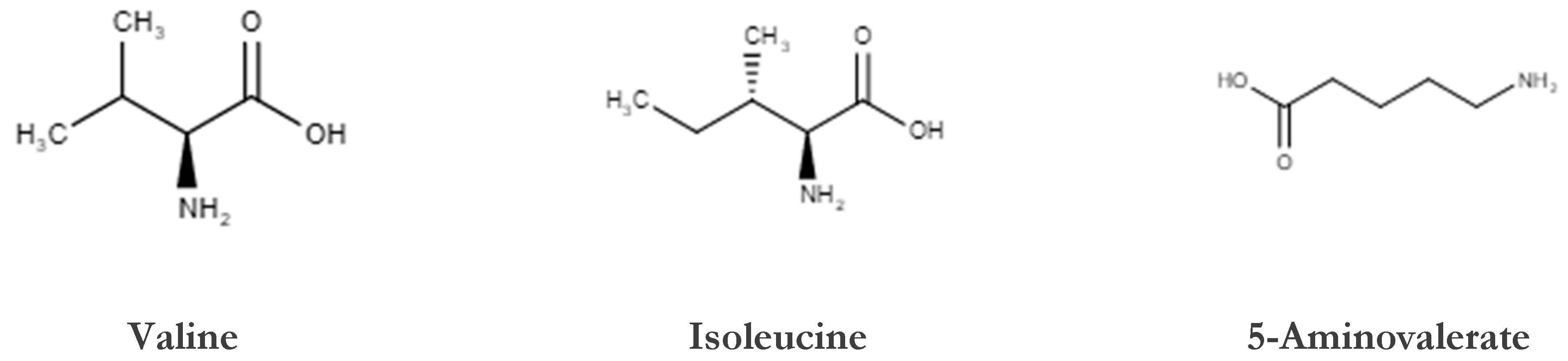
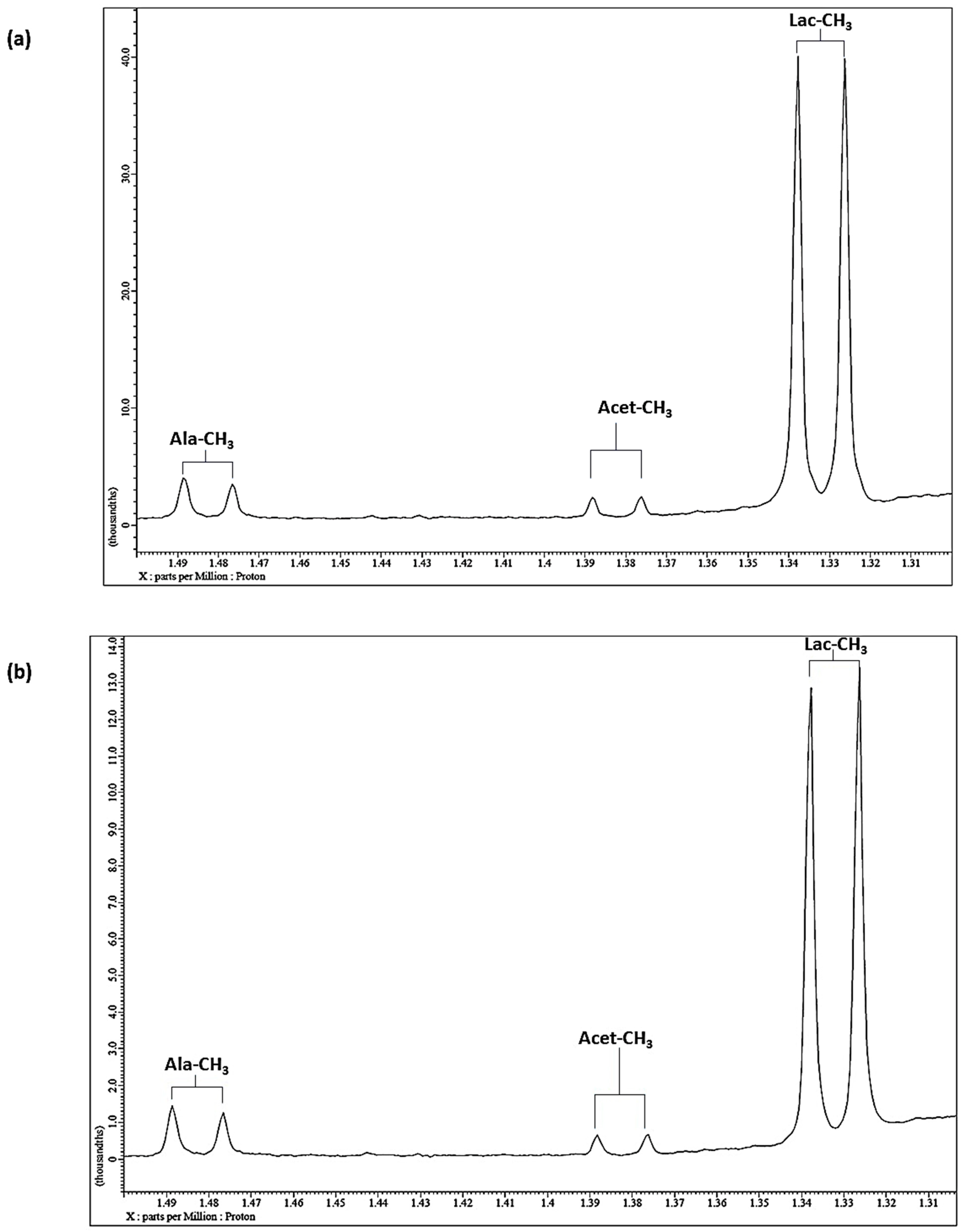
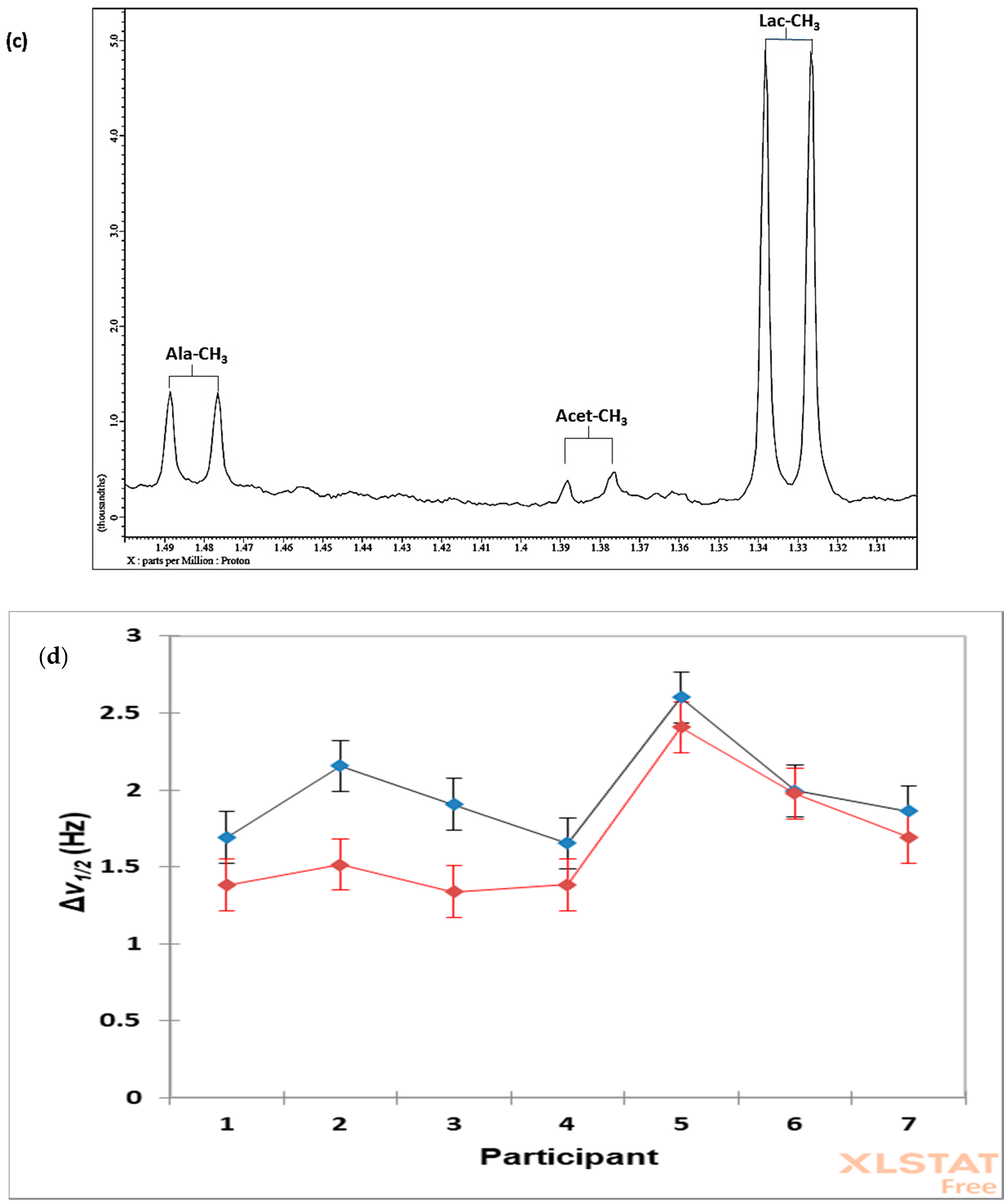
7.3. Case Study 1: Investigation of the Influences of Sample Donor, Metabolite Identity, and Doublet Resonance Line δ Value on the Δv1/2 Values of Lactate and Alanine in Single-Pulse 1H NMR Spectra Acquired on WMS Supernatants—A Design of Experiments Approach
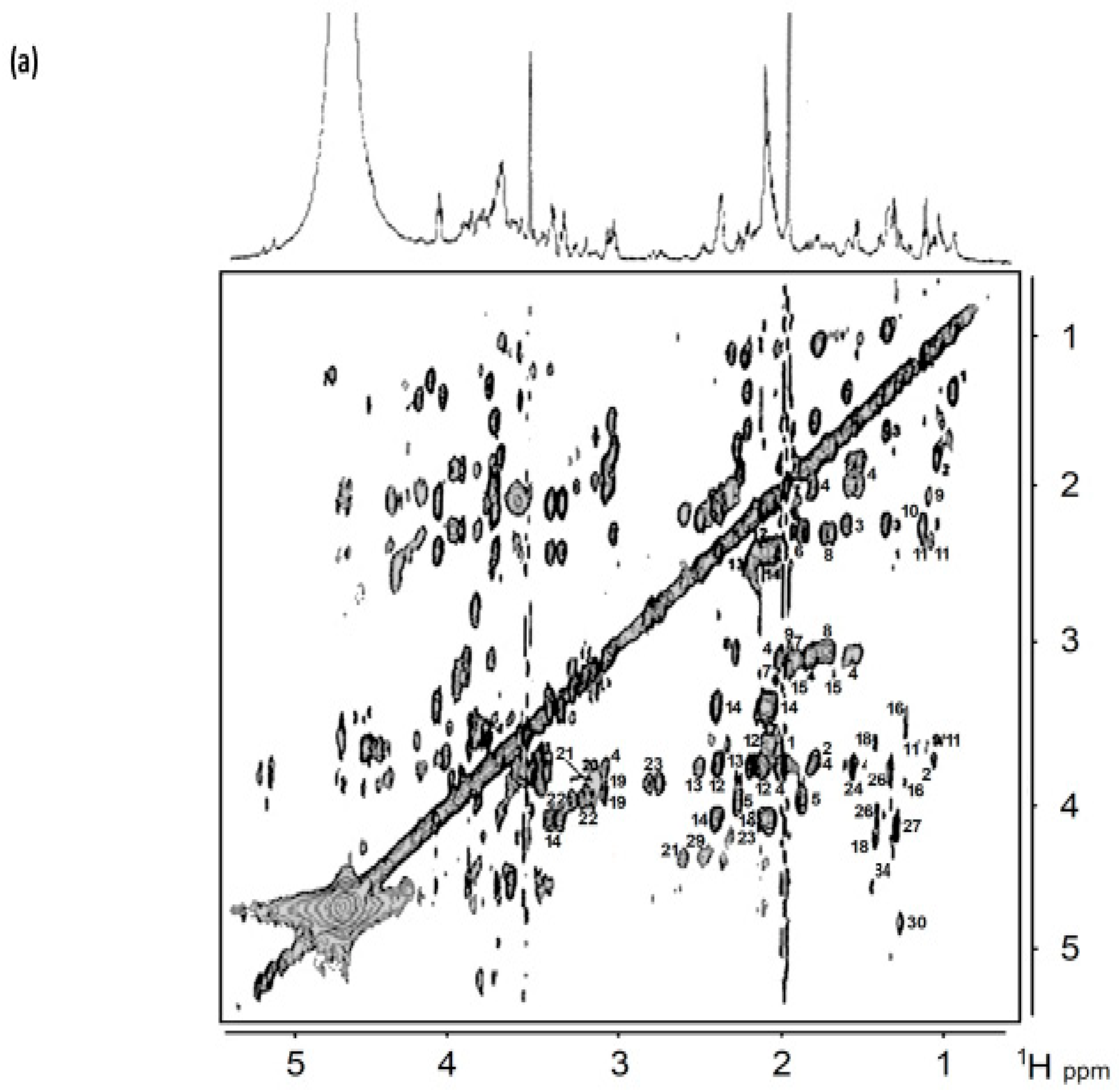
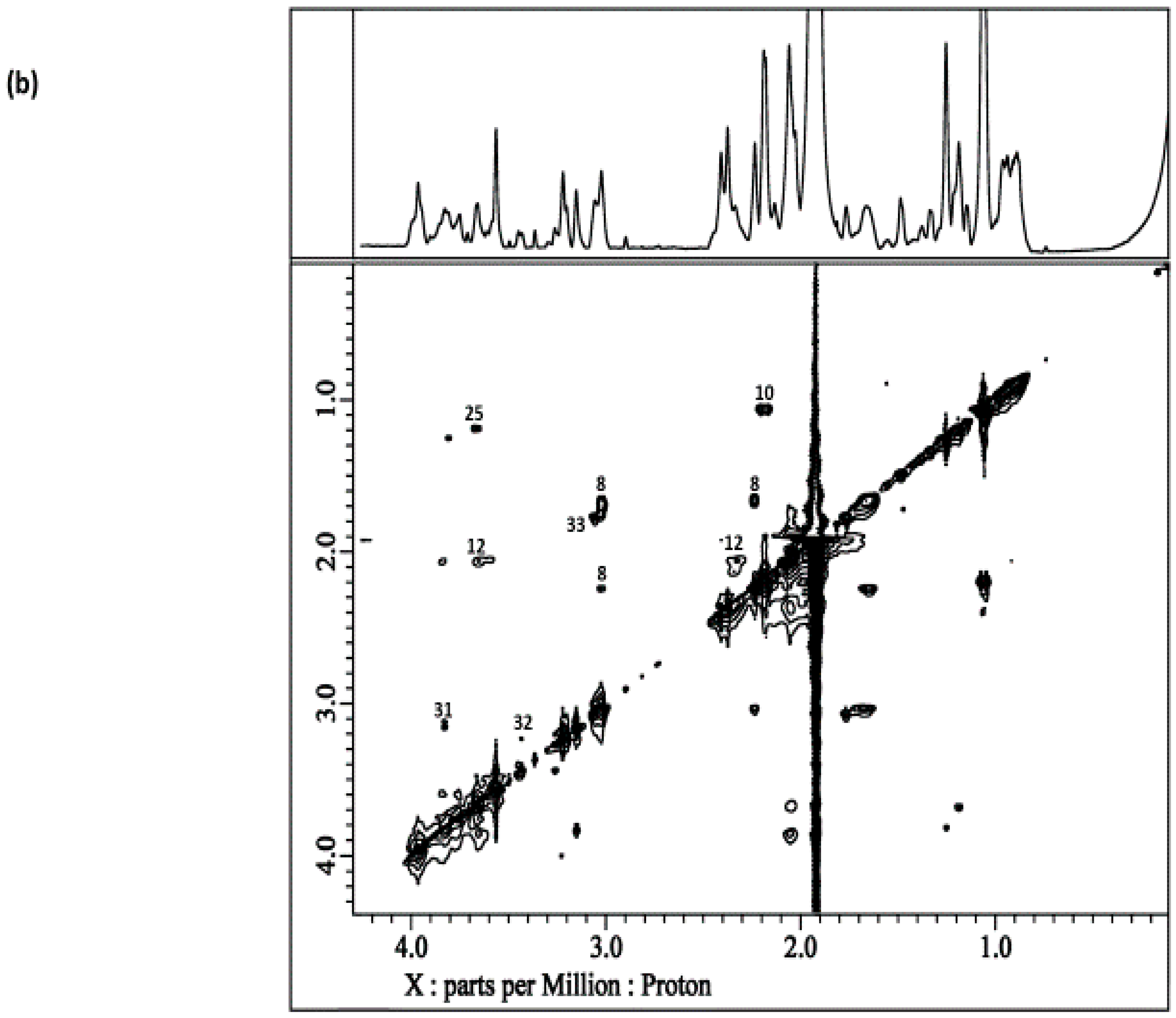
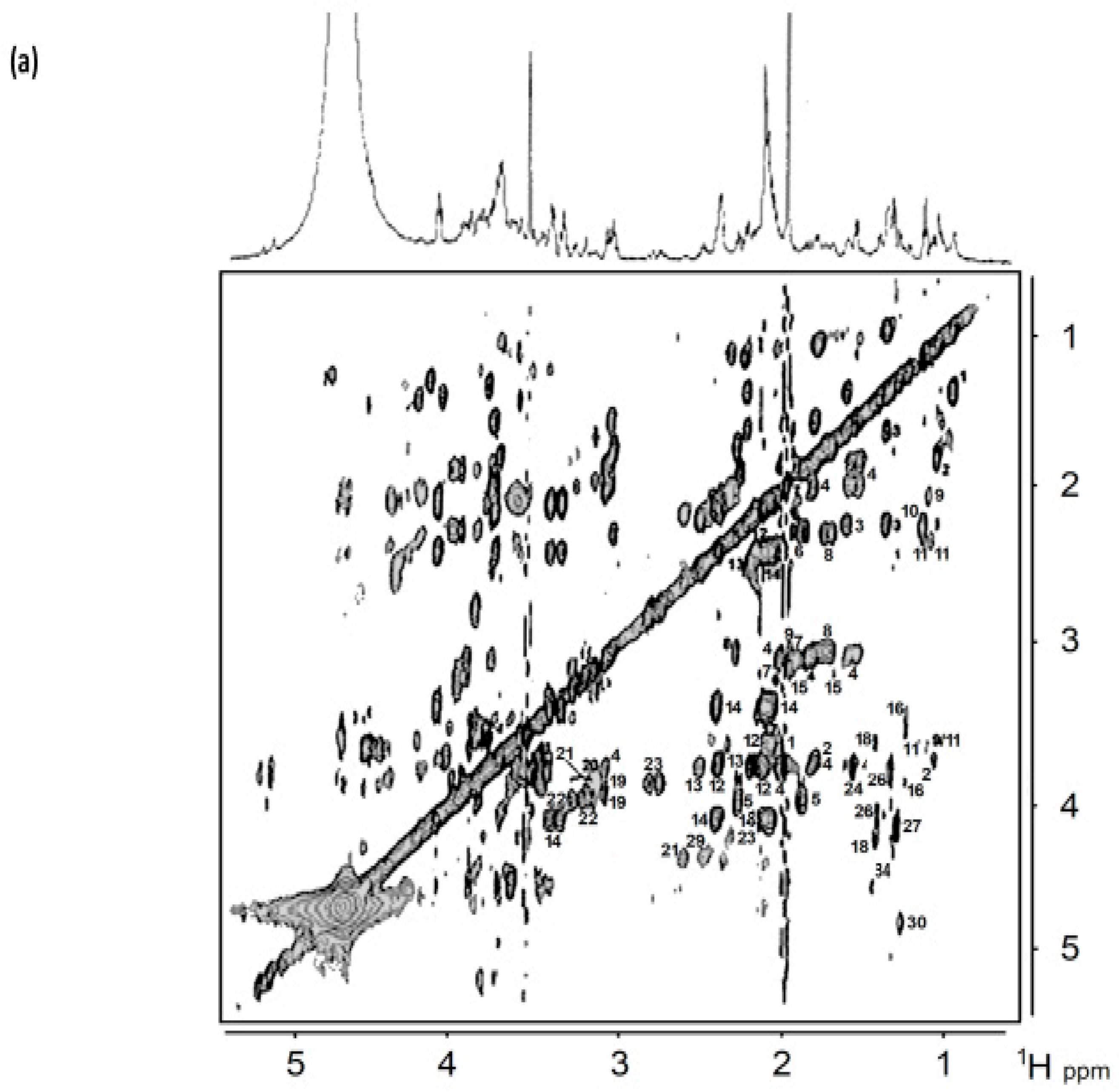
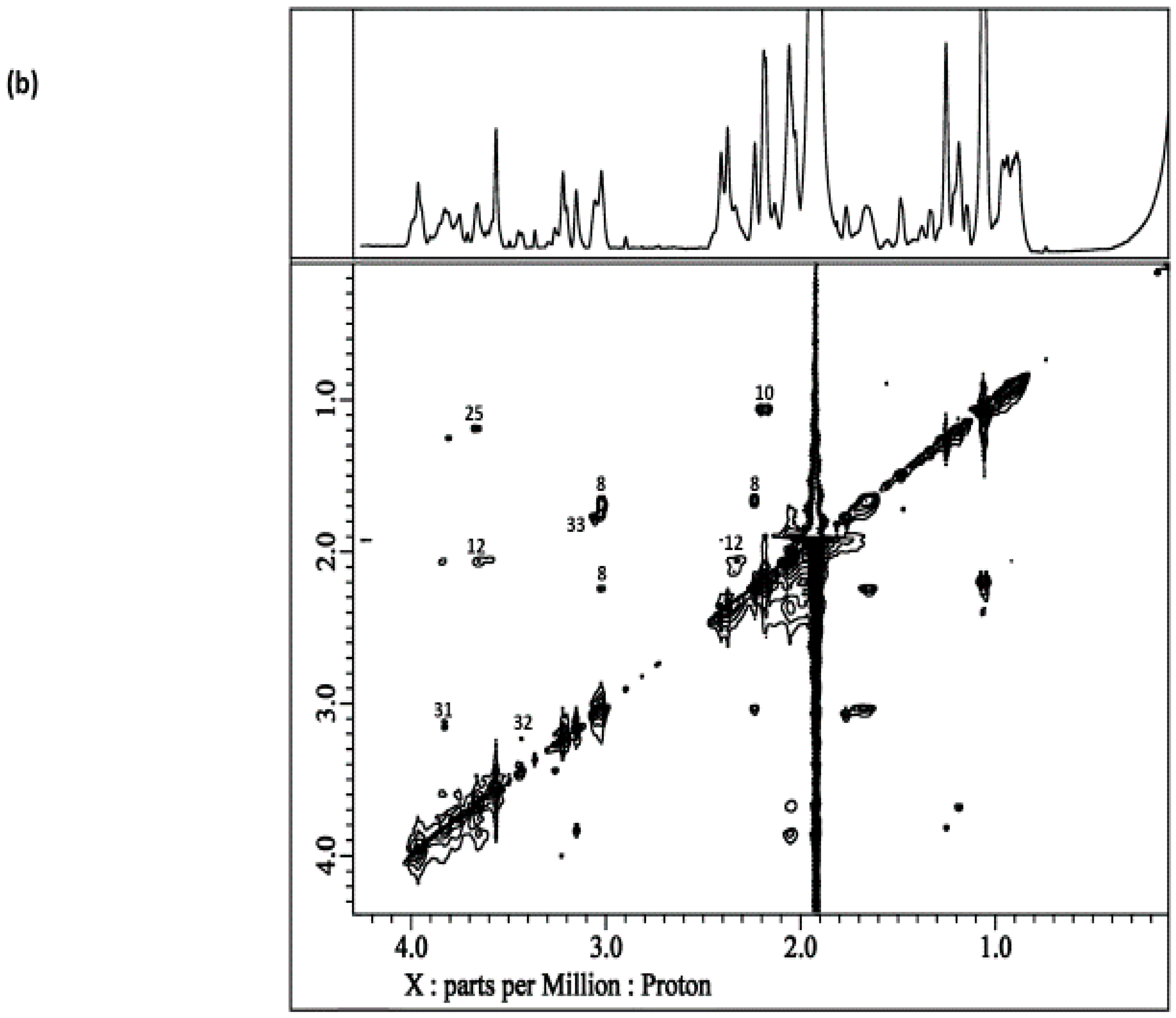
8. Homo- and Heteronuclear Two-Dimensional (2D) NMR Analysis of Human Saliva
8.1. Homonuclear 1H-1H Correlation and Total Correlation Spectroscopies
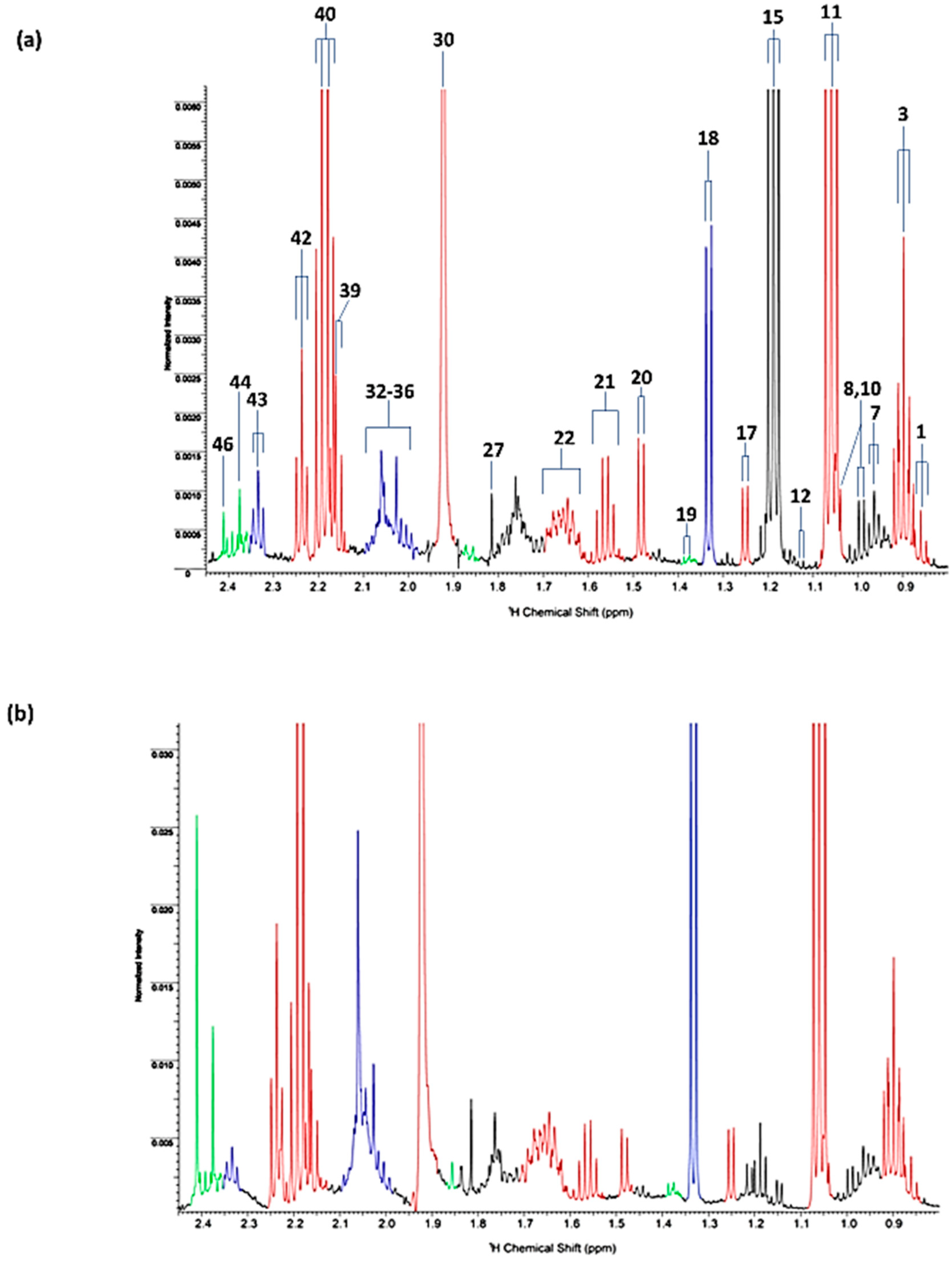
8.2. J-Resolved NMR Analysis
8.3. 1H-13C Heteronuclear Multiple Quantum Coherence and Heteronuclear Multiple Bond Coherence Spectroscopies
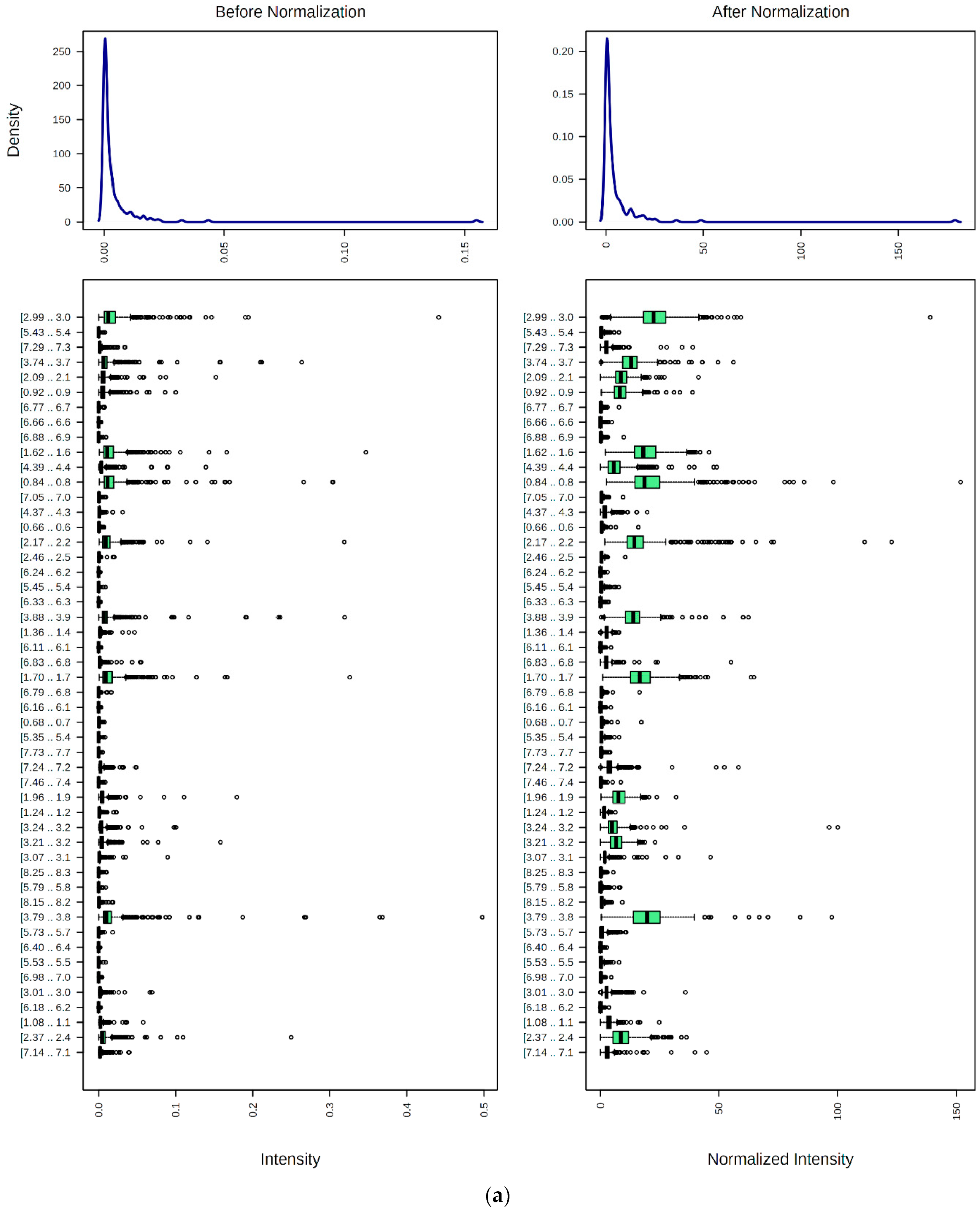
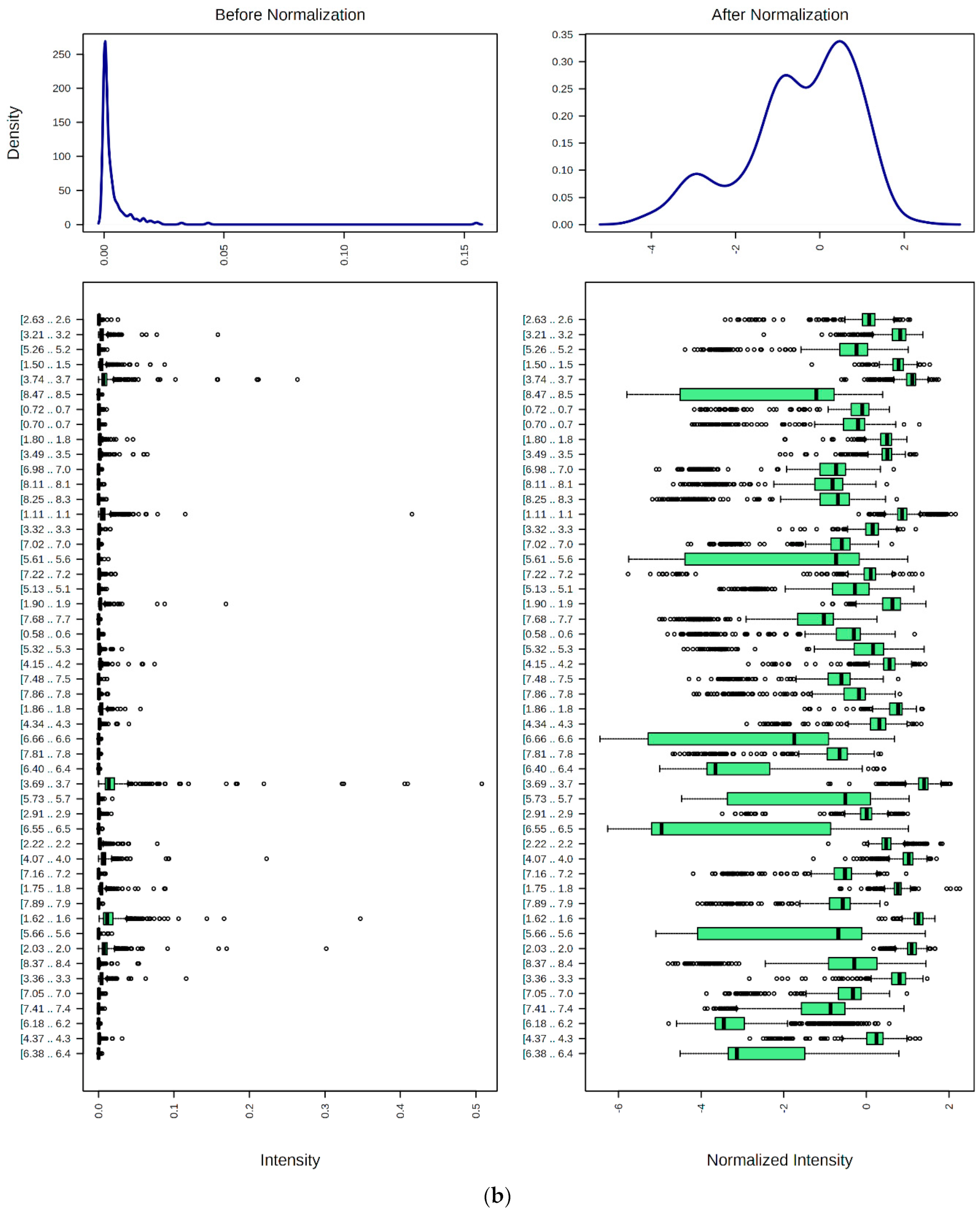
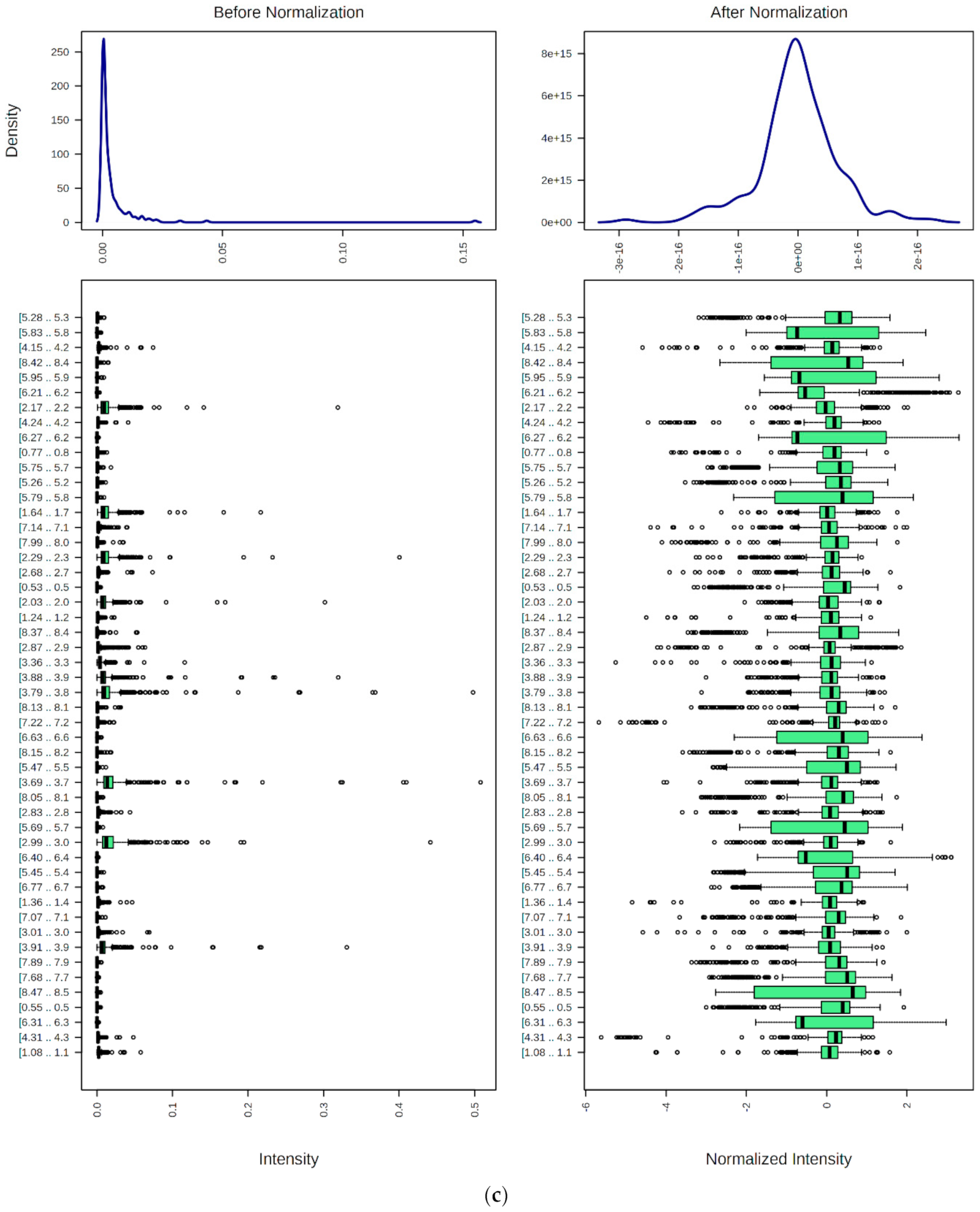
9. Preprocessing, Bucketing, Normalization, Transformation, and Scaling of 1H NMR Profiles of HSS Specimens: Bioanalytical Considerations for Metabolite Quantification and Metabolomics Analysis
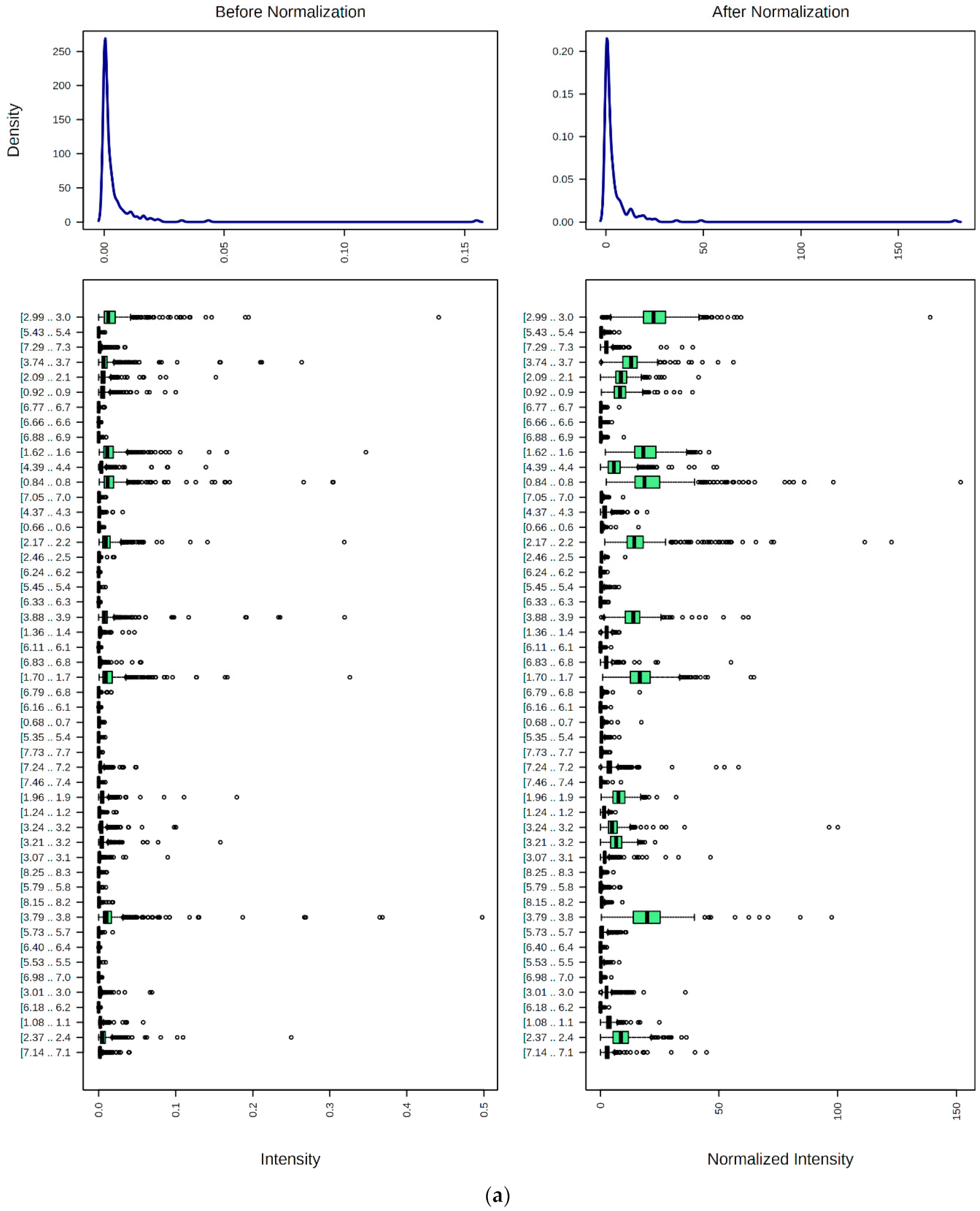
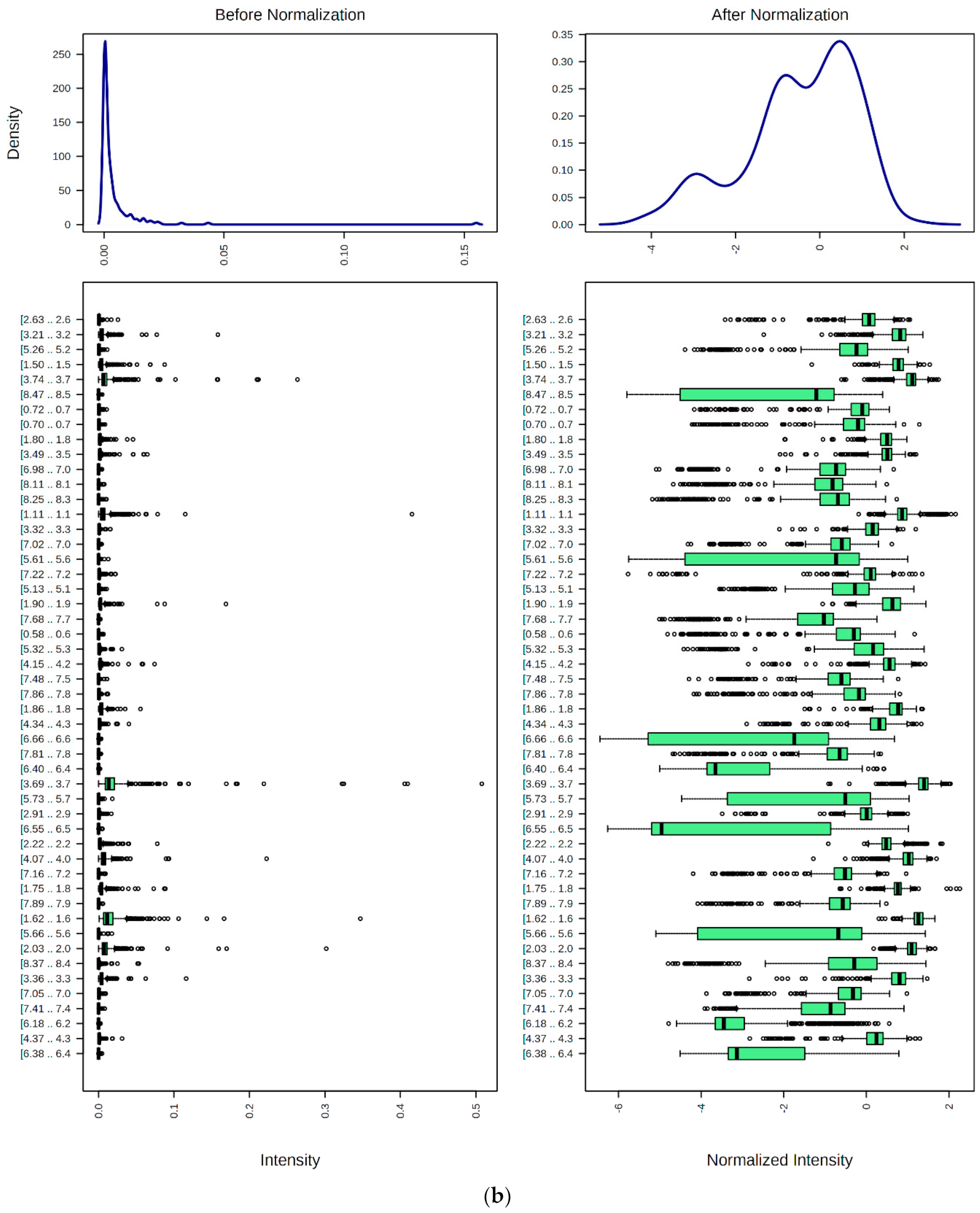
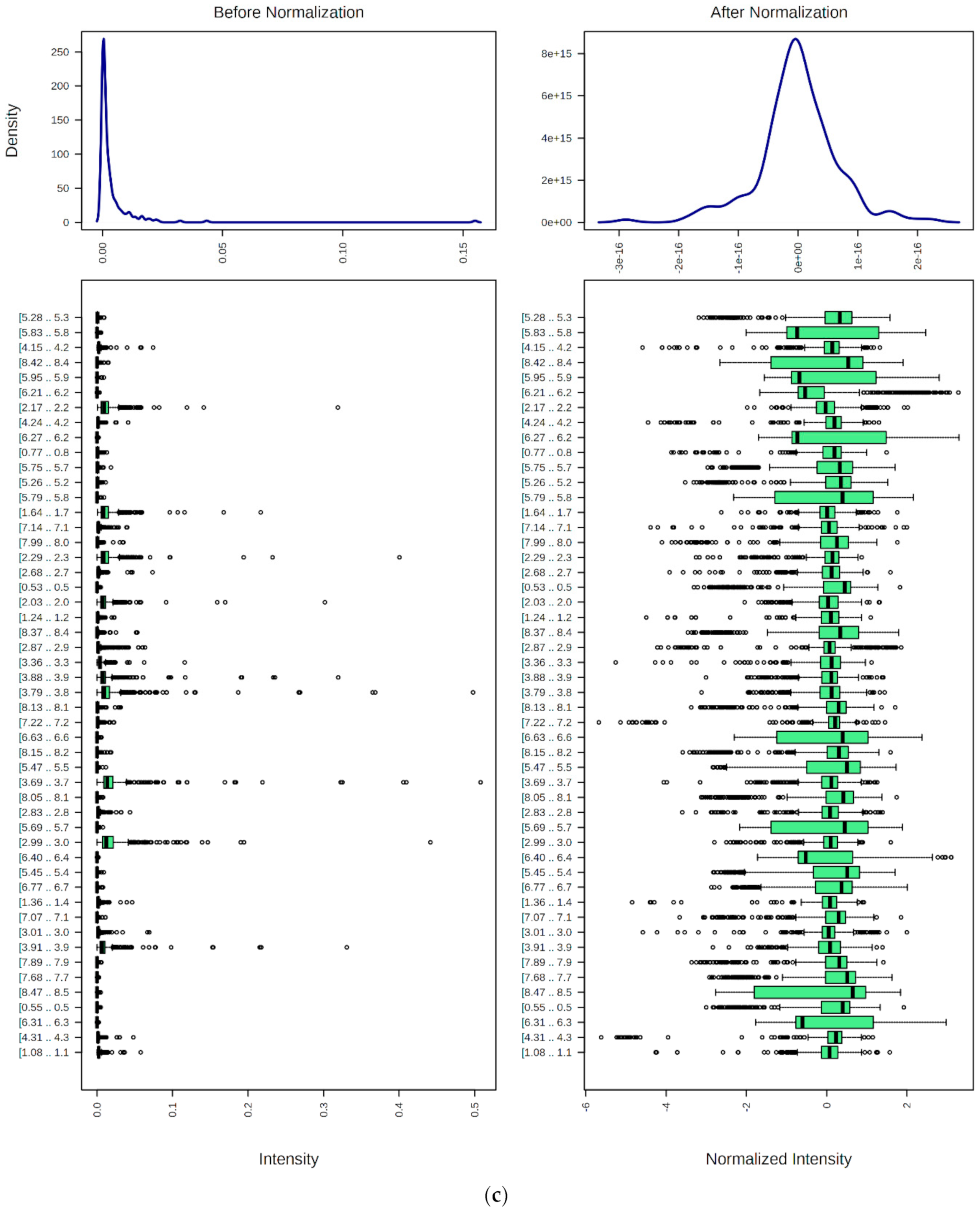
9.1. Spectral Bucketing
9.2. Data Normalization, Transformation, Scaling, and Dimensionality Reduction
9.3. Sample Size Considerations and MV Power Calculations
10. Significance of between-Participant Sources of Metabolite Level Variance in Healthy Humans: Salivary Phenotype Analysis
10.1. Case Study 2: Salivary Phenotype Analysis, and Identification of Differential Metabolite Pools within WMS Supernatants and Their Sources
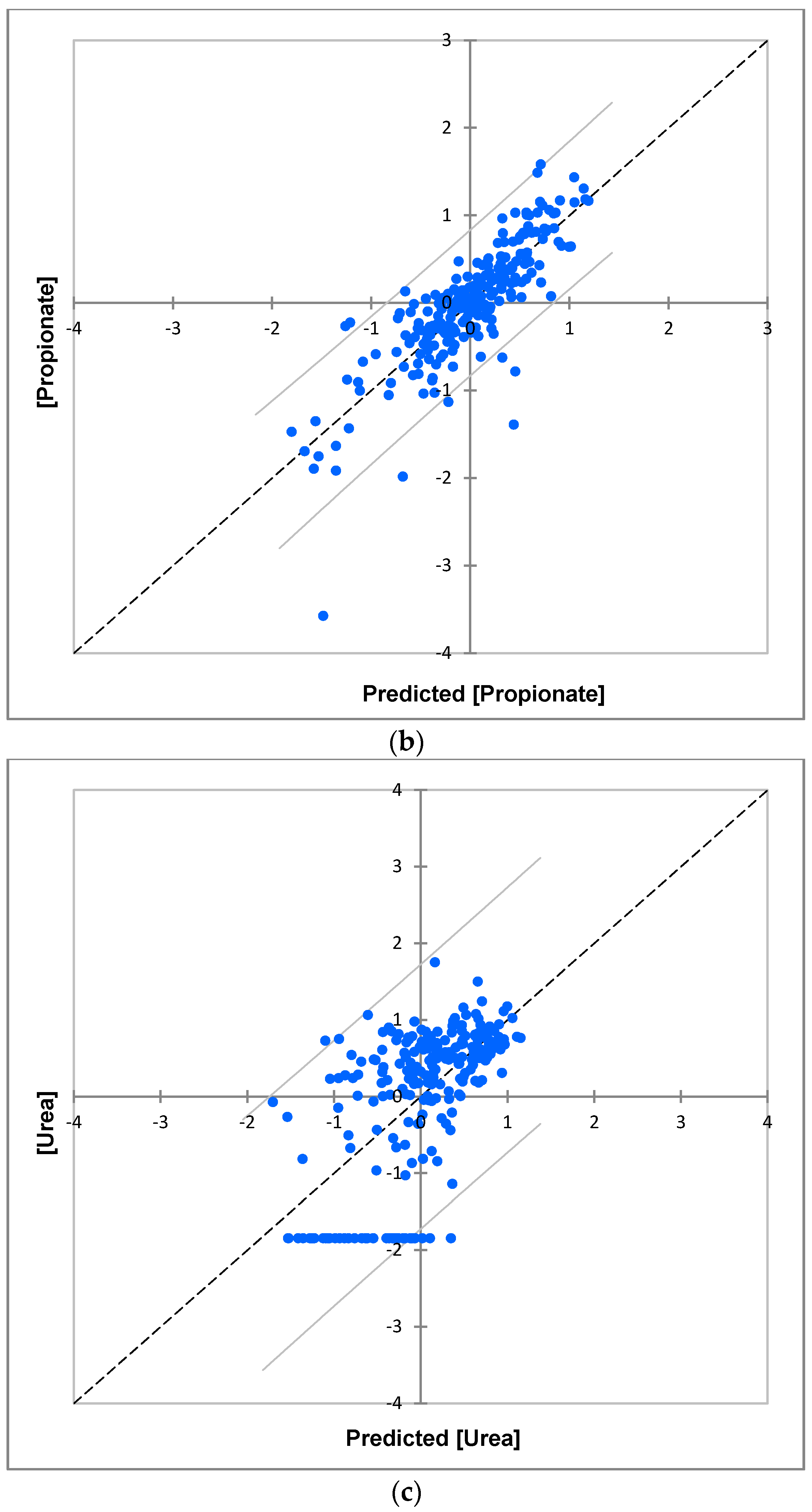
10.1.1. Univariate Experimental Design Model for Isolating between- and within-Participant Components of Variance for WMS Supernatant Metabolites in a Healthy Control Population
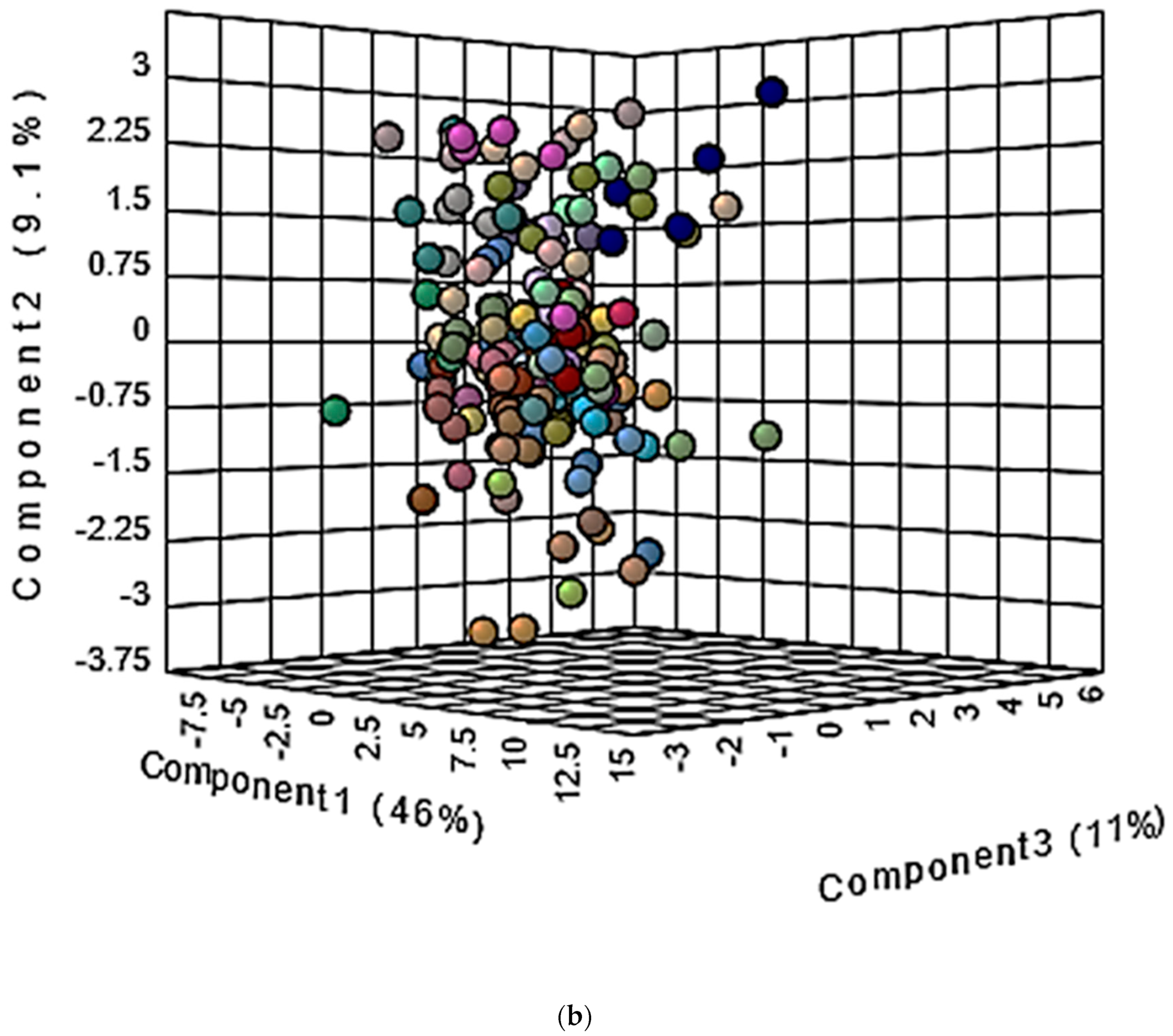
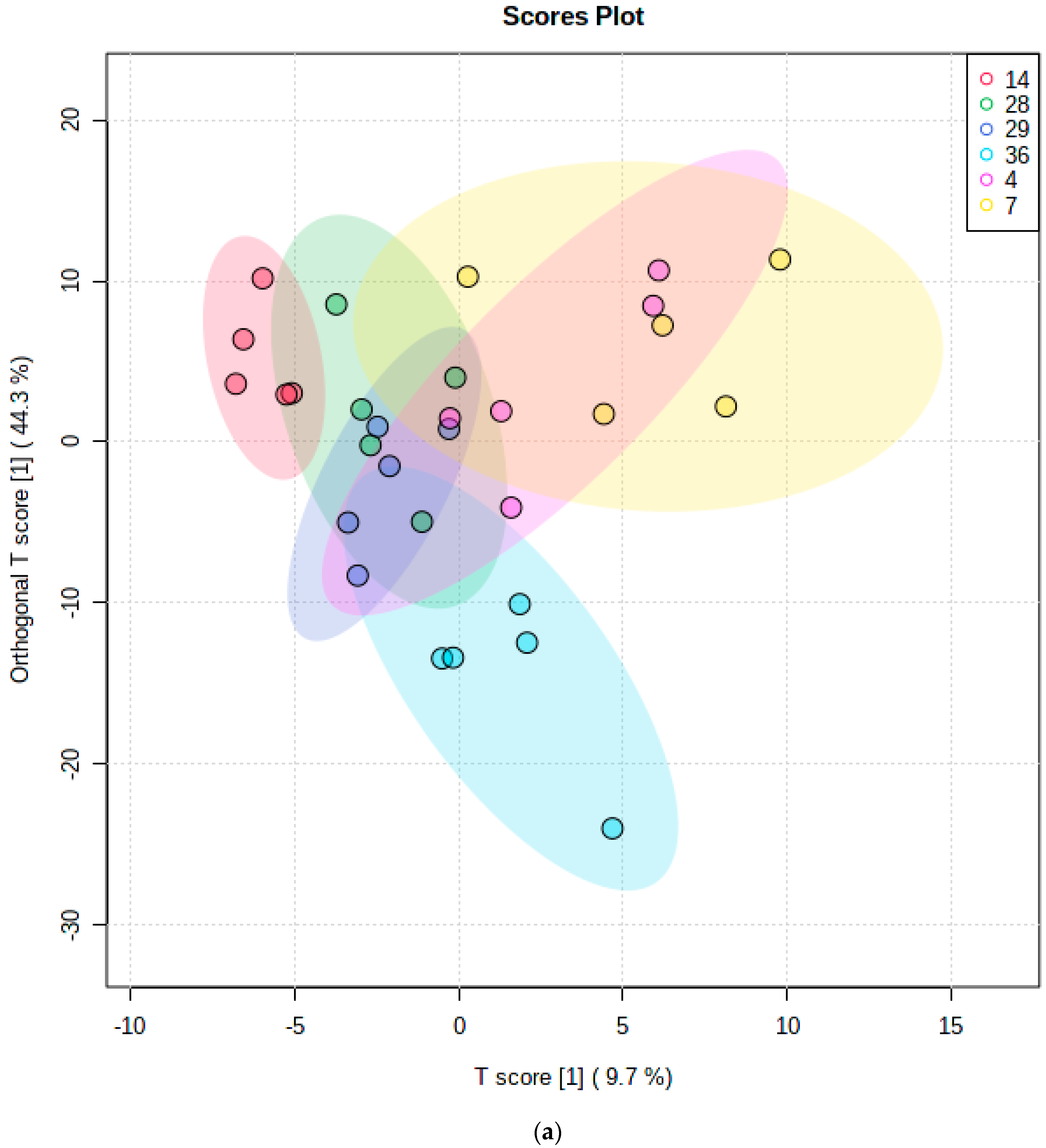
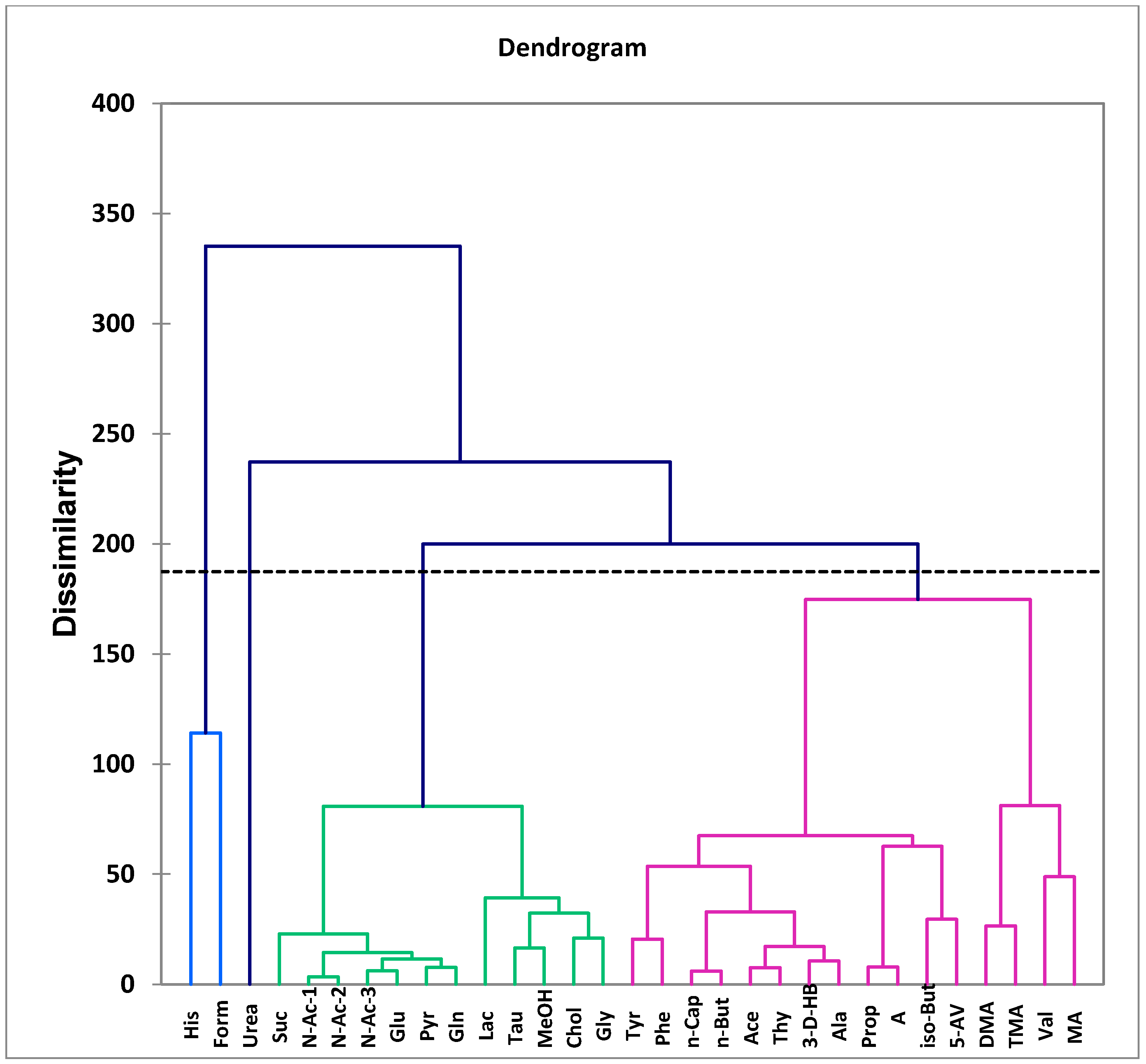
10.1.2. Multivariate Metabolomics Analysis of Distinctive Salivary Phenotypes
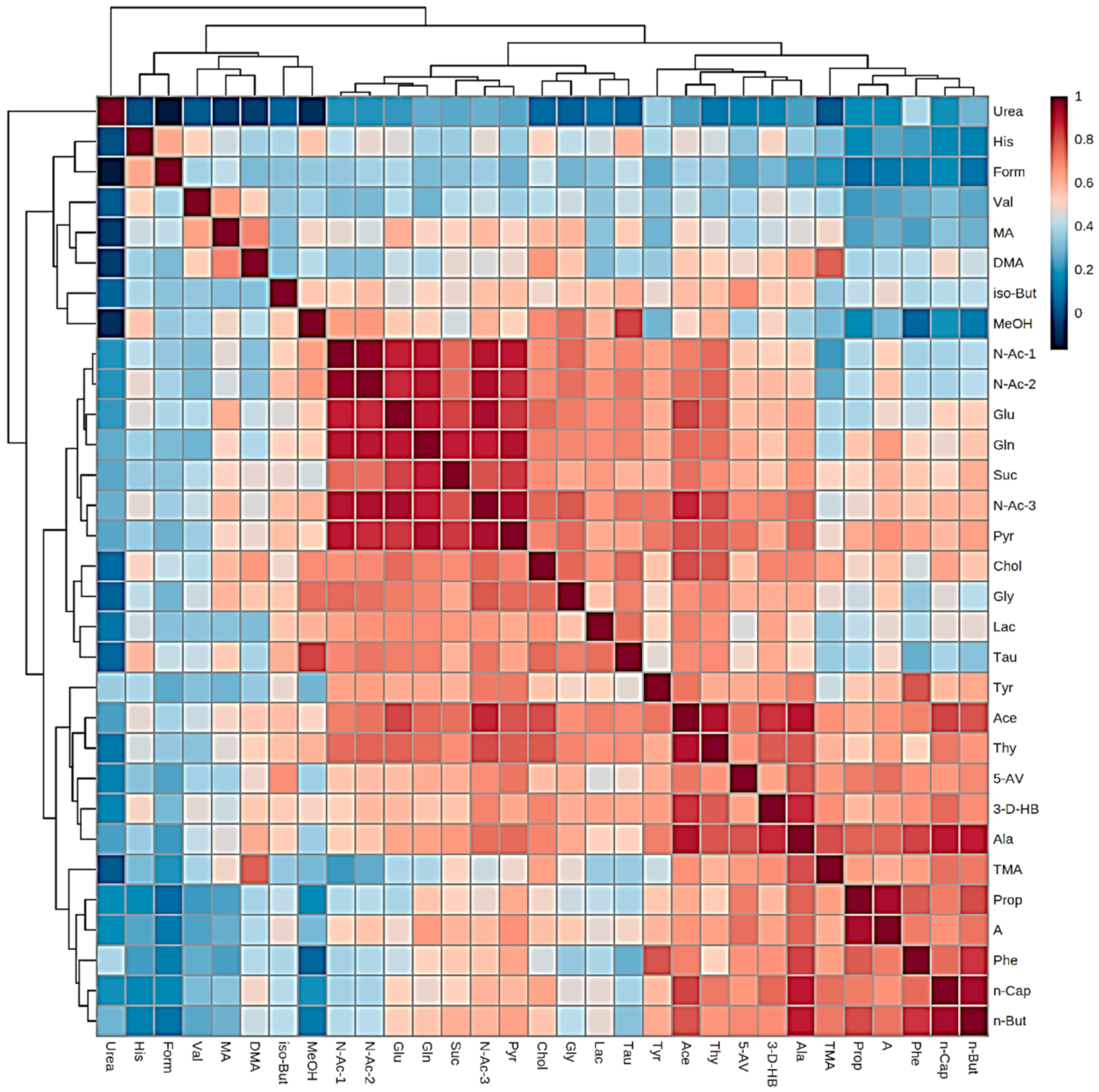
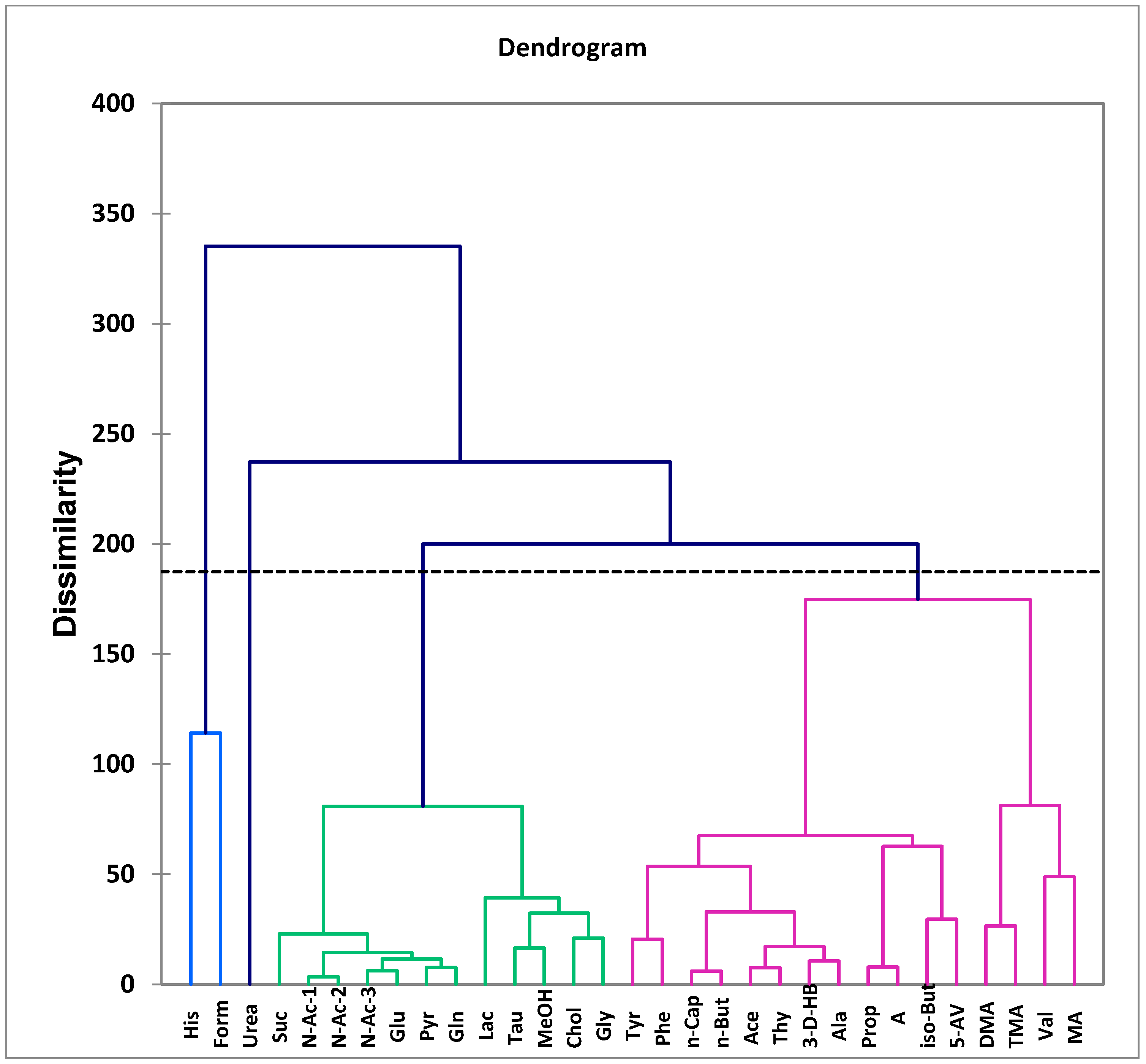
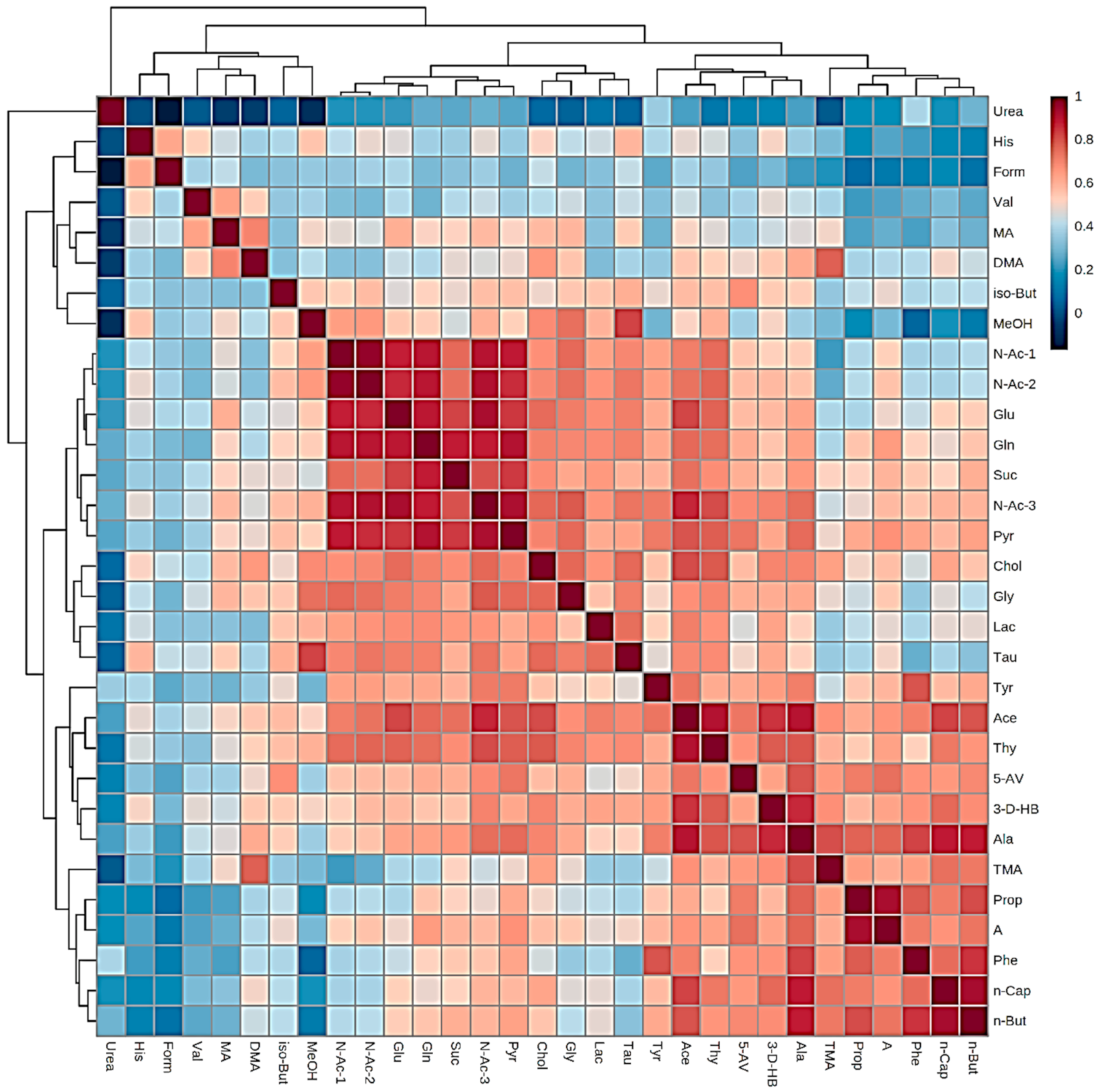
10.2. Investigation of Distinctive Metabolite Network Pools Putatively Arising from Host or Salivary Microbiome Sources: Factor Analysis of Component Loading Vectors and Patterns
11. Future Perspectives for Clinical Salivary NMR Bioanalysis and Monitoring with Low-Field Benchtop NMR Spectrometers
12. Concluding Remarks and Recommendations for Future Experimental Investigations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, P.B.; Grootveld, M. (Eds.) Computational Techniques for Analytical Chemistry and Bioanalysis; RSC the Chem Series; Royal Society of Chemistry: Cambridge, UK, 2020. [Google Scholar]
- Silwood, C.J.L.; Lynch, E.J.; Seddon, S.; Sheerin, A.; Claxson, A.W.D.; Grootveld, M.C. 1H NMR analysis of microbial-derived organic acids in primary root carious lesions and saliva. NMR Biomed. 1999, 12, 345–356. [Google Scholar] [CrossRef]
- Silwood, C.J.L.; Lynch, E.; Claxson, A.W.D.; Grootveld, M.C. 1H and 13C NMR spectroscopic analysis of human saliva. J. Dent. Res. 2002, 81, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Grootveld, M.; Silwood, C.J.L. 1H NMR analysis as an analytical probe for human saliva. Biochem. Biophys. Res. Commun. 2005, 329, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Silwood, C.J.L.; Grootveld, M.; Lynch, E. 1H NMR Investigations of the molecular nature of low-molecular-mass calcium ions in biofluids. J. Biol. Inorg. Chem. 2002, 7, 46–57. [Google Scholar] [CrossRef]
- Dame, Z.T.; Aziat, F.; Mandal, R.; Dame, Z.; Krishnamurthy, R.; Bouatra, S.; Borzouie, S.; Guo, A.; Sajed, T.; Deng, L.; et al. The human saliva metabolome. Metabolomics 2015, 11, 1864–1883. [Google Scholar] [CrossRef]
- Gardner, A.; Carpenter, G.; So, P.W. Salivary metabolomics: From diagnostic biomarker discovery to investigating biological function. Metabolites 2020, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, G.R.; Wongravee, K.; Silwood, C.J.L.; Grootveld, M.; Brereton, R.G. Self organising maps for variable selection: Application to human saliva analysed by nuclear magnetic resonance spectroscopy to investigate the effect of an oral healthcare product. Chemomet. Intell. Lab. Systems 2009, 98, 149–161. [Google Scholar] [CrossRef]
- Wongravee, K.; Lloyd, G.R.; Silwood, C.J.; Grootveld, M.; Brereton, R.G. Supervised Self Organizing Maps (SOMs) for classification and variable selection: Illustrated by application to NMR metabolomic profiling. Anal. Chem. 2010, 82, 628–638. [Google Scholar] [CrossRef]
- Lemanska, A.; Grootveld, M.C.; Silwood, C.J.L.; Brereton, R.G. Chemometric variance analysis of 1H NMR metabolomics data on the effects of oral rinse on saliva. Metabolomics 2011, 8, 64–80. [Google Scholar] [CrossRef]
- Carmona, C.J.; Ruiz-Rodado, V.; del Jesus, M.J.; Weber, A.; Grootveld, M.; González, P.; Elizondo, D. FuGePSD: Fuzzy genetic programming-base for subgroup discovery. A case study: Pathogenesis of acute sore throat conditions in humans. Informat. Sci. 2015, 298, 180–197. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, X. Saliva metabolomics opens door to biomarker discovery, disease diagnosis, and treatment. Appl. Biochem. Biotechnol. 2012, 168, 1718–1727. [Google Scholar] [CrossRef]
- Chang, H.; Tomoda, S.; Silwood, C.J.L.; Lynch, E.; Grootveld, M. 1H NMR investigations of the molecular nature of cobalt(II) ions in human saliva. Arch. Biochem. Biophys. 2012, 520, 51–65. [Google Scholar] [CrossRef]
- Dodds, M.; Roland, S.; Edgar, M.; Thornhill, M. Saliva A review of its role in maintaining oral health and preventing dental disease. BDJ Team 2015, 2, 15123. [Google Scholar] [CrossRef]
- Proctor, G.B. The physiology of salivary secretion. Periodontology 2016, 70, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Navazesh, M. Methods for collecting saliva. Ann. N. Y. Acad. Sci. 1993, 694, 72–77. [Google Scholar] [CrossRef]
- Baliga, S.; Muglikar, S.; Kale, R. Salivary pH: A diagnostic biomarker. J. Indian Soc. Periodontol. 2013, 17, 461–465. [Google Scholar] [CrossRef]
- Jenkins, G.N. The Physiology and Biochemistry of the Mouth, 4th ed.; Blackwell Scientific: Oxford, UK, 1978; p. 295. [Google Scholar]
- Lentner, C. (Ed.) Serum. In Geigy Scientific Tables, 8th ed.; Ciba-Geigy Ltd.: Basle, Switzerland, 1981; Volume 3, p. 141. [Google Scholar]
- Gardner, A.; Parkes, H.G.; So, P.-W.; Carpenter, G.H. Determining bacterial and host contributions to the human salivary metabolome. J. Oral Microbiol. 2019, 11, 1–1617014. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, A.; Geddes, T.; Han, B.; Bahado-Singh, R.O.; Wilson, G.D.; Imam, K.; Maddens, M.; Graham, S.F. Diagnostic biomarkers of Alzheimer’s disease as identified in saliva using 1H NMR-based metabolomics. J. Alzheimers Dis. 2017, 58, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Yamada-Nosaka, A.; Fukutomi, S.; Uemura, S.; Hashida, T.; Fujishita, M.; Kobayashi, Y.; Kyogoku, Y. Preliminary nuclear magnetic resonance studies on human saliva. Arch. Oral Biol. 1991, 36, 697–701. [Google Scholar] [CrossRef]
- Figueira, J.; Gouveia-Figueira, S.; Öhman, C.; Lif Holgerson, P.; Nording, M.L.; Öhman, A. Metabolite quantification by nmr and lc-ms/ms reveals differences between unstimulated, stimulated, and pure parotid saliva. J. Pharm. Biomed. Anal. 2017, 140, 295–300. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Fernández, J.; Redondo-Blanco, S.; Gutierrez-del-Rio, I.; Miguelez, E.M.; Villar, C.J.; Lombo, F. Colon microbiota fermentation of dietary prebiotics towards short-chain fatty acids and their roles as anti-inflammatory and antitumour agents: A review. J. Funct. Foods. 2016, 25, 511–522. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- van de Wouw, M.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. Microbiota-gut-brain axis: Modulator of host metabolism and appetite. J. Nutri. 2017, 147, 727–745. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N. Oral microbiome metabolism: From “who are they?” to “what are they doing?”. J. Dent. Res. 2015, 94, 1628–1637. [Google Scholar] [CrossRef]
- Wallner-Liebmann, S.; Tenori, L.; Mazzoleni, A.; Dieber-Rotheneder, M.; Konrad, M.; Hofmann, P.; Luchinat, C.; Turano, P.; Zatlouka, I.K. Individual human metabolic phenotype analyzed by 1H NMR of saliva samples. J. Proteome Res. 2016, 15, 1787–1793. [Google Scholar] [CrossRef]
- Gardner, A.; Parkes, H.G.; Carpenter, G.H.; So, P.-W. Developing and standardizing a protocol for quantitative proton nuclear magnetic resonance (1h nmr) spectroscopy of saliva. J. Proteome Res. 2018, 17, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar]
- Romano, K.A.; Martinez-Del Campo, A.; Kasahara, K.; Chittim, C.L.; Vivas, E.I.; Amador-Noguez, D.; Balskus, E.P.; Rey, F.E. Metabolic, epigenetic, and transgenerational effects of gut bacterial choline consumption. Cell Host Microbe 2017, 22, 279–290.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Jameson, E.; Crosatti, M.; Schafer, H.; Rajakumar, K.; Bugg, T.D.H.; Chen, Y. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 4268–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, T.P.N.; Ritari, J.; Boeren, S.; de Waard, P.; Plugge, C.M.; de Vos, W.M. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat. Commun. 2019, 6, 10062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. MBio 2014, 5, e00889-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, S. The scientific exploration of saliva in the post-proteomic era: From database back to basic function. Expert Rev. Proteom. 2012, 9, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleaver, L.M.; Moazzez, R.; Carpenter, G.H. Mixed aerobic-anaerobic incubation conditions induce proteolytic activity from in vitro salivary biofilms. J. Oral Microbiol. 2019, 11, 1643206. [Google Scholar] [CrossRef] [Green Version]
- Campo, J.; Perea, M.A.; Del Romero, J.; Cano, J.; Hernando, V.; Bascones, A. Oral transmission of HIV, reality or fiction? An update. Oral Dis. 2006, 12, 219–228. [Google Scholar] [CrossRef]
- Miller, S. Saliva testing—A non traditional diagnostic tool. Clin. Lab. Sci. 1994, 7, 39–44. [Google Scholar]
- Challacombe, S.J.; Percival, R.S.; Marsh, P.D. Age-related changes in immunoglobulin isotypes in whole and parotid saliva and serum in healthy individuals. Oral Microbiol. Immunol. 1995, 10, 202–207. [Google Scholar] [CrossRef]
- Lee, Y.H.; Wong, D.T. Saliva: An emerging biofluid for early detection of diseases. Am. J. Dent. 2009, 22, 241–248. [Google Scholar]
- Haeckel, R.; Hanecke, P. The application of saliva, sweat, and tear fluid for diagnostic purposes. Ann. Biol. Clin. 1993, 51, 903–910. [Google Scholar]
- Yoshizawa, J.M.; Schafer, C.A.; Schafer, J.J.; Farrell, J.J.; Paster, B.J.; Wong, D.T. Salivary biomarkers: Toward future clinical and diagnostic utilities. Clin. Microbiol. Rev. 2013, 2013 26, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Drobitch, R.K.; Svensson, C.K. Therapeutic drug monitoring in saliva: An update. Clin. Pharmacokinet. 1992, 23, 365–379. [Google Scholar] [CrossRef]
- Jusko, W.J.; Milsap, R.L. Pharmacokinetic principles of drug distribution in saliva. Ann. N. Y. Acad. Sci. 1993, 694, 36–47. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0—The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46, D608–D6117. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted metabolomics strategies-challenges and emerging directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current analytical platforms and methodologies. Trac-Trend Anal. Chem. 2005, 24, 285–294. [Google Scholar]
- Lynch, E.; Sheerin, A.; Claxson, A.W.D.; Atherton, M.D.; Rhodes, C.J.; Silwood, C.J.L.; Naughton, D.P.; Grootveld, M. Multicomponent spectroscopic investigations of salivary antioxidant consumption by an oral rinse preparation containing the stable free radical species chlorine dioxide (ClO2∙). Free Rad. Res. 1997, 26, 209–237. [Google Scholar] [CrossRef]
- Lynch, E.; Sheerin, A.; Silwood, C.J.; Grootveld, M. Multicomponent evaluations of the oxidising actions and status of a peroxoborate-containing tooth-whitening system in whole human saliva using high resolution proton NMR spectroscopy. J. Inorg. Biochem. 1999, 73, 65–84. [Google Scholar] [CrossRef]
- Dona, A.C.; Jiménez, B.; Schäfer, H.; Humpfer, E.; Spraul, M.; Lewis, M.R.; Pearce, J.T.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Precision high-throughput proton nmr spectroscopy of human urine, serum, and plasma for large-scale metabolic phenotyping. Anal. Chem. 2014, 86, 9887–9894. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.C.Y.; Pasikanti, K.K.; Nicholson, J.K. Global urinary metabolic profiling procedures using gas chromatography–mass spectrometry. Nat. Protoc. 2011, 6, 1483. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Sánchez, B.; Priego-Capote, F.; Luque de Castro, M.D. Study of sample preparation for metabolomic profiling of human saliva by liquid chromatography–time of flight/mass spectrometry. J. Chromatogr. A 2012, 1248, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.A.D.; Elliottt, J.C. Basic and Applied Dental Biochemistry; Churchill Livingstone: Edinburgh, UK, 1979. [Google Scholar]
- Percival, B.C.; Wann, A.; Masania, J.; Sinclair, J.; Sullo, N.; Grootveld, M. Detection and determination of methanol and further potential toxins in human saliva collected from cigarette smokers: A 1H NMR investigation. JSM Biotechnol. Biomed. Eng. 2018, 5, 1081–1088. [Google Scholar]
- Ishikawa, S.; Sugimoto, M.; Kitabatake, K.; Sugano, A.; Nakamura, M.; Kaneko, M.; Ota, S.; Hiwatari, K.; Enomoto, A.; Soga, T.; et al. Identification of salivary metabolomic biomarkers for oral cancer screening. Sci. Rep. 2016, 6, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Duarte, D.; Castro, B.; Pereira, J.L.; Marques, J.F.; Costa, A.L.; Gil, A.M. Evaluation of saliva stability for nmr metabolomics: Collection and handling protocols. Metabolites 2020, 10, 515. [Google Scholar] [CrossRef]
- De Oliveira, L.R.P.; Martins, C.; Fidalgo, T.K.S.; Freitas-Fernandes, L.B.; De Oliveira Torres, R.; Soares, A.L.; Almeida, F.C.L.; Valente, A.P.; De Souza, I.P.R. Salivary metabolite fingerprint of type 1 diabetes in young children. J. Proteome Res. 2016, 15, 2491–2499. [Google Scholar] [CrossRef]
- Gawron, K.; Wojtowicz, W.; Łazarz-Bartyzel, K.; Łamasz, A.; Qasem, B.; Mydel, P.; Chomyszyn-Gajewska, M.; Potempa, J.; Mlynarz, P. Metabolomic status of the oral cavity in chronic periodontitis. In Vivo 2019, 33, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Freitas-Fernandes, L.B.; Fidalgo, T.K.S.; de Almeida, P.A.; Souza, I.P.R.; Valente, A.P. Salivary metabolome of children and adolescents under peritoneal dialysis. Clin. Oral Investig. 2021, 25, 2345–2351. [Google Scholar] [CrossRef]
- Morgana, I.; Gonçalves, F.; Pessoa, M.B.; Leitão, A.D.S.; Godoy, P.; Francisco, C.; Nonaka, W.; Alves, P.M. Salivary and Serum Biochemical Analysis from Patients with Chronic Renal Failure in Hemodialysis: A Cross-Sectional Study. Pesqui. Bras. Em Odontopediatria E Clínica Integr. 2021, 21, 1–8.e0036. [Google Scholar]
- Gilany, K.; Mohamadkhani, A.; Chashmniam, S.; Shahnazari, P.; Amini, M.; Arjmand, B.; Malekzadeh, R.; Ghoochani, B.F.N.M. Metabolomics analysis of the saliva in patients with chronic hepatitis by using nuclear magnetic resonance: A pilot study. Iran. J. Basic Med. Sci. 2019, 22, 1044–1049. [Google Scholar]
- Schulz, A.; Lang, R.; Behr, J.; Hertel, S.; Reich, M.; Kümmerer, K.; Hannig, M.; Hannig, C.; Hofmann, T. Targeted metabolomics of pellicle and saliva in children with different caries activity. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liebsch, C.; Pitchika, V.; Pink, C.; Samietz, S.; Kastenmüller, G.; Artati, A.; Suhre, K.; Adamski, J.; Nauck, M.; Völzke, H.; et al. The saliva metabolome in association to oral health status. J. Dent. Res. 2019, 98, 642–651. [Google Scholar] [CrossRef]
- Liang, Q.; Liu, H.; Zhang, T.; Jiang, Y.; Xing, H.; Zhang, A.H. Metabolomics-based screening of salivary biomarkers for early diagnosis of Alzheimer’s disease. RSC Adv. 2015, 5, 96074–96079. [Google Scholar] [CrossRef]
- Song, X.; Yang, X.; Narayanan, R.; Shankar, V.; Ethiraj, S.; Wang, X.; Duan, N.; Ni, Y.H.; Hu, Q.; Zare, R.N. Oral squamous cell carcinoma diagnosed from saliva metabolic profiling. Proc. Natl. Acad. Sci. USA 2020, 117, 16167–16173. [Google Scholar] [CrossRef]
- Murata, T.; Yanagisawa, T.; Kurihara, T.; Kaneko, M.; Ota, S.; Enomoto, A.; Tomita, M.; Sugimoto, M.; Sunamura, M.; Hayashida, T.; et al. Salivary metabolomics with alternative decision tree-based machine learning methods for breast cancer discrimination. Breast Cancer Res. Treat. 2019, 177, 591–601. [Google Scholar] [CrossRef]
- Romano, F.; Meoni, G.; Manavella, V.; Baima, G.; Tenori, L.; Cacciatore, S.; Aimetti, M. Analysis of salivary phenotypes of generalized aggressive and chronic periodontitis through nuclear magnetic resonance-based metabolomics. J. Periodontol. 2018, 89, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, S.; Huan, T.; Tran, T.; Zheng, J.; Camicioli, R.; Li, L.; Dixon, R.A. Alzheimer’s biomarkers from multiple modalities selectively discriminate clinical status: Relative importance of salivary metabolomics panels, genetic, lifestyle, cognitive, functional health and demographic risk markers. Front. Aging Neurosci. 2018, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Schulte, F.; King, O.D.; Paster, B.J.; Moscicki, A.B.; Yao, T.J.; Van Dyke, R.B.; Shiboski, C.; Ryder, M.; Seage, G.; Hardt, M. Salivary metabolite levels in perinatally HIV-infected youth with periodontal disease. Metabolomics 2020, 16, 98. [Google Scholar] [CrossRef]
- Kageyama, G.; Saegusa, J.; Irino, Y.; Tanaka, S.; Tsuda, K.; Takahashi, S.; Sendo, S.; Morinobu, A. Metabolomics analysis of saliva from patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2015, 182, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Xie, G.; Zhou, Z.; Shi, P.; Qiu, Y.; Zheng, X.; Chen, T.; Su, M.; Zhao, A.; Jia, W. Salivary metabolite signatures of oral cancer and leukoplakia. Int. J. Cancer 2011, 129, 2207–2217. [Google Scholar] [CrossRef]
- Assad, D.X.; Acevedo, A.C.; Mascarenhas, E.C.P.; Normando, A.G.C.; Pichon, V.; Chardin, H.; Guerra, E.N.S.; Combes, A. Using an untargeted metabolomics approach to identify salivary metabolites in women with breast cancer. Metabolites 2020, 10, 506. [Google Scholar] [CrossRef]
- Singh, M.P.; Saxena, M.; Saimbi, C.S.; Siddiqui, M.H.; Roy, R. Post-periodontal surgery propounds early repair salivary biomarkers by 1H NMR based metabolomics. Metabolomics 2019, 15, 141. [Google Scholar] [CrossRef]
- Sugimoto, M.; Wong, D.T.; Hirayama, A.; Soga, T.; Tomita, M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 2010, 6, 78–95. [Google Scholar] [CrossRef] [Green Version]
- Kawanishi, N.; Hoshi, N.; Masahiro, S.; Enomoto, A.; Ota, S.; Kaneko, M.; Soga, T.; Tomita, M.; Kimoto, K. Effects of inter-day and intra-day variation on salivary metabolomic profiles. Clin. Chim. Acta 2019, 489, 41–48. [Google Scholar] [CrossRef]
- Turunen, S.; Puurunen, J.; Auriola, S.; Kullaa, A.M.; Kärkkäinen, O.; Lohi, H.; Hanhineva, K. Metabolome of canine and human saliva: A non-targeted metabolomics study. Metabolomics 2020, 16, 90. [Google Scholar] [CrossRef]
- Almeida, P.A.; Fidalgo, T.K.S.; Freitas-Fernandes, L.B.; Almeida, F.C.L.; Souza, I.P.R.; Valente, A.P. Salivary metabolic profile of children and adolescents after hemodialysis. Metabolomics 2017, 13, 141. [Google Scholar] [CrossRef]
- Ishikawa, S.; Sugimoto, M.; Edamatsu, K.; Sugano, A.; Kitabatake, K.; Iino, M. Discrimination of oral squamous cell carcinoma from oral lichen planus by salivary metabolomics. Oral Dis. 2020, 26, 35–42. [Google Scholar] [CrossRef]
- Jo, J.; Seo, S.-H.; Park, S.-E.; Kim, H.-W.; Kim, E.-J.; Na, C.-S.; Cho, K.-M.; Kwon, S.-J.; Moon, Y.-H.; Son, H.-S. Identification of Salivary Microorganisms and Metabolites Associated with Halitosis. Metabolites 2021, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Sugimoto, M.; Kitabatake, K.; Tu, M.; Sugano, A.; Yamamori, I.; Iba, A.; Yusa, K.; Kaneko, M.; Ota, S.; et al. Effect of timing of collection of salivary metabolomic biomarkers on oral cancer detection. Amino Acids 2017, 49, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, T.K.S.; Freitas-Fernandes, L.B.; Almeida, F.C.L.; Valente, A.P.; Souza, I.P.R. Longitudinal evaluation of salivary profile from children with dental caries before and after treatment. Metabolomics 2015, 11, 583–593. [Google Scholar] [CrossRef]
- Gardner, A.; So, P.W.; Carpenter, G. Endogenous salivary citrate is associated with enhanced rheological properties following oral capsaicin stimulation. Exp. Physiol. 2020, 105, 96–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, I.; Stretch, C.; Barnaby, P.; Bhatnager, K.; Rankin, K.; Fu, H.; Weljie, A.; Jha, N.; Slupsky, C. Understanding the human salivary metabolome. NMR Biomed. 2009, 22, 577–584. [Google Scholar] [CrossRef]
- Lalue Sanches, M.; Sforça, M.L.; Guimarães Lo Turco, E.; Faber, J.; Smith, R.L.; de Moraes, L.O.C. 1H-NMR-Based salivary metabolomics from females with temporomandibular disorders—A pilot study. Clin. Chim. Acta 2020, 510, 625–632. [Google Scholar] [CrossRef]
- Kumari, S.; Goyal, V.; Kumaran, S.S.; Dwivedi, S.N.; Srivastava, A.; Jagannathan, N.R. Quantitative metabolomics of saliva using proton NMR spectroscopy in patients with Parkinson’s disease and healthy controls. Neurol. Sci. 2020, 41, 1201–1210. [Google Scholar] [CrossRef]
- Wijnant, K.; Van Meulebroek, L.; Pomian, B.; De Windt, K.; De Henauw, S.; Michels, N.; Vanhaecke, L. Validated ultra-high-performance liquid chromatography hybrid high-resolution mass spectrometry and laser-assisted rapid evaporative ionization mass spectrometry for salivary metabolomics. Anal. Chem. 2020, 92, 5116–5124. [Google Scholar] [CrossRef] [Green Version]
- Prodan, A.; Imangaliyev, S.; Brand, H.S.; Rosema, M.N.A.; Levin, E.; Crielaard, W.; Keijser, B.J.F.; Veerman, E.C.I. Effect of experimental gingivitis induction and erythritol on the salivary metabolome and functional biochemistry of systemically healthy young adults. Metabolomics 2016, 12, 147. [Google Scholar] [CrossRef]
- Herrala, M.; Mikkonen, J.J.W.; Pesonen, P.; Lappalainen, R.; Tjäderhane, L.; Niemelä, R.K.; Seitsalo, H.; Salo, T.; Myllymaa, S.; Kullaa, A.M. Variability of salivary metabolite levels in patients with Sjögren’s syndrome. J. Oral Sci. 2021, 63, 22–26. [Google Scholar] [CrossRef]
- Santone, C.; Dinallod, V.; Paci, M.; D’Ottavio, S.; Barbato, G.; Bernardini, S. Saliva metabolomics by NMR for the evaluation of sport performance. J. Pharm. Biomed. Anal. 2014, 88, 441–446. [Google Scholar] [CrossRef]
- Mikkonen, J.J.W.; Singh, S.P.; Akhi, R.; Salo, T.; Lappalainen, R.; González-Arriagada, W.A.; Lopes, M.A.; Kullaa, A.M.; Myllymaa, S. Potential role of nuclear magnetic resonance spectroscopy to identify salivary metabolite alterations in patients with head and neck cancer. Oncol. Lett. 2018, 16, 6795–6800. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, S.; Hiraka, T.; Kirii, K.; Sugimoto, M.; Shimamoto, H.; Sugano, A.; Kitabatake, K.; Toyoguchi, Y.; Kanoto, M.; Nemoto, K.; et al. Relationship between standard uptake values of positron emission tomography/computed tomography and salivary metabolites in oral cancer: A pilot study. J. Clin. Med. 2020, 9, 3958. [Google Scholar] [CrossRef]
- Troisi, J.; Belmonte, F.; Bisogno, A.; Pierri, L.; Colucci, A.; Scala, G.; Cavallo, P.; Mandato, C.; Nuzzi, A.D.; Michele, L.D.; et al. Metabolomic salivary signature of pediatric obesity related liver disease and metabolic syndrome. Nutrients 2019, 11, 274. [Google Scholar] [CrossRef] [Green Version]
- Bardow, A.; Moe, D.; Nyvad, B.; Nauntofte, B. The buffer capacity and buffer systems of human whole saliva measured without loss of CO2. Arch. Oral Biol. 2000, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, P.A.; Morelatto, R.A.; Benavidez, T.E.; Baruzzi, A.M.; Lopez de Blanc, S.A. Effect of two mouthwashes on salivary pH. Acta Odontológica Latinoam. 2014, 27, 66–71. [Google Scholar]
- Zappacosta, B.; Manni, A.; Persichilli, S.; Boari, A.; Scribano, D.; Minucci, A.; Raffaelli, L.; Giardina, B.; De Sole, P. Salivary thiols and enzyme markers of cell damage in periodontal disease. Clin. Biochem. 2007, 40, 661–665. [Google Scholar] [CrossRef]
- Perera, A.; Parkes, H.; Herz, H.; Haycock, R.; Blake, D.R.; Grootveld, M.C. High resolution 1H NMR investigations of the reactivities of a-keto acid anions with hydrogen peroxide. Free. Radic. Res. 1997, 26, 145–157. [Google Scholar] [CrossRef]
- Grootveld, M.; Herz, H.; Haywood, R.; Hawkes, G.E.; Naughton, D.; Perera, A.; Knappitt, J.; Blake, D.R.; Claxson, A.W.D. Multicomponent analysis of radiolytic products in human body fluids using high field proton nuclear magnetic resonance (NMR) spectroscopy. Radiat. Phys. Chem. 1993, 43, 445–453. [Google Scholar] [CrossRef]
- Chao, C.K.; Zeisel, S.H. Formation of trimethylamine from dietary choline by Streptococcus sanguis I, which colonizes the mouth. J. Nutr. Biochem. 1990, 1, 89–97. [Google Scholar] [CrossRef]
- Park, Y.D.; Jang, J.H.; Oh, Y.J.; Kwon, H.J. Analyses of organic acids and inorganic anions and their relationship in human saliva before and after glucose intake. Arch. Oral Biol. 2014, 59, 1–11. [Google Scholar] [CrossRef]
- Disler, W.; Kröncke, A. The lactate metabolism of the oral bacterium Veillonella from human saliva. Arch. Oral Biol. 1981, 26, 657–661. [Google Scholar] [CrossRef]
- Barnes, V.M.; Teles, R.; Trivedi, H.M.; Devizio, W.; Xu, T.; Mitchell, M.W.; Milburn, M.V.; Guo, L. Acceleration of purine degradation by periodontal diseases. J. Dent. Res. 2009, 88, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Lichstein, H.C.; Soule, M.H. Studies of the effect of sodium azide on microbic growth and respiration: I. The action of sodium azide on microbic growth. J. Bacteriol. 1944, 47, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, M.J.; Yamashita, E.; Inoue, N.; Yao, M.; Yamaguchi, H.; Tsukihara, T.; Shinzawa-Itoh, K.; Nakashima, R.; Yoshikawa, S. X-ray structure of azide-bound fully oxidized cytochrome c oxidase from bovine heart at 2.9 Å resolution. Acta Crystallogr. D 2000, 56, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, S.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yamashita, E.; Inoue, N.; Yao, M.; Fei, M.J.; Libeu, C.P.; Mizushima, T.; et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 1998, 280, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Hyndman, D.J.; Milgrom, Y.M.; Bramhall, E.A.; Cross, R.L. Nucleotide-binding sites on Escherichia coli F1-ATPase. Specificity of noncatalytic sites and inhibition at catalytic sites by MgADP. J. Biol. Chem. 1994, 269, 28871–28877. [Google Scholar] [CrossRef]
- Marquis, R.E. Antimicrobial actions of fluoride for oral bacteria. Can. J. Microbiol. 1995, 41, 955–964. [Google Scholar] [CrossRef]
- Radić, N.; Komljenović, J. Kinetic-potentiometric determination of Fe(III) using a fluoride ion-selective electrode. Croatica Chemica Acta 1991, 64, 679–687. [Google Scholar]
- Kleinhofs, A.; Owais, W.M.; Nilan, R.A. Azide. Mutat. Res./Rev. Genetic Toxicol. 1978, 55, 165–195. [Google Scholar] [CrossRef]
- Esser, D.; Alvarez-Llamas, G.; De Vries, M.P.; Weening, D.; Vonk, R.J.; Roelofsen, H. Sample Stability and Protein Composition of Saliva: Implications for Its Use as a Diagnostic Fluid. Biomark. Insights 2008, 3, 25–37. [Google Scholar] [CrossRef]
- Fluoride Vol. 28 No. 2 71-74 1995 Research Report 71 Effect of Fluoride on Human Salivary Amylase Activity; Haraa, K.; Yu, M.H. (Eds.) Bellingham: Washington, DC, USA, 1995. [Google Scholar]
- Yu, M.; Sumway, M.; Brockbank, A. Effects of NaF on amylase in mung bean seedlings. J. Fluorine Chem. 1988, 41, 95–100. [Google Scholar] [CrossRef]
- Mautner, F.A.; Krischner, H. Preparation and structure determination of tetraethylammonium calcium azide, [N(C2H5)4]Ca(N3)3. Z. Krist.-Cryst. Mater. 1986, 175, 105–110. [Google Scholar] [CrossRef]
- Hoult, D. Solvent peak saturation with single phase and quadrature Fourier transformation. J. Magn. Reson. 1976, 21, 337. [Google Scholar] [CrossRef]
- McKay, R.T. How the 1D-NOESY suppresses solvent signal in metabolomics NMR spectroscopy: An examination on the pulse sequence components and evolution. Concepts Magn. Reson. 2011, 38A, 197–220. [Google Scholar] [CrossRef]
- Bell, J.D.; Sadler, P.J.; Macleod, A.F.; Turner, P.R.; La Ville, A. 1H NMR studies of human blood plasma. Assignment of resonances for lipoproteins. FEBS Lett. 1987, 219, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Wang, Y.; Nicholson, J.K.; Lindon, J.C. Use of relaxation edited one-dimensional and two-dimensional nuclear magnetic resonance spectroscopy to improve detection of small metabolites in blood plasma. Anal. Biochem. 2004, 325, 260–272. [Google Scholar] [CrossRef]
- Grootveld, M.; Claxson, A.W.D.; Chander, C.L.; Haycock, P.; Blake, D.R.; Hawkes, G.E. High resolution proton NMR investigations of rat blood plasma. Assisgnment of resonances for the molecularly mobile carbohydrate side-chains of ‘acute-phase’ glycoproteins. FEBS Lett. 1993, 322, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Aiello, F.; Gerretzen, J.; Simons, M.G.; Davies, A.N.; Dani, P. A multivariate approach to investigate the NMR CPMG pulse sequence for analysing low MW species in polymers. Magn. Reson. Chem. 2021, 59, 172–186. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.D.; Brown, J.C.C.; Kubal, G.; Sadler, P.J. NMR-invisible lactate in blood plasma. FEBS Lett. 1988, 235, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Carr, Y.; Purcell, E.M. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954, 94, 630. [Google Scholar] [CrossRef]
- Hahn, E.L. Spin echoes. Phys. Rev. 1950, 80, 580. [Google Scholar] [CrossRef]
- Aguilar, J.A.; Cassani, J.; Probert, F.; Palace, J.; Claridge, T.D.W.; Botana, A.; Kenwright, A.M. Reliable, high-quality suppression of NMR signals arising from water and macromolecules: Application to bio-fluid analysis. Analyst 2019, 144, 7270. [Google Scholar] [CrossRef]
- Martineau, E.; Giraudeau, P. Fast quantitative 2D NMR for untargeted and targeted metabolomics. Methods Mol. Biol. 2019, 2037, 365–383. [Google Scholar] [CrossRef]
- Martineau, E.; Dumez, J.N.; Giraudeau, P. Fast quantitative 2D NMR for metabolomics and lipidomics: A tutorial. Magn. Reson. Chem. 2020, 58, 390–403. [Google Scholar] [CrossRef]
- Jenzano, J.W.; Hogan, S.L.; Noyes, C.M.; Featherstone, G.L.; Lundblad, R.L. Comparison of five techniques for the determination of protein content in mixed human saliva. Anal. Biochem. 1986, 159, 370–376. [Google Scholar] [CrossRef]
- Lindon, J.C.; Nicholson, J.K.; Everett, J.R. NMR spectroscopy of biofluids. Ann. Rep. NMR Spec. 1999, 38, 1–88. [Google Scholar]
- Available online: https://r.search.yahoo.com/_ylt=AwrIS.g0h.Bh0tgAQRV3Bwx.;_ylu=Y29sbwMEcG9zAzEEdnRpZAMEc2VjA3Ny/RV=2/RE=1642133429/RO=10/RU=https%3a%2f%2fscrippslabs.com%2fprotein-levels-in-human-plasma%2f/RK=2/RS=mwRICqA1HOncIk9beChKyDbv7X8- (accessed on 31 December 2021).
- De Meyer, T.; Sinnaeve, D.; Van Gasse, B.; Tsiporkova, E.; Rietzschel, E.R.; De Buyzere, M.L.; Gillebert, T.C.; Bekaert, S.; Martins, J.C.; Van Criekinge, W. NMR-based characterization of metabolic alterations in hypertension using an adaptive, intelligent binning algorithm. Anal. Chem. 2008, 80, 3783–3790. [Google Scholar] [CrossRef]
- Sysi-Aho, M.; Katajamaa, M.; Yetukuri, L.; Oresic, M. IDconverter and IDClight: Conversion and annotation of gene and protein IDs. BMC Bioinform. 2007, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Grootveld, M. Metabolic Profiling: Disease and Xenobiotics; Royal Society of Chemistry: Cambridge, UK, 2014; 395p, ISBN 1849731632. [Google Scholar]
- Percival, B.C.; Gibson, M.; Wilson, P.B.; Platt, F.M.; Grootveld, M. Metabolomic Studies of Lipid Storage Disorders, with Special Reference to Niemann-Pick Type C Disease: A Critical Review with Future Perspectives. Int. J. Mol. Sci. 2020, 21, 2533. [Google Scholar] [CrossRef] [Green Version]
- Pena-Reyes, C.A.; Sipper, M. Evolutionary computation in medicine: An overview. Artific. Intell. Med. 2000, 19, 1–23. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billoir, E.; Navratil, V.; Blaise, B.J. Sample size calculation in metabolic phenotyping studies. Brief. Bioinform. 2014, 16, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Nyamundanda, G.; Gormley, I.C.; Fan, Y.; Gallagher, W.M.; Brennan, L. MetSizeR: Selecting the optimal sample size for metabolomic studies using an analysis based approach. BMC Bioinform. 2013, 14, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Iterson, M.; ’t Hoen, P.A.C.; Pedotti, P.; Hooiveld, G.J.; den Dunnen, J.T.; van Ommen, G.J.; Boer, J.M.; Menezes, R.X. Relative power and sample size analysis on gene expression profiling data. BMC Genomics. 2009, 10, 439. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://bioconductor.org/biocLite.R (accessed on 31 December 2021).
- Miller, B.F.; Muntz, J.A.; Bradel, S. Decomposition of carbohydrate substrates by dental plaque material. J. Dent. Res. 1940, 19, 473–478. [Google Scholar] [CrossRef]
- Takahashi, N.; Washio, J.; Mayanagi, G. Metabolomics of supragingival plaque and oral bacteria. J. Dent. Res. 2010, 89, 1383–1388. [Google Scholar] [CrossRef]
- Takahashi, N.; Washio, J. Metabolomic effects of xylitol and fluoride on plaque biofilm in vivo. J. Dent. Res. 2011, 90, 1463–1468. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, M.; Saruta, J.; Matsuki, C.; To, M.; Onuma, H.; Kaneko, M.; Soga, T.; Tomita, M.; Tsukinoki, K. Physiological and environmental parameters associated with mass spectrometry-based salivary metabolomic profiles. Metabolomics 2013, 9, 454–463. [Google Scholar] [CrossRef]
- Humphrey, S.P.; Williamson, R.T. A review of saliva: Normal composition, flow, and function. J. Prosth. Dent. 2001, 85, 162–169. [Google Scholar] [CrossRef]
- Kopstein, J.; Wrong, O.M. The origin and fate of salivary urea and ammonia in man. Clin. Sci. Mol. Med. 1977, 52, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Lemos, J.A.; Abranches, J.; Burne, R.A. Responses of cariogenic streptococci to environmental stresses. Curr. Issues Molec. Biol. 2005, 7, 95–107. [Google Scholar]
- Loesche, W.J. Role of Streptococcus mutans in human dental decay. Microbiol. Rev. 1986, 50, 353–380. [Google Scholar] [CrossRef]
- Burne, R.A.; Marquis, R.E. Alkali production by oral bacteria and protection against dental caries. FEMS Microbiol. Lett. 2000, 193, 1–6. [Google Scholar] [CrossRef]
- Griswold, A.R.; Jameson-Lee, M.; Burne, R.A. Regulation and physiologic significance of the agmatine deiminase system of Streptococcus mutans UA159. J. Bacteriol. 2006, 188, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Reyes, E.; Martin, J.; Moncada, G.; Neira, M.; Palma, P.; Gordan, V.; Oyarzo, J.F.; Yevenes, I. Caries-free subjects have high levels of urease and arginine deiminase activity. J. Appl. Oral Sci. 2014, 22, 235–240. [Google Scholar] [CrossRef]
- Zarling, E.J.; Ruchim, M.A. Protein origin of the volatile fatty acids isobutyrate and isovalerate in human stool. J. Lab. Clin. Med. 1987, 109, 566–570. [Google Scholar]
- Arnstein, H. The effect of vitamin B12 on the metabolism of formate and certain formate precursors by the rat. Biochem. J. 1960, 74, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Pietzke, M.; Meiser, J.; Vazquez, A. Formate metabolism in health and disease. Molec. Metab. 2020, 33, 23–37. [Google Scholar] [CrossRef]
- Ripps, H.; Shen, W. Review: Taurine: A “very essential” amino acid. Mol. Vis. 2012, 18, 2673–2686. [Google Scholar]
- Schuller-Levis, G.B.; Park, E. Taurine: New implications for an old amino acid. FEMS Microbiol. Lett. 2003, 226, 195–202. [Google Scholar] [CrossRef]
- Percival, B.C.; Grootveld, M.; Gibson, M.; Osman, Y.; Molinari, M.; Jafari, F.; Sahota, T.; Martin, M.; Casanova, F.; Mather, M.L.; et al. Low-field, benchtop NMR spectroscopy as a potential tool for point-of-care diagnostics of metabolic conditions: Validation, protocols and computational models. High-Throughput 2019, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Elipe, M.V.S.; Milburn, R.R. Monitoring chemical reactions by low-field benchtop NMR at 45 MHz: Pros and cons. Magn. Reson. Chem. 2016, 4, 437–443. [Google Scholar] [CrossRef]
- Edgar, M.; Percival, B.C.; Gibson, M.; Masania, J.; Beresford, K.; Wilson, P.B.; Grootveld, M. Benchtop NMR spectroscopy and spectral analysis of the cis- and trans-stilbene products of the Wittig reaction. J. Chem. Educ. 2019, 96, 1938–1947. [Google Scholar] [CrossRef]
- Isaac-Lam, M.F. Analysis of bromination of ethylbenzene using a 45 MHz NMR spectrometer: An undergraduate organic chemistry laboratory experiment. J. Chem. Educ. 2014, 91, 1264–1266. [Google Scholar] [CrossRef]
- Hoppenbrouwers, P.M.; Driessens, F.C. The effect of lactic and acetic acid on the formation of artificial caries lesions. J. Dent. Res. 1988, 67, 1466–1467. [Google Scholar] [CrossRef]
- Niederman, R.; Zhang, J.; Kashket, S. Short-chain carboxylic-acid-stimulated, PMN-mediated gingival inflammation. Crit. Rev. Oral Biol Med. 1997, 8, 269–290. [Google Scholar] [CrossRef]
- Niederman, R.; Buyle-Bodin, Y.; Lu, B.Y.; Naleway, C.; Robinson, P.; Kent, R. The relationship of gingival crevicular fluid short chain carboxylic acid concentration to gingival inflammation. J. Clin. Periodontol. 1996, 23, 743–749. [Google Scholar] [CrossRef]
- Bose, S.; Ramesh, V.; Locasale, J.W. Acetate metabolism in physiology, cancer, and beyond. Trends Cell Biol. 2019, 29, 695–703.167. [Google Scholar] [CrossRef]
- Tam, Y.C.; Chan, E.C. Purification and characterization of hyaluronidase from oral Peptostreptococcus species. Infect. Immun. 1985, 47, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Last, K.S.; Embery, G. Hyaluronic acid and hyaluronidase activity in gingival exudate from sites of acute ulcerative gingivitis in man. Arch. Oral Biol. 1987, 32, 811–815. [Google Scholar] [CrossRef]
- Mitchell, S. Trimethylaminuria (fish-odour syndrome) and oral malodour. Oral Dis. 2005, 11 (Suppl. 1), 10–13. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Dong, L.L.; Kong, J.J. Impact of volatile sulphur compounds in periodontal pockets on initial periodontal therapy. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2008, 37, 418–421. (In Chinese) [Google Scholar] [PubMed]
- Mercier, P.; Lewis, M.J.; Chang, D.; Baker, D.; Wishart, D.S. Towards automatic metabolomic profiling of high-resolution one-dimensional proton NMR spectra. J. Biomol. NMR. 2011, 49, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Astle, W.; De Iorio, M.; Ebbels, T.M.D. BATMAN-an R package for the automated quantification of metabolites from nuclear magnetic resonance spectra using a Bayesian model. Bioinformatics 2012, 28, 2088–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]







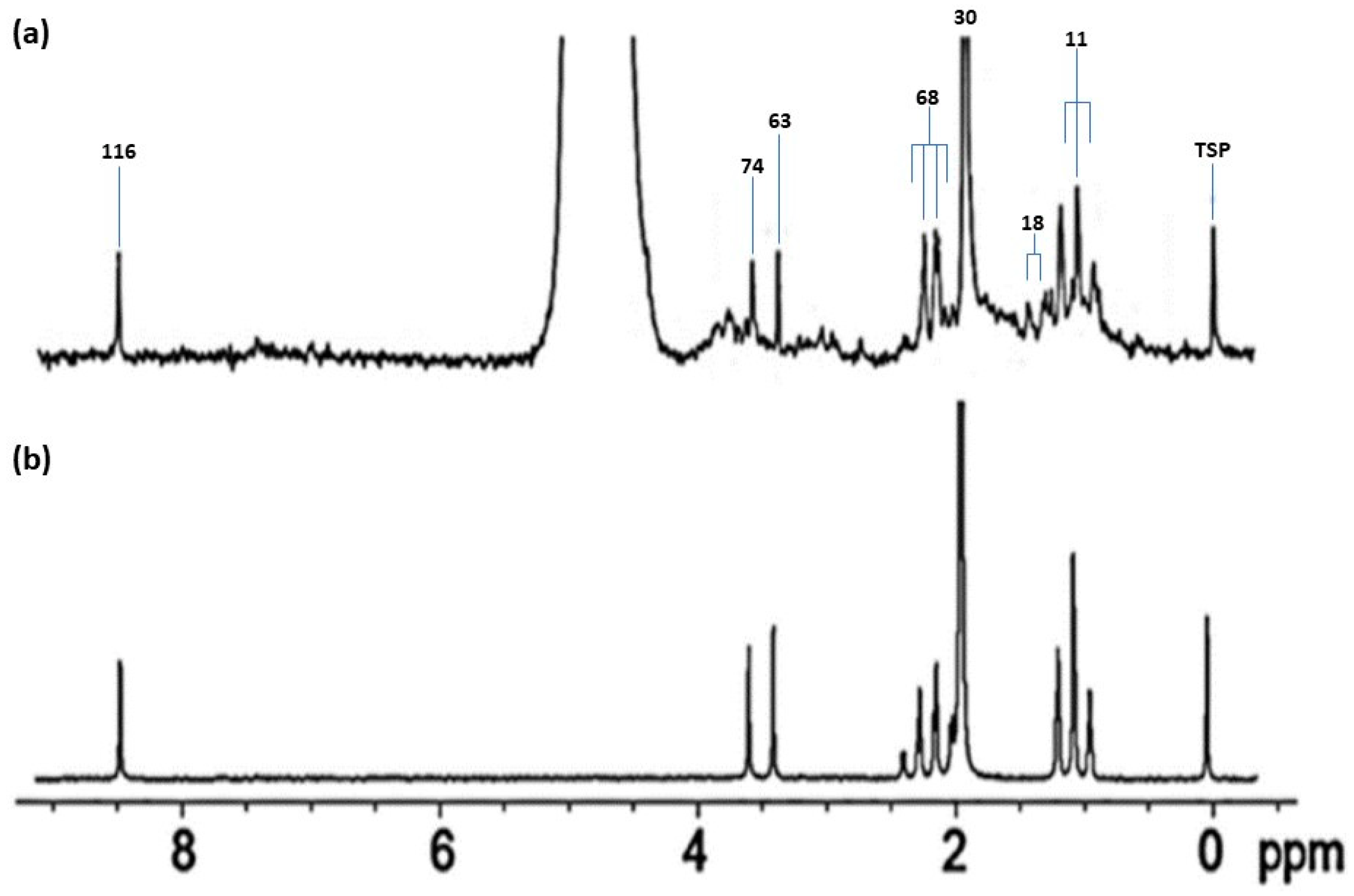

{kind=link}
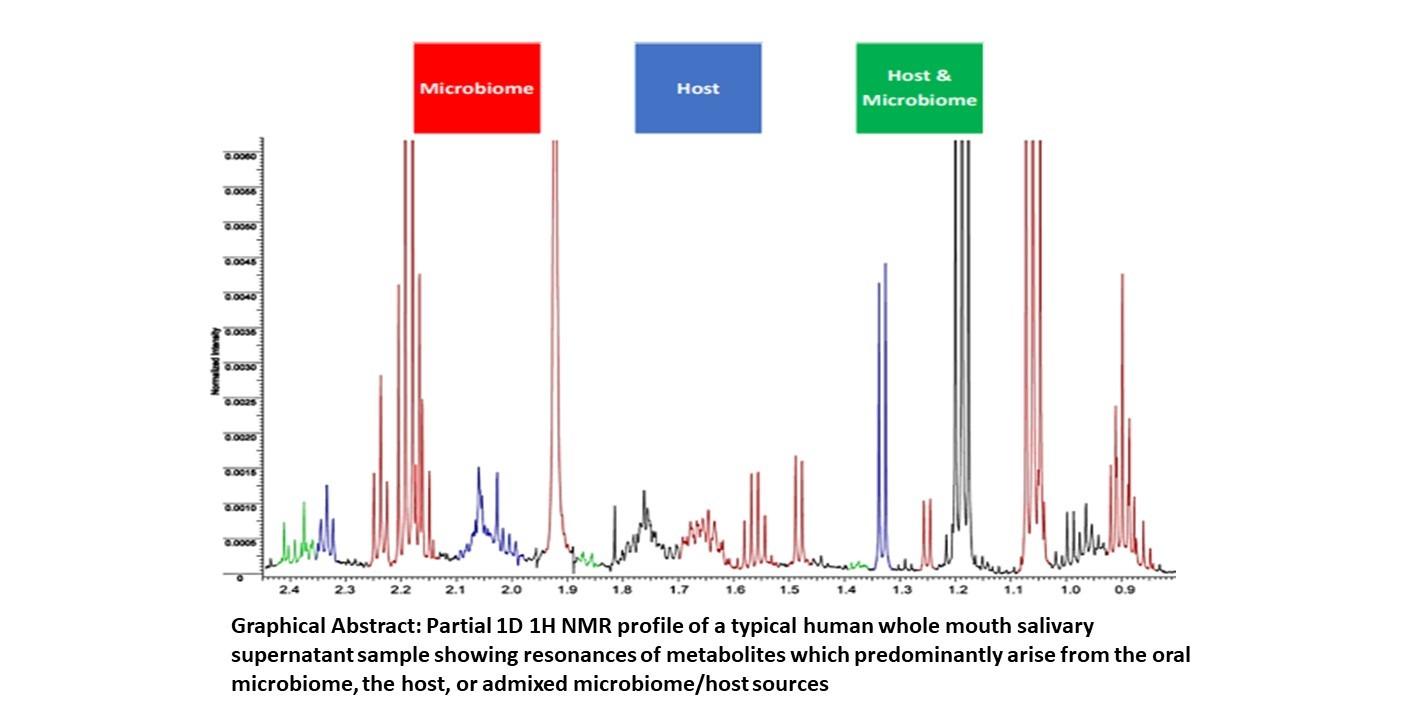
{kind=link}
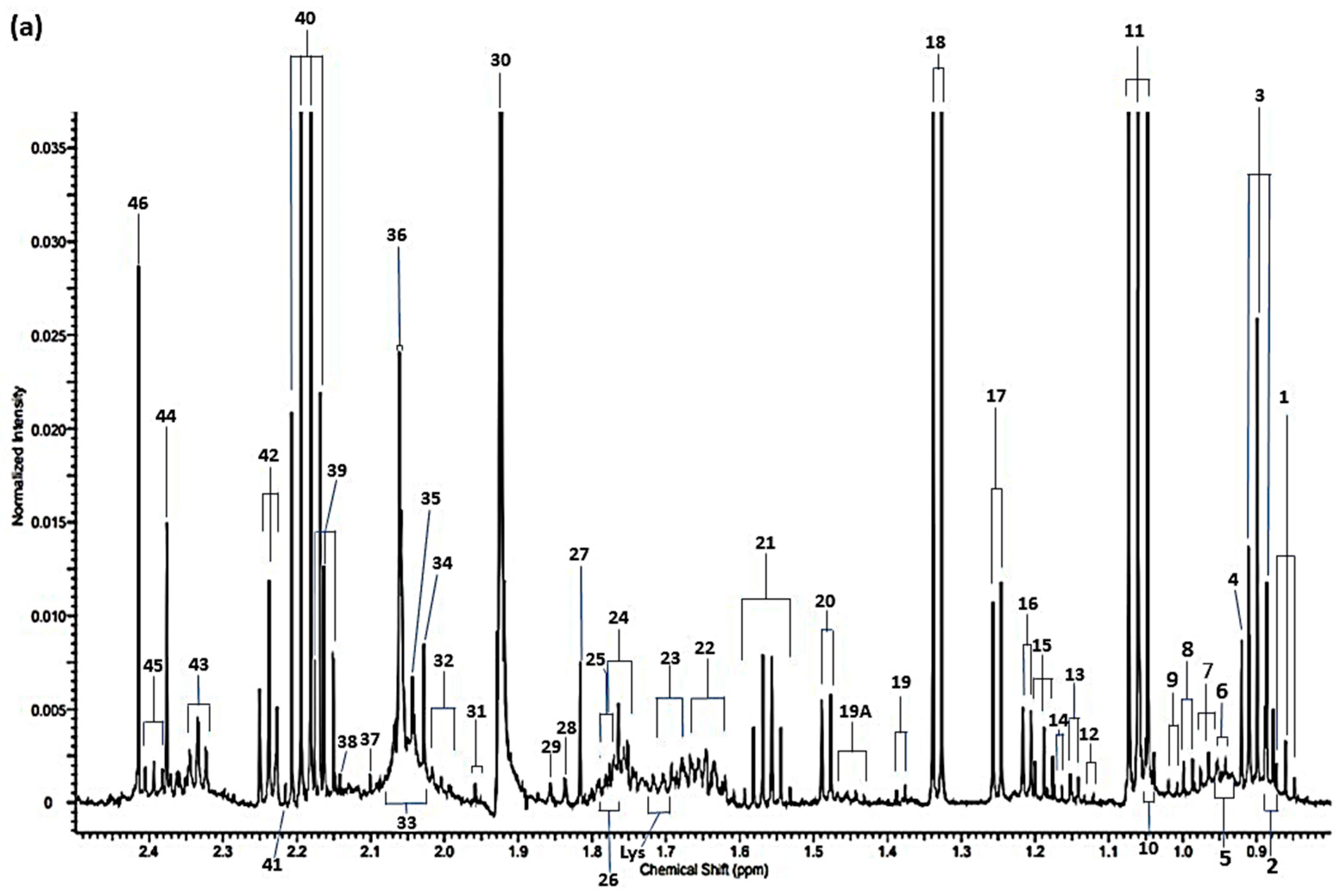
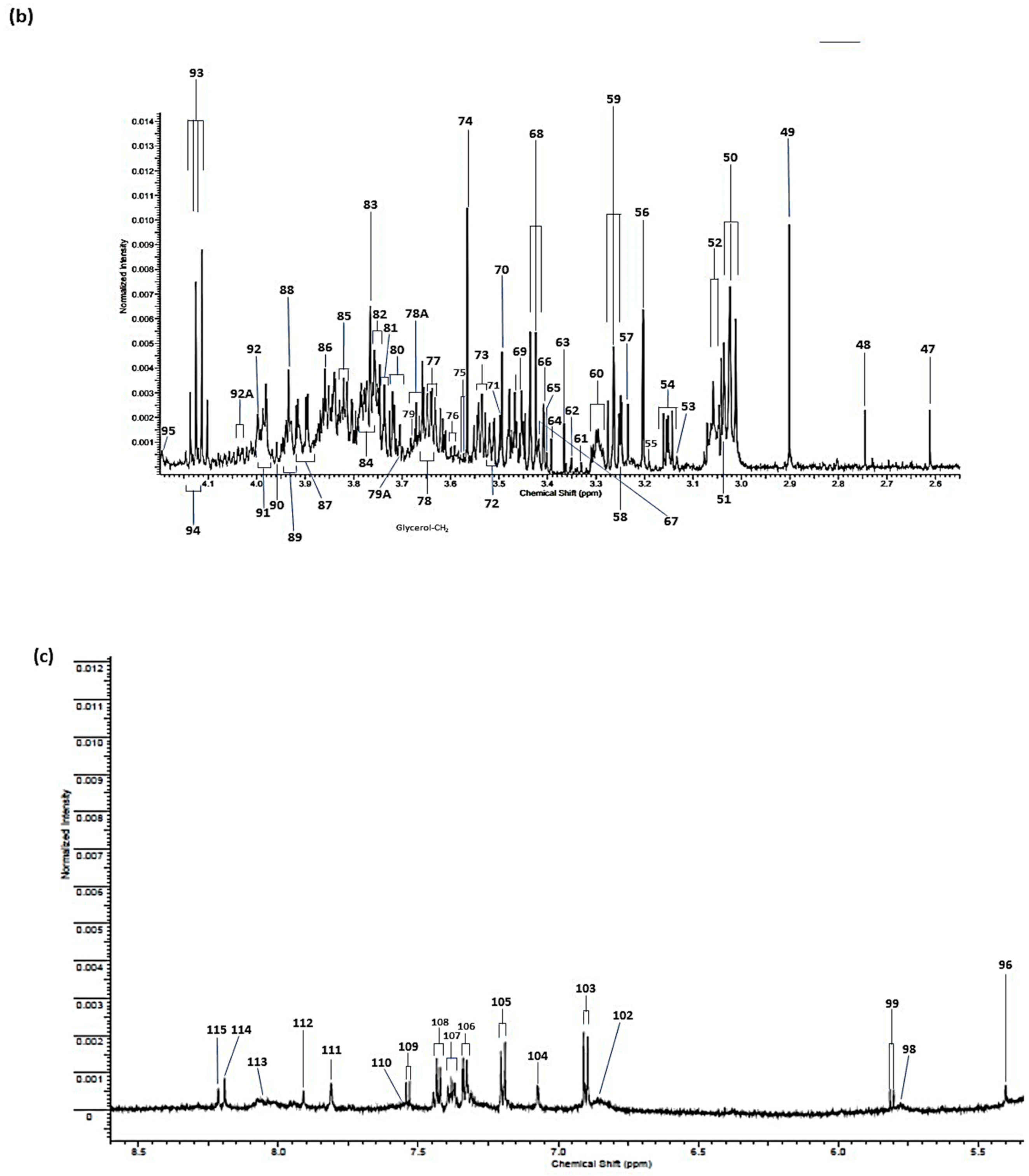{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
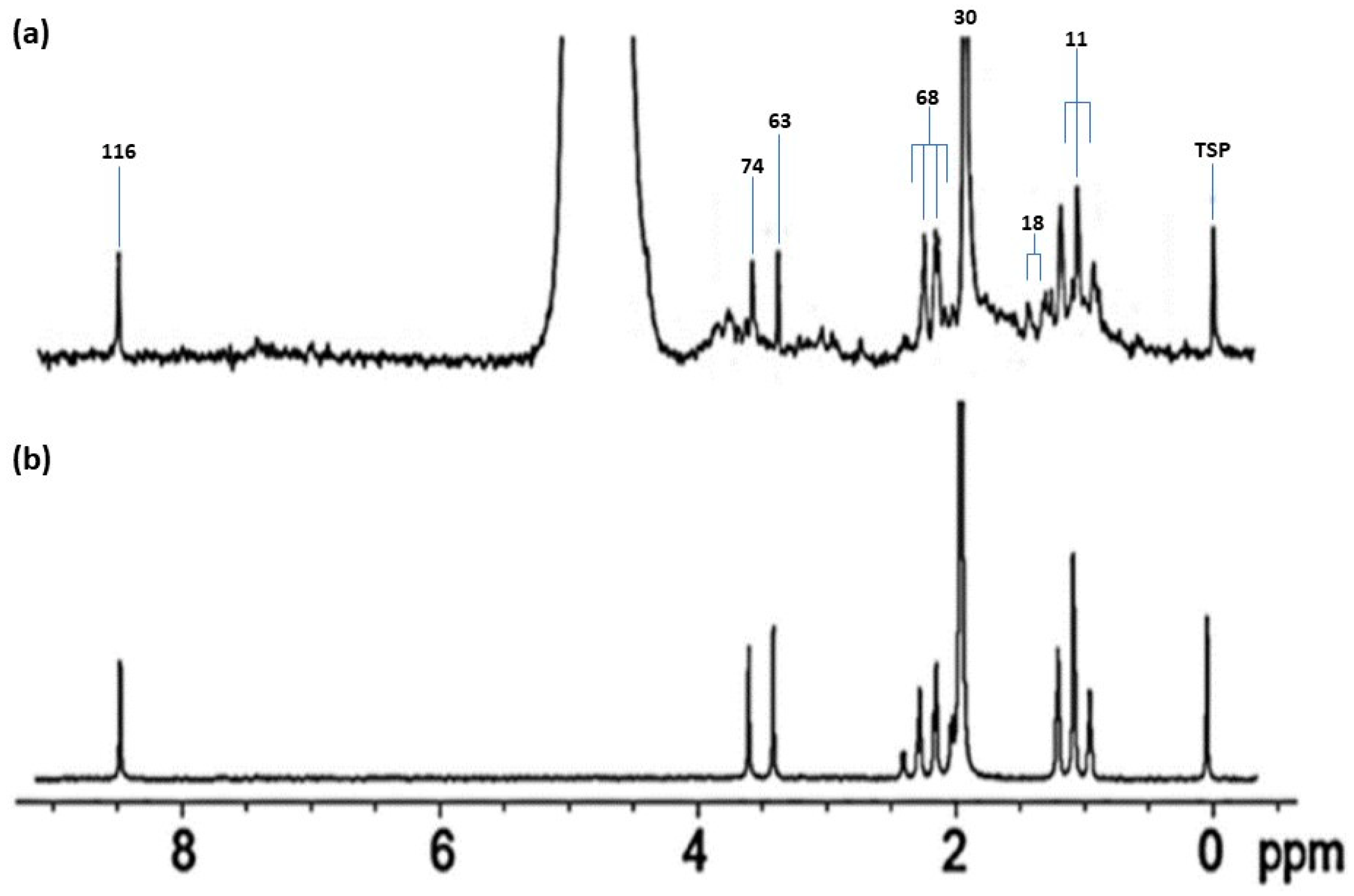{kind=link}
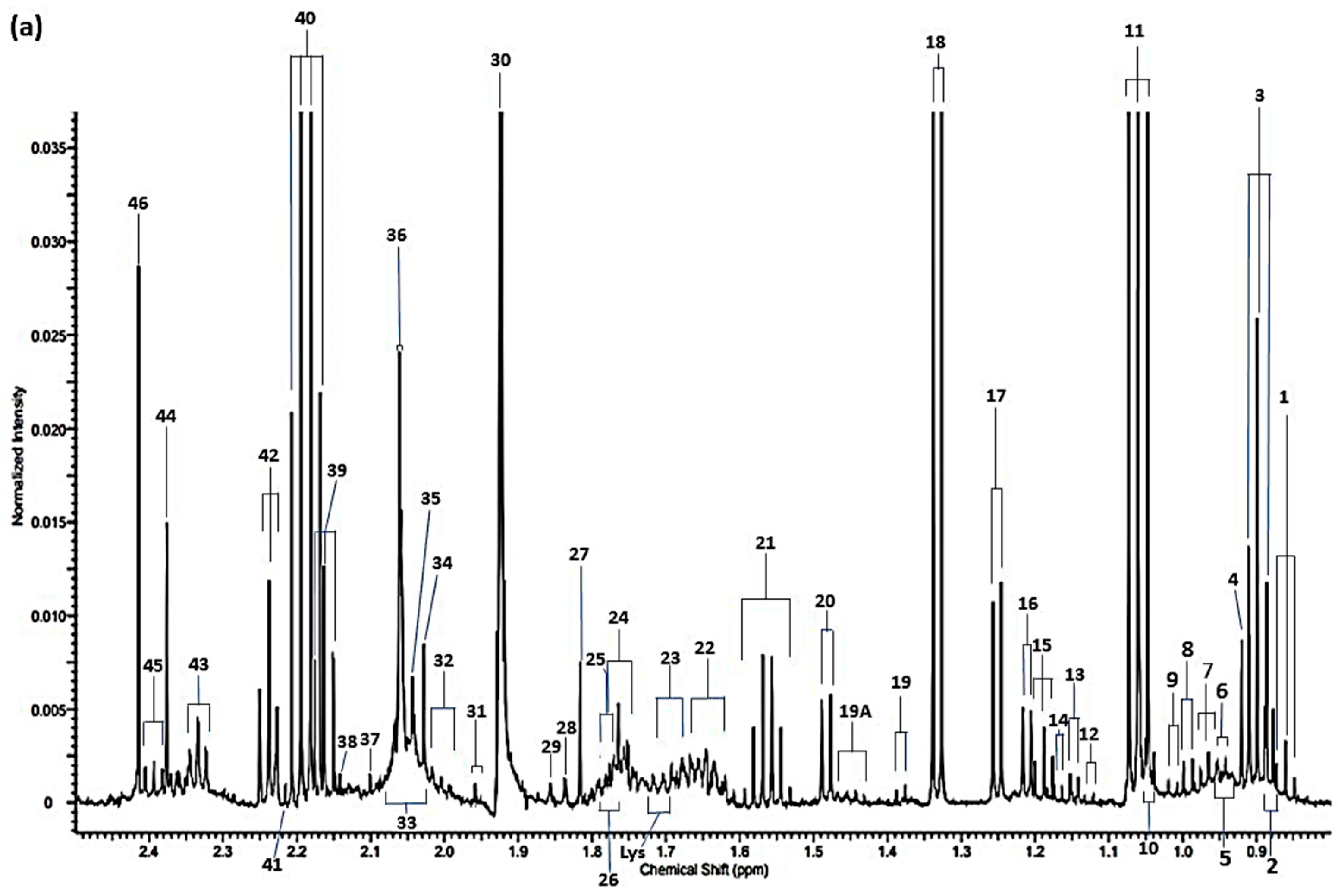
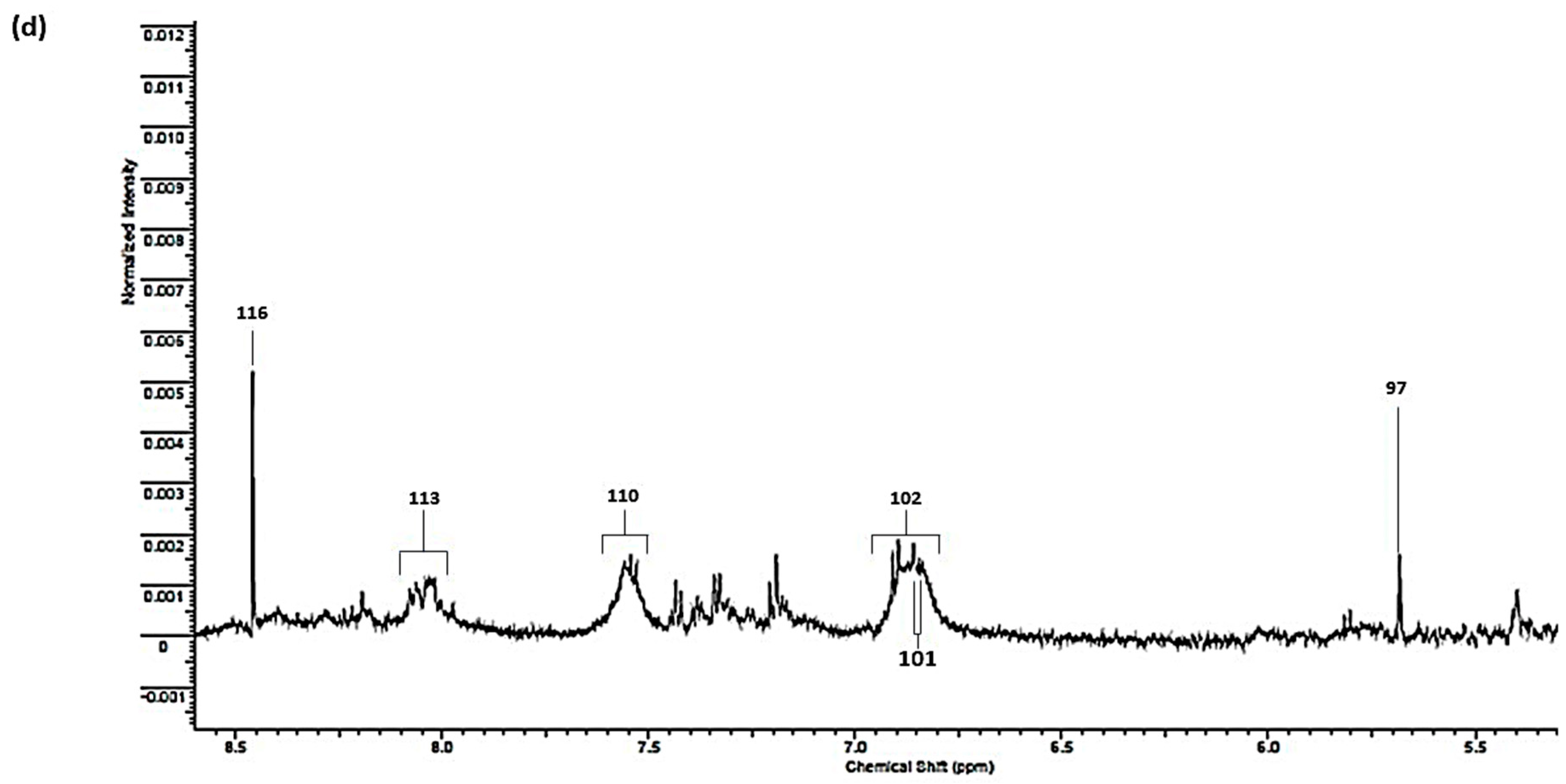
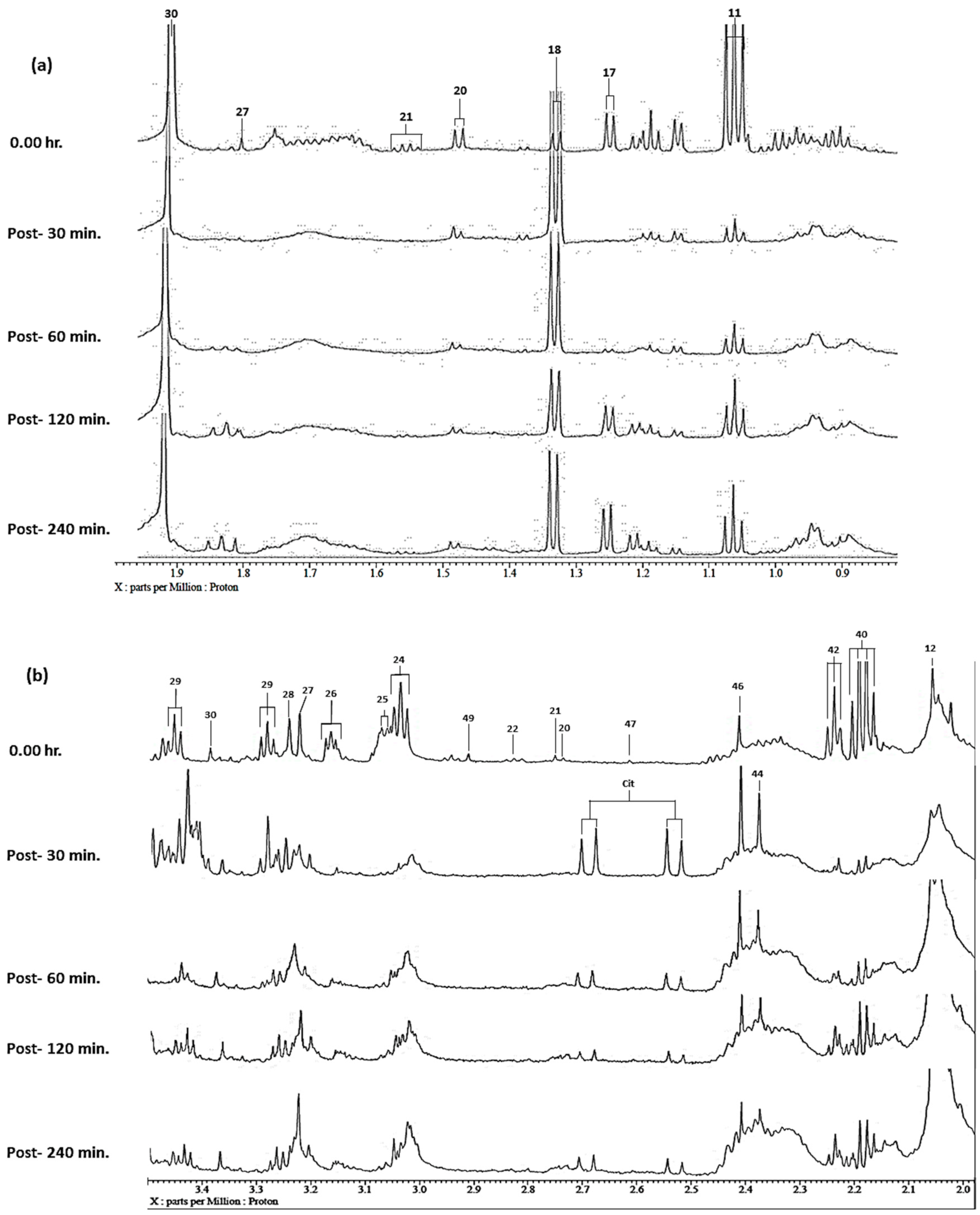
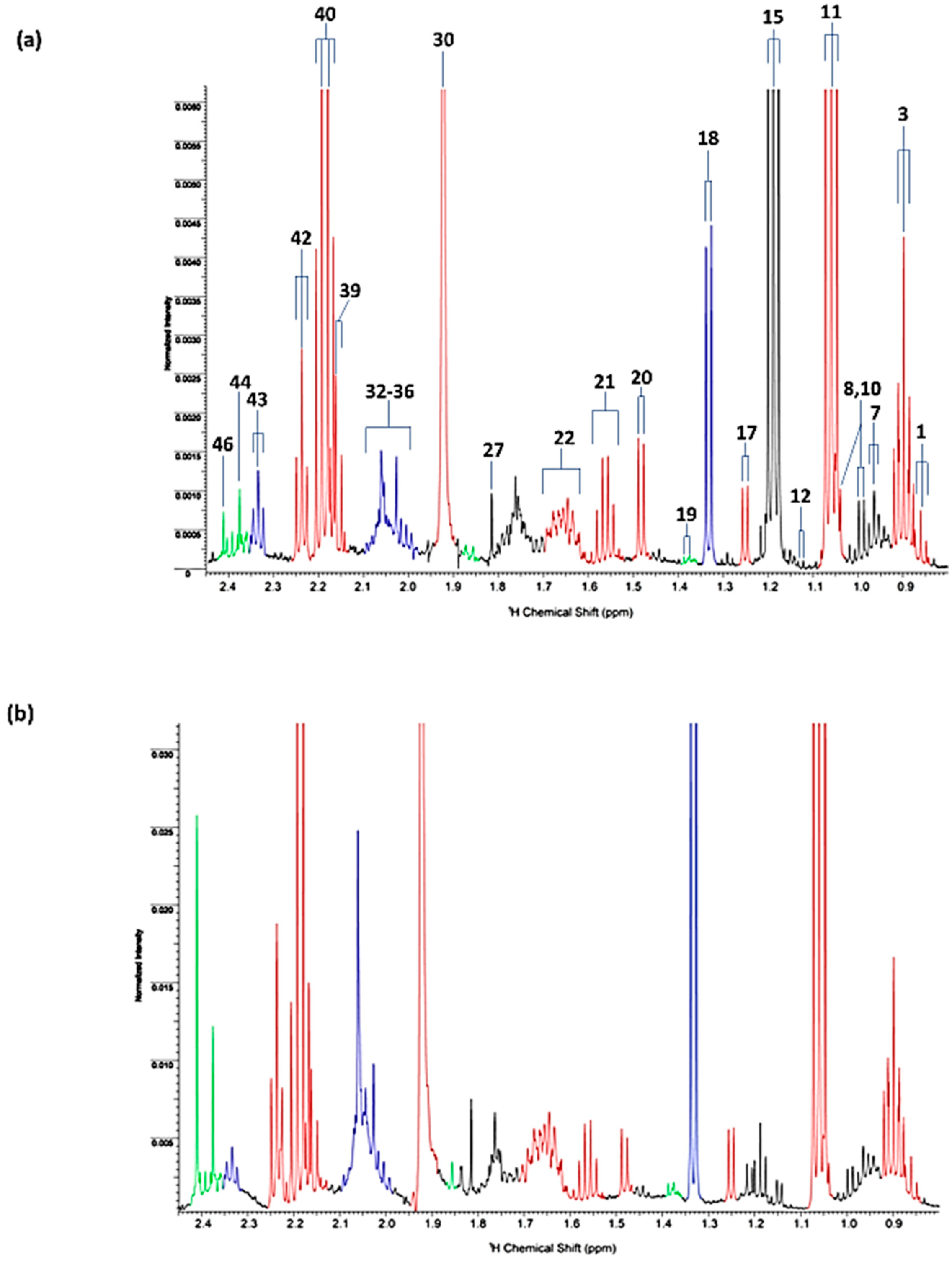
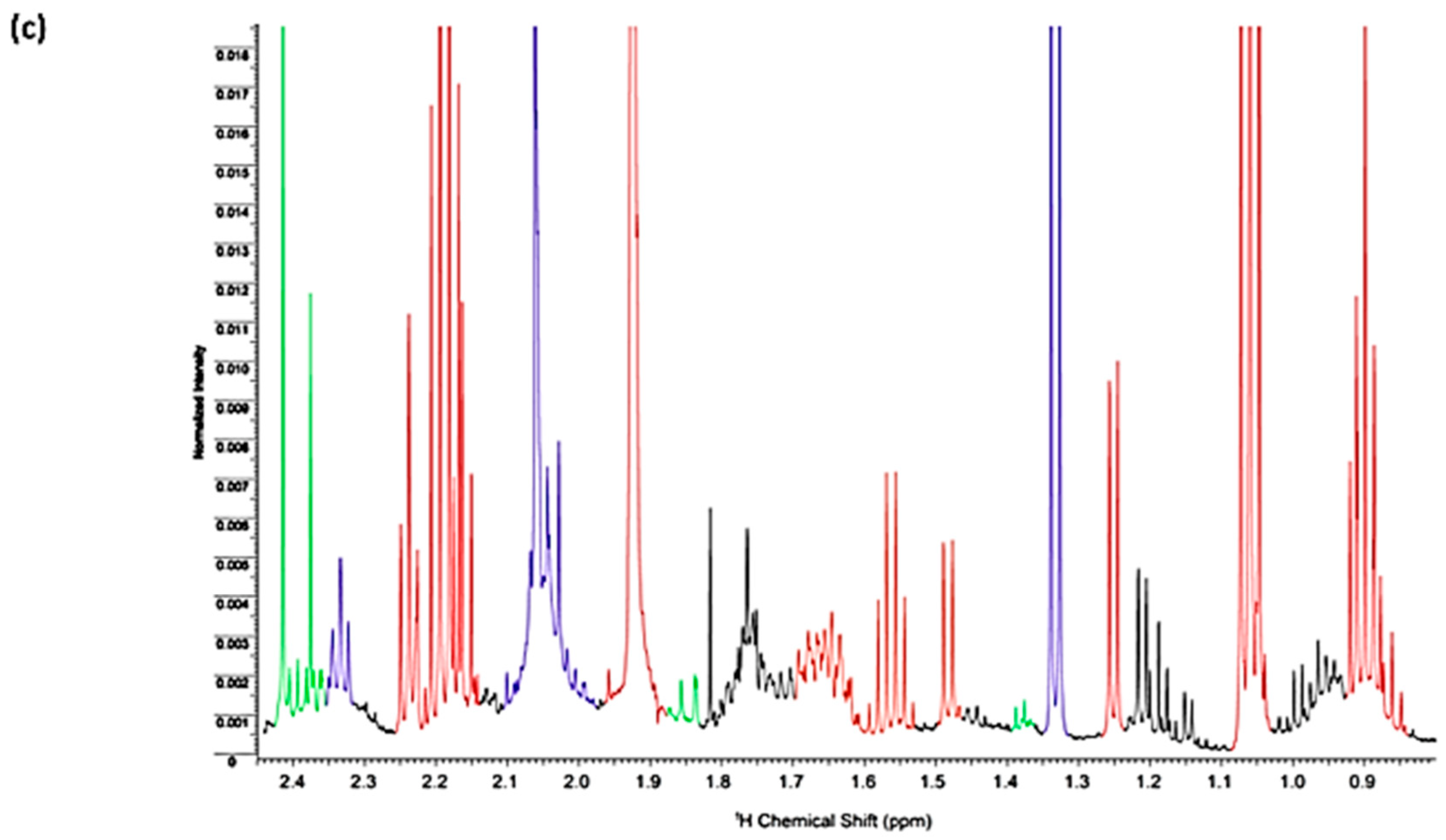
| Assignment | Assignment Code | Chemical Shift (δ/ppm) | Coupling Pattern |
|---|---|---|---|
| n-Caproate-CH3 | 1 | 0.861 | t |
| iso-Valerate-CH3′s | 2 | 0.885 | d |
| n-Butyrate-CH3 | 3 | 0.893 | t |
| iso-Caproate-CH3 | 4 | 0.912 | ½ d |
| Isoleucine-CH3 | n/a | 0.921 | t |
| * Long-chain fatty acid-/Triacylglycerol acyl chain-terminal-CH3 | 5 | 0.94 | broad |
| Hydroxy-iso-caproate-CH3s | 6 | 0.946 | d |
| Leucine-CH3 | 7 | 0.962 | t |
| Valine-CH3/Isoleucine-CH3 | 8 | 0.982/0.996 | 2 × d |
| Methylsuccinate-CH3 | 9 | 1.014 | d |
| Valine-CH3 | 10 | 1.035 | d |
| Propioniate-CH3 | 11 | 1.058 | t |
| Unassigned | n/a | 1.10 | d |
| iso-Butyrate-CH3 | 12 | 1.125 | d |
| 3-Amino-iso-butyrate-CH3 | 13 | 1.150 | d |
| iso-Propyl alcohol-CH3s | 14 | 1.168 | d |
| Ethanol-CH3 | 15 | 1.183 | t |
| Methylmalonate-CH3/α-Fucose-CH3 | 16 | 1.211 | d |
| 3-D-hydroxybutyrate-CH3/β-Fucose-CH3 | 17 | 1.242 | d |
| Lactate-CH3 | 18 | 1.330 | d |
| Acetoin-CH3 | 19 | 1.371 | d |
| Unassigned | 19A | 1.454 | m |
| Alanine-CH3 | 20 | 1.486 | d |
| n-Butyrate-β-CH2 | 21 | 1.551 | q |
| 5-Aminovalerate-β,γ-CH2′s | 22 | 1.641 | m |
| Leucine-CH2 | 23 | 1.685 | m |
| Lysine-δ-CH2 | Lys | 1.71 | m |
| Arginine-γ-CH2 | 24 | 1.72 | m |
| Putrescine-β-CH2 | 25 | 1.76 | m |
| Ornithine-β-CH2/Propane-1,3-diol-β-CH2 | 26 | 1.77 | m |
| Senicioate-CH3 | 27 | 1.81 | s |
| * Glutarate-β-CH2 | 28 | 1.832 | m |
| Thymine-CH3 | 29 | 1.87 | s |
| Acetate-CH3 | 30 | 1.92 | s |
| * 2-Hydroxyglutarate-γ-CH2 | 31 | 1.954 | m |
| Proline-γ-CH2/N-Acetylneuraminate-C3H | 32 | 2.005 | m |
| Hyaluronate/Glycoprotein carbohydrate side-chain N-acetylsugar-NHCOCH3 functions | 33 | 2.02–2.080 (3 partially resolved signals) | broad |
| N-Acetylglutamate-/N-Acetylaspartate--NHCOCH3 (2 signals) | 34 | 2.025/2.030 | s |
| N-Acetylglucosamine-/N-Acetylneuraminate--NHCOCH3 | 35 | 2.040 | s |
| * Unassigned N-Acetylated metabolite-NHCOCH3 (2 signals) | 36 | 2.053/2.058 | 2 × s |
| Dimethylsulphide-S-CH3 | 37 | 2.098 | s |
| Methionone-S-CH3 | 38 | 2.140 | s |
| n-Butyrate-α-CH2 | 39 | 2.164 | t |
| Propioniate-CH2 | 40 | 2.193 | q |
| Acetone-CO-CH3 | 41 | 2.215 | s |
| 5-Aminovalerate-α-CH2 | 42 | 2.235 | t |
| Glutamate-γ-CH2 | 43 | 2.334 | m |
| Pyruvate-CH3 | 44 | 2.377 | s |
| Glutamine-γ-CH2 | 45 | 2.393 | m |
| Succinate-CH2 | 46 | 2.415 | s |
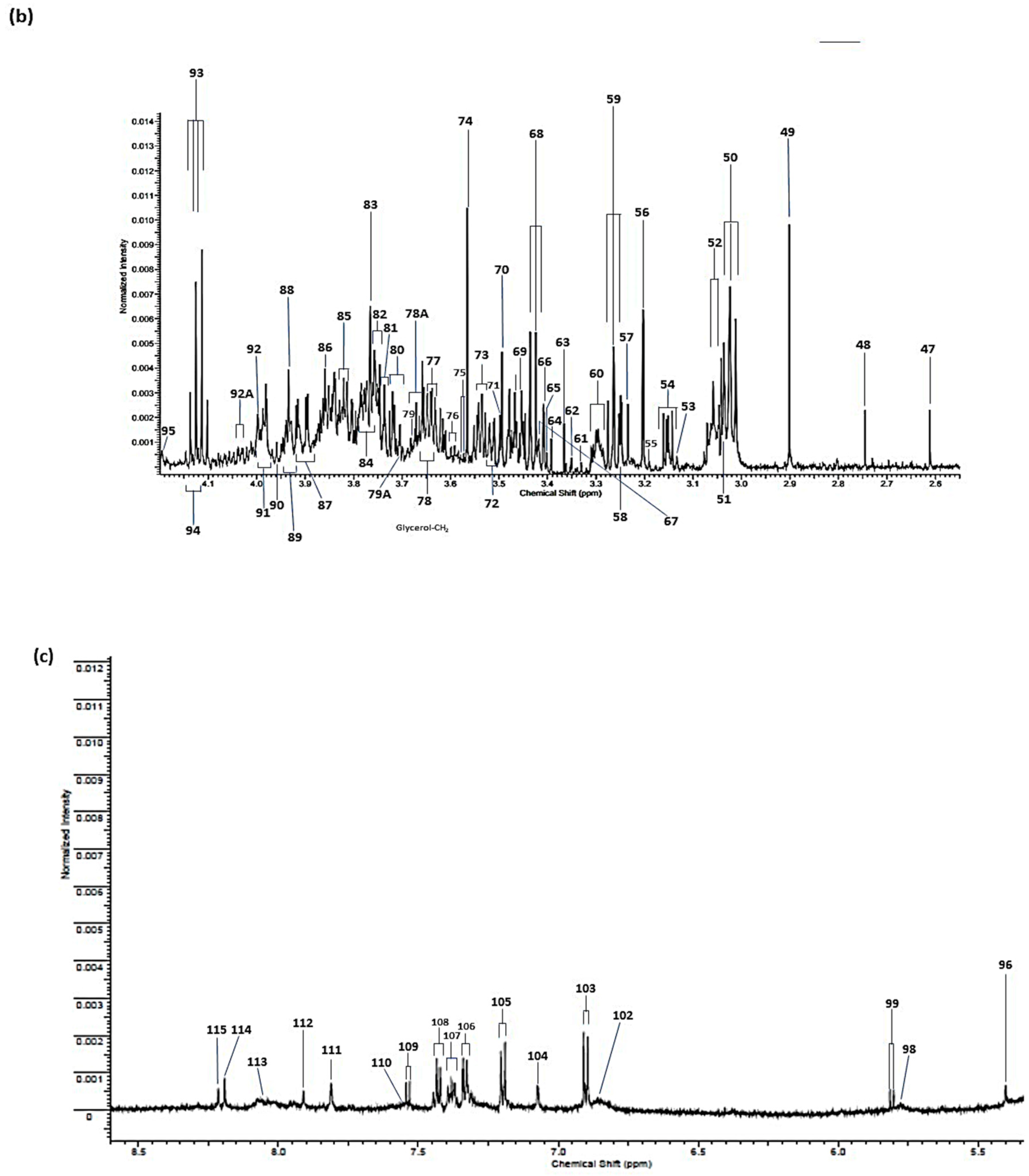
| Methylamine-N(CH3) | 47 | 2.610 | s |
| Citrate-CH2CO2− | Cit | 2.611 | AB coupling pattern |
| Dimethylamine-N(CH3)2 | 48 | 2.745 | s |
| Trimethylamine-N(CH3)3 | 49 | 2.900 | s |
| 5-Aminovalerate-δ-CH2/Lysine-ε-CH2 | 50 | 3.023 | t/t |
| Creatine-N(CH3) | 51 | 3.037 | s |
| Putrescine-α-CH2 | 52 | 3.072 | t |
| Dimethylsulphone-SO(CH3)2 | 53 | 3.13 | s |
| ½ Histidine-/Phenylalanine-β-CH2 | 54 | 3.148 | m |
| * 1,9-Dimethylurate-N1(CH3) | 55 | 3.190 | s |
| Choline-N(CH3)3+ | 56 | 3.201 | s |
| Trimethylamine-N-oxide-N(CH3)3 | 57 | 3.232 | s |
| Betaine-N(CH3)3+ | 58 | 3.248 | s |
| Taurine-CH2NH3+ | 59 | 3.263 | t |
| ½ Phenylalanine-β-CH2/Tryptophan-β-CH2 | 60 | 3.293 | m |
| * Paraxanthine-N1(CH3) | 61 | 3.331 | s |
| * 1,3,7-Trimethylurate-N7(CH3) | 62 | 3.351 | s |
| Methanol-CH3 | 63 | 3.367 | s |
| * 1,3-Dimethyluracil-N1(CH3) | 64 | 3.392 | s |
| * 3,7-Dimethylurate-N7(CH3) | 65 | 3.402 | s |
| * Carnitine-β-CH2 | 66 | 3.409 | m |
| Proline-α-C1H | 67 | 3.419 | dd |
| Taurine-CH2SO3− | 68 | 3.423 | t |
| * 4-Hydroxyphenylacetate-CH2/β-Fucose-C2H | 69 | 3.453 | s/dd |
| * 1-Methylguanine-N1(CH3) | 69A | 3.464 | s |
| Unassigned | 70 | 3.491 | s |
| Phenylacetate-CH2 | 71 | 3.499 | s |
| Choline-C2-CH2 | 72 | 3.511 | dd |
| ½ Glycerol-CH2OH | 73 | 3.536 | m |
| Glycine-CH2 | 74 | 3.564 | s |
| Threonine-α-CH | 75 | 3.575 | d |
| Valine-α-CH | 76 | 3.597 | d |
| * Propane-1,3-diol-CH2OH/β-Fucose-C3H | 77 | 3.637 | t/dd |
| ½ Glycerol-CH2OH | 78A | 3.649 | m |
| Ethanol-CH2 | 78 | 3.654 | q |
| 1-Methylhistidine-N1(CH3) | 79 | 3.677 | s |
| 3-Methylhistidine-N3(CH3) | 79A | 3.709 | s |
| Lysine-α-CH | 80 | 3.718 | m |
| Leucine-α-CH | 81 | 3.733 | m |
| * Gluconate-C4/C5-CHOH | 82 | 3.741 | m |
| Guanidoacetate-CH2 | 83 | 3.760 | s |
| Alanine-α-CH/Glycerol-CHOH | 84 | 3.771 | m/m |
| Ethanolamine-CH2OH/β-Fucose-C5H | 85 | 3.814 | t/m |
| * α-Fucose-C3H | 86 | 3.852 | m |
| Serine-α-CH | 87 | 3.901 | m |
| Glycolate-CH2OH | 88 | 3.929 | s |
| Tyrosine-α-CH | 89 | 3.934 | m |
| Creatine-N-CH2 | 90 | 3.958 | s |
| Histidine/Phenylalanine-α-CH | 91 | 3.985 | m |
| 4-Hydroxyphenylpyruvate-CH2 | 92 | 3.996 | s |
| Tryptophan-α-CH | 92A | 4.032 | dd |
| Lactate-CH | 93 | 4.120 | q |
| 3-D-Hydroxybutyrate-CHOH | 94 | 4.132 | m |
| α-Fucose-C5H | 95 | 4.198 | m |
| Threonine-β-CHOH | 95A | 4.234 | m |
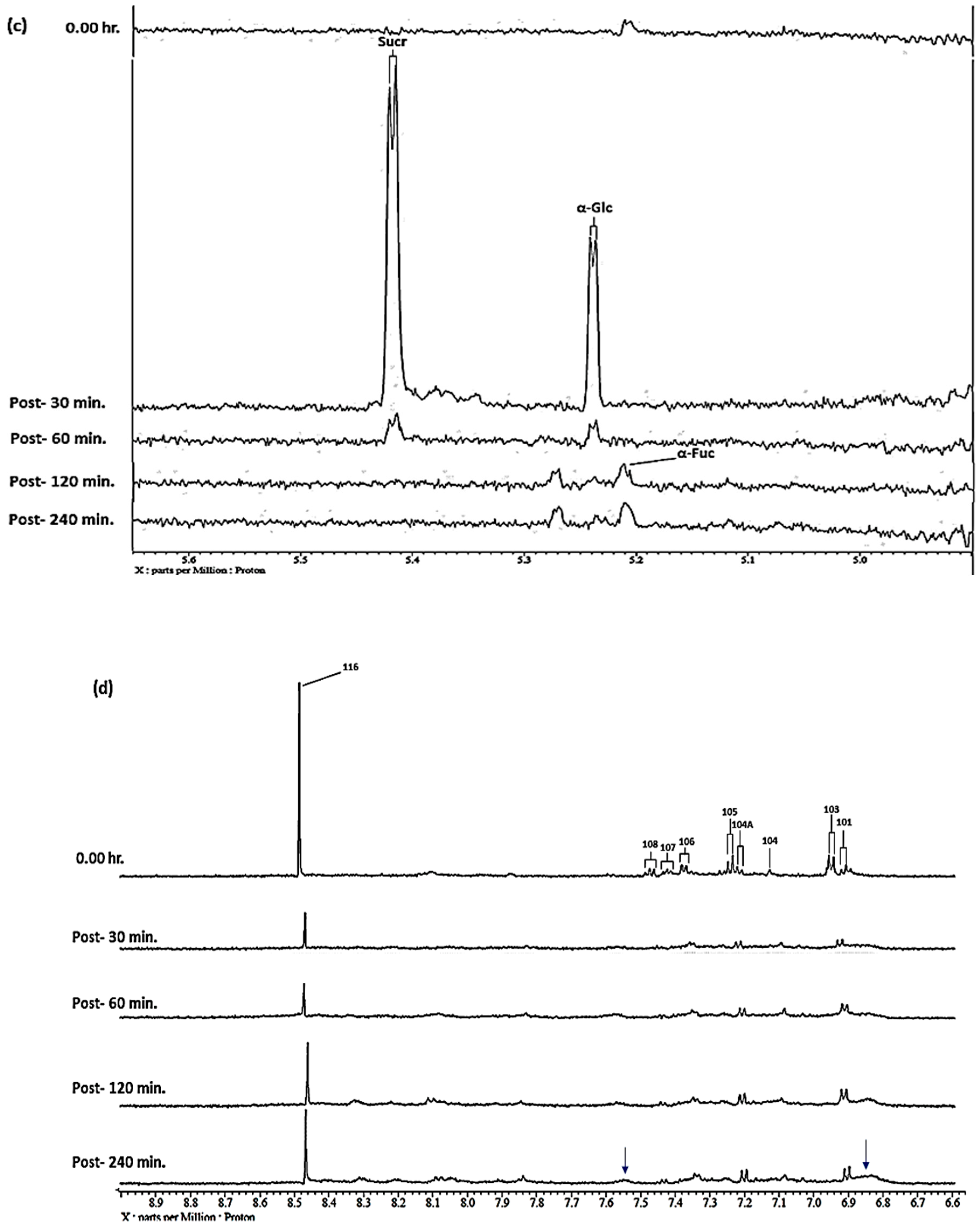
| α-Fucose-C1H | α-Fuc | 5.209 | d |
| α-Glucose-C1H | α-Glc | 5.232 | d |
| Sucrose-C1H | Sucr | 5.414 | d |
| Allantoin-CH | 96 | 5.392 | s |
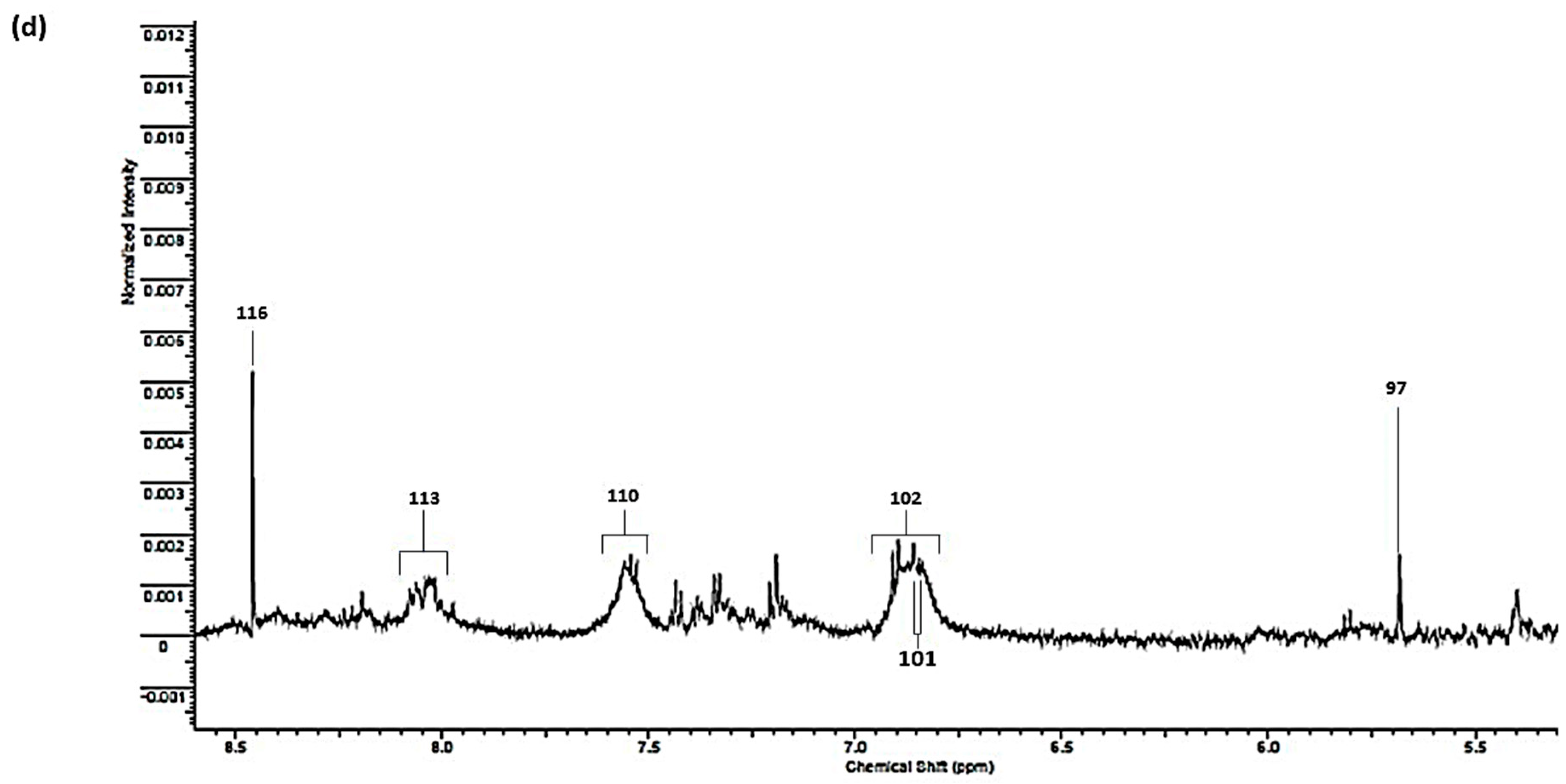
| Senecioate-α-CH=C< | 97 | 5.666 | s |
| Urea-CO-NH2 | 98 | 5.79 | broad |
| Uracil-C2H | 99 | 5.800 | d |
| Fumarate-CH=CH- | 100 | 6.52 | s |
| 4-Hydroxyphenylacaetate aromatic ring-C3H/C5H | 101 | 6.84 | d |
| * Protein aromatic amino acid residue(s) | 102 | 6.85 | broad |
| Tyrosine aromatic ring-C2H/C6H | 103 | 6.880 | d |
| Histidine imidazole ring-C5H | 104 | 7.071 | s |
| 4-hydroxyphenylacetate aromatic ring-C2H/C6H | 104A | 7.155 | d |
| Tyrosine aromatic ring-C3H/C5H | 105 | 7.237 | d |
| Phenylalanine aromatic Ring-C2H/C6H | 106 | 7.320 | m |
| Phenylalanine aromatic ring-C4H | 107 | 7.375 | m |
| Phenylalanine aromatic ring-C3H/C5H | 108 | 7.43 | m |
| Uracil-C1H | 109 | 7.533 | d |
| * Protein aromatic amino acid residue(s) | 110 | 7.552 | m |
| Histidine imidazole ring-C2H | 111 | 7.812 | s |
| 3-Methylhisitidine imidazole ring-C2H | 112 | 7.913 | s |
| * Protein aromatic amino acid residue(s) | 113 | 8.05 | 2 × broad signals |
| Hypoxanthine-C8H | 114 | 8.175 | s |
| Hypoxanthine-C3H | 115 | 8.219 | s |
| Formate-CH | 116 | 8.456 | s |
| * trans-2-Alkenal-CHO Function | 117 | 9.57 | d |
| Code | Disease(s) Studied/Notes | Bioanalytical Techique(s) Employed | Sample Sizes: Diseased (n1)/Controls (n2)) | Protocol: Pre-Collection Abstention/Fasting Limit Specified (h) | Reference | Date Accessed |
|---|---|---|---|---|---|---|
| 1 | Oral cancer | CE-TOF-MS | 24/44 | 1.5 | [59] | 19/07/21 |
| 2 | Saliva stability study | 1H NMR | na/5 | 1.5 | [60] | 19/07/21 |
| 3 | Salivary metabolome contributions | 1H NMR | na/11 | 1 | [20] | 19/07/21 |
| 4 | Type 1 diabetes | 1H NMR | 34/34 | 1 | [61] | 19/07/21 |
| 5 | Chronic periodontitis | 1H NMR | 30/15 | Unspecified | [62] | 19/07/21 |
| 6 | Peritoneal dialysis treatment | 1H NMR | 12/31 | 1 | [63] | 19/07/21 |
| 7 | Chronic renal failure in hemodialysis | Spectrophotometry | 57/na | 1.5 | [64] | 19/07/21 |
| 8 | Chronic hepatitis | 1H NMR | 20/16 | 8 | [65] | 19/07/21 |
| 9 | Dental caries | UPLC-MSGC/EI-MS | 21/36 | Unspecified | [66] | 19/07/21 |
| 10 | Oral health status (periodontitis) | UHPLC-MS | na/909 | Unspecified | [67] | 19/07/21 |
| 11 | Alzheimer’s disease | FUPLC-MS | 256/218 | 2 | [68] | 19/07/21 |
| 12 | Oral squamous cell carcinoma | CPSI-MS | 249/124 | 1 | [69] | 19/07/21 |
| 13 | Breast cancer | CE-LC-MS | 124/42 | 1 | [70] | 19/07/21 |
| 14 | Aggressive and chronic periodontitis | 1H NMR | 61/39 | 1 | [71] | 19/07/21 |
| 15 | Alzheimer’s disease | LC-MS | 47/35 | 1 | [72] | 19/07/21 |
| 16 | Perinatally hiv-infected youths with periodontal disease | GC-MS | 12/21 | 2 | [73] | 20/07/21 |
| 17 | Sjögren’s syndrome | LC-MS | 20/20 | 1.5 | [74] | 20/07/21 |
| 18 | Oral cancer and leukoplakia | UPLC-QTOF-MS | 69/34 | 1.5 | [75] | 20/07/21 |
| 19 | Breast cancer | LC-QTOF-MS | 23/35 | 1 | [76] | 20/07/21 |
| 20 | Post-periodontal surgery (early repair markers) | 1H NMR | 124/52 | 0 (no fasting) | [77] | 20/07/21 |
| 21 | Oral, breast, and pancreatic cancer | CE-TOF-MS | 128/87 | 1 | [78] | 20/07/21 |
| 22 | Effects of inter-day and intra-day variation | CE-TOF-MS | na/13 | 2 | [79] | 20/07/21 |
| 23 | Metabolome of canine and human saliva | UHPLC-QTOF-MS | na/14 (human); na/13 (dog) | 1.5 (human)/12 (dogs) | [80] | 20/07/21 |
| 24 | Children and adolescents after hemodialysis | 1H NMR | 30/40 | 1 | [81] | 20/07/21 |
| 25 | Oral squamous cell carcinoma and oral lichen planus | CE-TOF-MS | 60/na | 1.5 | [82] | 20/07/21 |
| 26 | Halitosis | GC-MS | 22/30 | Variable | [83] | 20/07/21 |
| 27 | Effect of timing of collection of salivary metabolomic biomarkers on oral cancer detection. | CE-TOF-MS | 22/44 | Variable | [84] | 20/07/21 |
| 28 | Children with dental caries before and after treatment. | 1H NMR | 20/10 | 2 | [85] | 20/07/21 |
| 29 | Endogenous salivary citrate | 1H NMR | na/10 | 1 | [86] | 20/07/21 |
| 30 | Understanding the human salivary metabolome | 1H NMR | 23 (smokers)/23 (non-smokers) | 1 | [87] | 20/07/21 |
| 31 | Females with temporomandibular disorders | 1H NMR | 26/27 | 1 | [88] | 20/07/21 |
| 32 | Parkinson’s disease | 1H NMR | 76/37 | 12 | [89] | 20/07/21 |
| 33 | Salivary metabolomics | UHPLC-HR-MSLA-REI-MS | na/140 | 3 | [90] | 20/07/21 |
| 34 | Effect of experimental gingivitis induction and erythritol | UPLC-MS | na/61 | 2 | [91] | 20/07/21 |
| 35 | Sjögren’s syndrome. | 1H NMR | 14/15 | 1 | [92] | 20/07/21 |
| 36 | Evaluation of sport performance | 1H NMR | na/14 | 3 | [93] | 20/07/21 |
| 37 | Head and neck cancer | 1H NMR | 45/30 | 1.5 | [94] | 20/07/21 |
| 38 | Oral cancer | CE-TOF-MS | 26/44 | 1.5 | [95] | 20/07/21 |
| 39 | Pediatric obesity- related liver disease and metabolic syndrome | GC-MS | 23/18 | 1 | [96] | 20/07/21 |
| Normalization, Transformation, and Scaling Methods Applied | FDR Correction | Significantly Upregulated ↑ | Not Significant | Significantly Downregulated ↓ |
|---|---|---|---|---|
| None | No | 68 | 141 | 0 |
| Yes | 6 | 203 | 0 | |
| Glog-/Pareto- only | No | 64 | 132 | 13 |
| Yes | 23 | 179 | 7 | |
| PQN plus Glog-/Pareto- | No | 78 | 114 | 17 |
| Yes | 55 | 141 | 13 | |
| CSN plus Glog-/Pareto- | No | 55 | 108 | 46 |
| Yes | 32 | 140 | 37 |
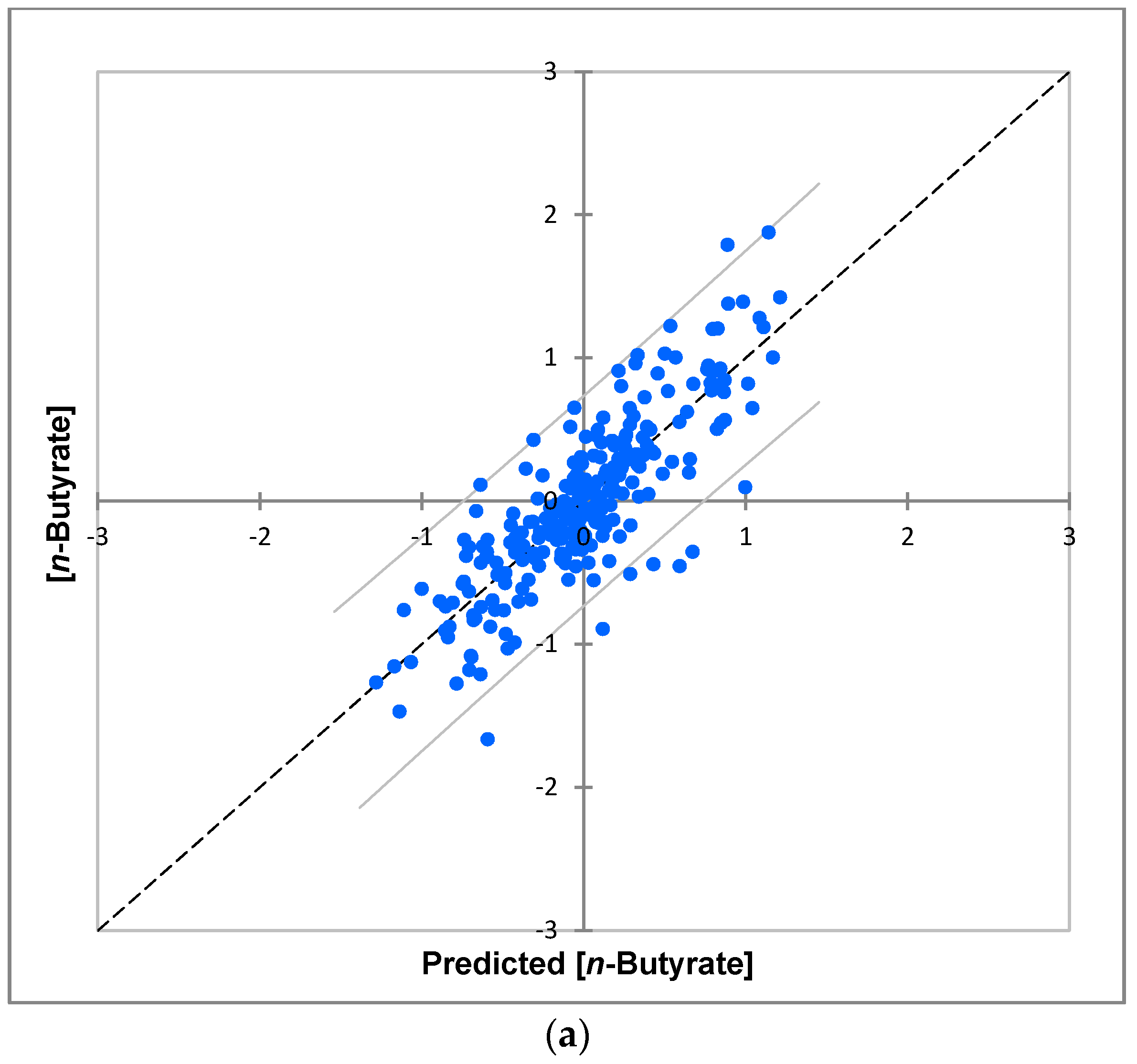
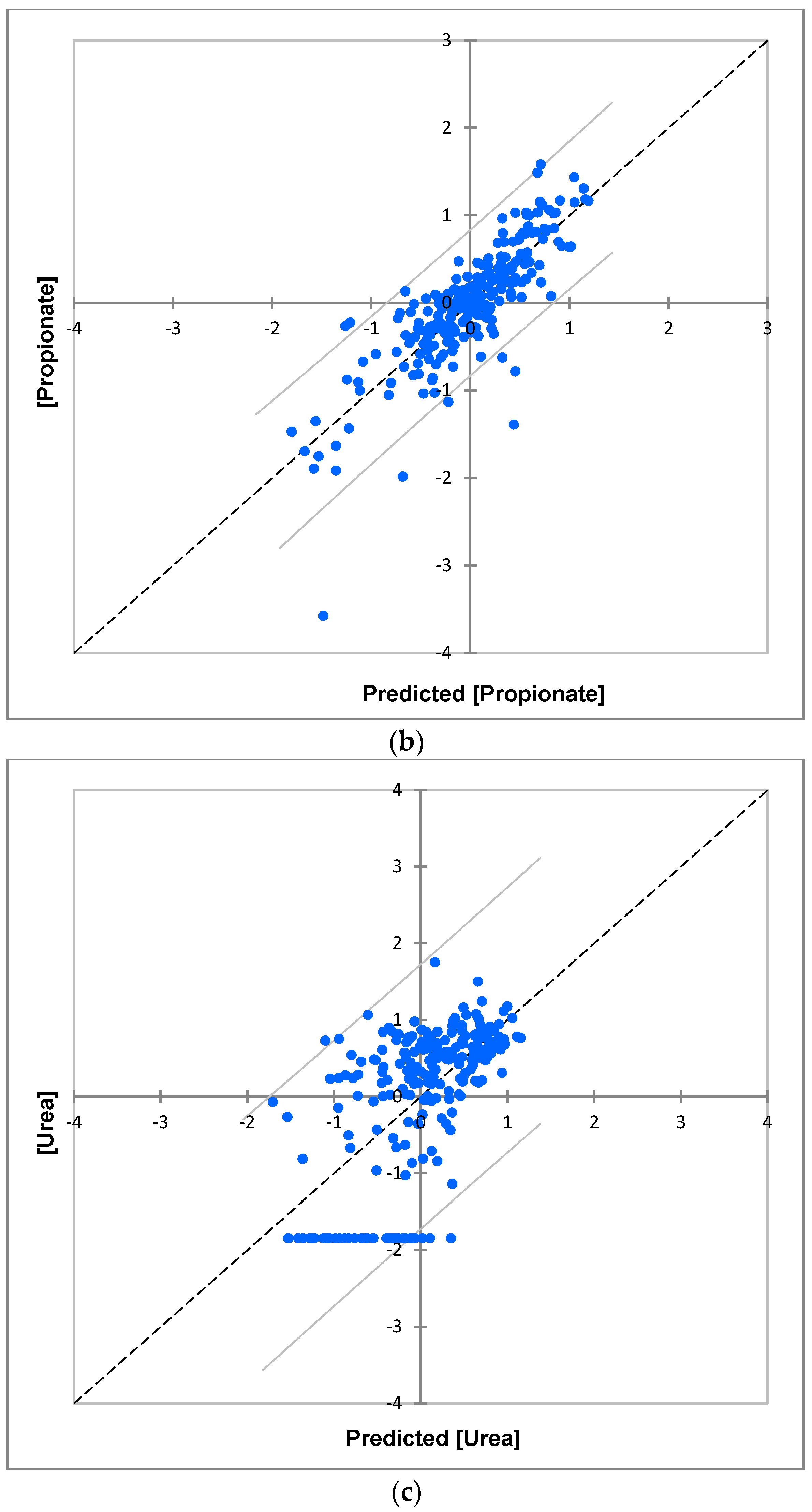
| Metabolite Variable | R2 | Q2 | σi2/σ(i)j2 * | Significance σi2 | Significance σ(i)j2 |
|---|---|---|---|---|---|
| n-Caproate | 0.72 | 0.55 | 184 | <10−6 | ns |
| n-Butyrate | 0.70 | 0.52 | 61 | <10−6 | 0.06 |
| Propionate | 0.69 | 0.50 | 321 | <10−6 | <0.01 |
| Valine | 0.38 | −0.01 | n/a (σ(i)j2 ≈ 0) | <10−4 | ns |
| iso-Butyrate | 0.74 | 0.58 | n/a (σ(i)j2 ≈ 0) | <10−6 | ns |
| 3-D-Hydroxybutyrate | 0.66 | 0.44 | 885 | <10−6 | ns |
| Lactate | 0.68 | 0.47 | 126 | <10−6 | ns |
| Acetoin | 0.69 | 0.50 | 63 | <10−6 | ns |
| Alanine | 0.70 | 0.50 | 75 | <10−6 | ns |
| Thymine | 0.65 | 0.43 | 85 | <10−6 | ns |
| Acetate | 0.63 | 0.39 | 24 | <10−6 | <0.01 |
| N-Ac-1 | 0.71 | 0.55 | 33 | <10−6 | <0.01 |
| N-Ac-2 | 0.68 | 0.48 | 50 | <10−6 | <0.05 |
| N-Ac-3 | 0.66 | 0.44 | 27 | <10−6 | <0.01 |
| 5-Aminovalerate | 0.63 | 0.40 | n/a (σ(i)j2 ≈ 0) | <10−6 | ns |
| Glutamate | 0.65 | 0.43 | 49 | <10−6 | ns |
| Pyruvate | 0.68 | 0.48 | 21 | <10−6 | <10−3 |
| Glutamine | 0.62 | 0.38 | 14 | <10−6 | <10−3 |
| Succinate | 0.58 | 0.32 | 27 | <10−6 | 0.04 |
| Methylamine | 0.39 | 0.38 | 259 | <10−4 | ns |
| Dimethylamine | 0.39 | 0.42 | 76 | <10−6 | ns |
| Trimethylamine | 0.60 | 0.35 | n/a (σ(i)j2 ≈ 0) | <10−6 | ns |
| Choline | 0.64 | 0.41 | 118 | <10−6 | ns |
| Taurine | 0.67 | 0.47 | 144 | <10−6 | ns |
| Methanol | 0.67 | 0.47 | n/a (σ(i)j2 ≈ 0) | <10−6 | ns |
| Glycine | 0.60 | 0.35 | 64 | <10−6 | ns |
| Urea | 0.39 | 0 | 19 | <10−4 | ns |
| Tyrosine | 0.56 | 0.28 | 66 | <10−4 | ns |
| Histidine | 0.53 | 0.24 | n/a (σ(i)j2 ≈ 0) | <10−6 | ns |
| Phenylalanine | 0.64 | 0.29 | 20 | <10−6 | <0.01 |
| Formate | 0.53 | 0.24 | n/a (σ(i)j2 ≈ 0) | <10−4 | ns |
| PC1 | PC2 | PC3 | PC4 | Mean Salivary Level (µmol./L) * | Mean Blood Level (µmol./L) * | |
|---|---|---|---|---|---|---|
| n-Cap | 0.19 | 0.85 | 0.23 | 0.12 | 7–8 | 17 ** |
| n-But | 0.28 | 0.91 | 0.14 | 0.00 | 12–1470 | 1.0 ** |
| Prop | 0.21 | 0.84 | 0.02 | 0.12 | 38–6420 | 0.9 ** |
| Val | 0.11 | 0.17 | 0.60 | 0.31 | 4–48 | 130–266 |
| iso-But | 0.26 | 0.38 | 0.08 | 0.56 | 117–885 | 2.3 ** |
| 3-D-HB | 0.21 | 0.67 | 0.29 | 0.48 | 3–16 | 36–323 |
| Lac | 0.42 | 0.34 | 0.08 | 0.59 | 96–20,310 | 740–2980 |
| Ace | 0.48 | 0.67 | 0.30 | 0.42 | na | 1 (WB) |
| Ala | 0.30 | 0.86 | 0.32 | 0.24 | 14–145 | 200–585 |
| Thy | 0.46 | 0.54 | 0.24 | 0.52 | 2–9 | na |
| A | 0.31 | 0.77 | 0.01 | 0.25 | 112–14,640 | 27–69 |
| N-Ac-1 | 0.84 | 0.18 | 0.12 | 0.47 | n/a | n/a |
| N-Ac-2 | 0.79 | 0.22 | 0.09 | 0.54 | n/a | n/a |
| N-Ac-3 | 0.76 | 0.39 | 0.28 | 0.42 | n/a | n/a |
| 5-AV | 0.32 | 0.68 | 0.19 | 0.31 | 74–470 | na |
| Glu | 0.78 | 0.26 | 0.34 | 0.37 | 12–14 | 24–177 |
| Pyr | 0.76 | 0.47 | 0.23 | 0.30 | 9–5350 | 34–67 |
| Gln | 0.82 | 0.35 | 0.17 | 0.35 | 5–42 | 491–658 |
| Suc | 0.69 | 0.40 | 0.31 | 0.25 | 9–2260 | 6–16 |
| MA | 0.30 | 0.10 | 0.82 | 0.27 | 101 ** | 19 * |
| DMA | 0.08 | 0.39 | 0.81 | 0.20 | 11–65 | 1–48 |
| TMA | −0.03 | 0.75 | 0.57 | 0.15 | 21–98 | 0.4–27 |
| Chol | 0.39 | 0.42 | 0.43 | 0.56 | 9–33 | 6–28 |
| Tau | 0.39 | 0.21 | 0.21 | 0.80 | 25–106 | 42–162 |
| MeOH | 0.27 | 0.02 | 0.25 | 0.86 | 5–133 | 47–468 |
| Gly | 0.47 | 0.30 | 0.36 | 0.55 | 17–188 | 213–330 |
| Urea | 0.36 | 0.23 | −0.14 | −0.20 | 277–617 | 4010–6500 |
| Tyr | 0.51 | 0.58 | 0.08 | 0.22 | 5–36 | 55–143 |
| Phe | 0.30 | 0.88 | 0.09 | −0.05 | 6–44 | 52–166 |
| His | 0.12 | 0.08 | 0.38 | 0.64 | 6–25 | 82–143 |
| Form | 0.13 | −0.01 | 0.38 | 0.46 | 35–600 | 12–225 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grootveld, M.; Page, G.; Bhogadia, M.; Edgar, M. Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part I: Emerging Platforms and Perspectives. Appl. Sci. 2022, 12, 1235. https://doi.org/10.3390/app12031235
Grootveld M, Page G, Bhogadia M, Edgar M. Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part I: Emerging Platforms and Perspectives. Applied Sciences. 2022; 12(3):1235. https://doi.org/10.3390/app12031235
Chicago/Turabian StyleGrootveld, Martin, Georgina Page, Mohammed Bhogadia, and Mark Edgar. 2022. "Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part I: Emerging Platforms and Perspectives" Applied Sciences 12, no. 3: 1235. https://doi.org/10.3390/app12031235
APA StyleGrootveld, M., Page, G., Bhogadia, M., & Edgar, M. (2022). Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part I: Emerging Platforms and Perspectives. Applied Sciences, 12(3), 1235. https://doi.org/10.3390/app12031235