
CRISPR-Based Multi-Gene Integration Strategies to Create Saccharomyces cerevisiae Strains for Consolidated Bioprocessing



Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids, Microbial Strains, and Primers Used
2.2. Microbial Cultivation
2.3. Plasmid Preparation and PCR Amplification of Repair Templates
2.4. Yeast Transformation
2.5. PCR Confirmation of Gene Integration and Positioning
2.6. Activity Screening and Quantitative Enzyme Assays
2.7. qPCR Gene Copy Number Analysis
3. Results
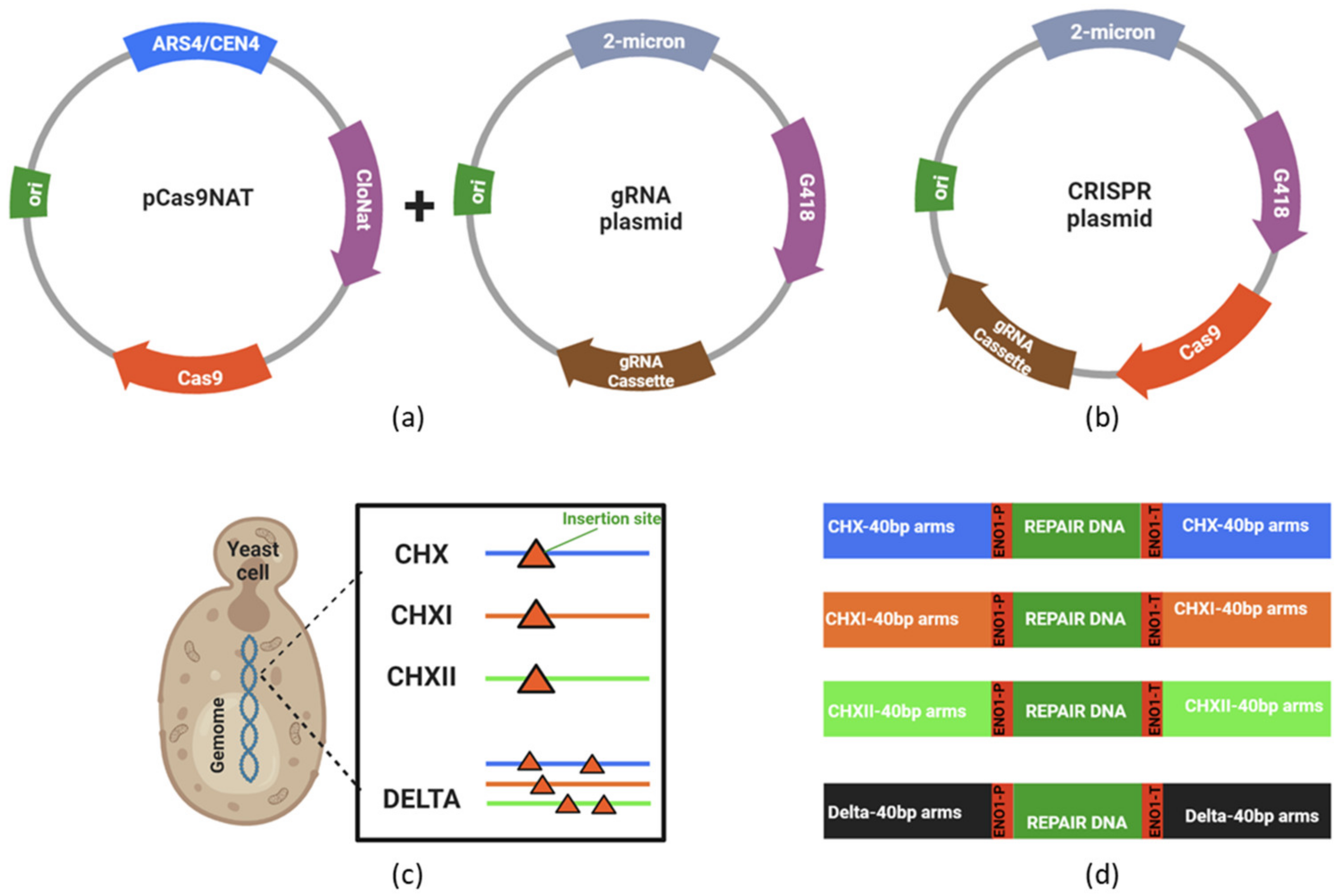
3.1. Construction and Evaluation of Recombinants Strains Created via CRISPR-Cas9
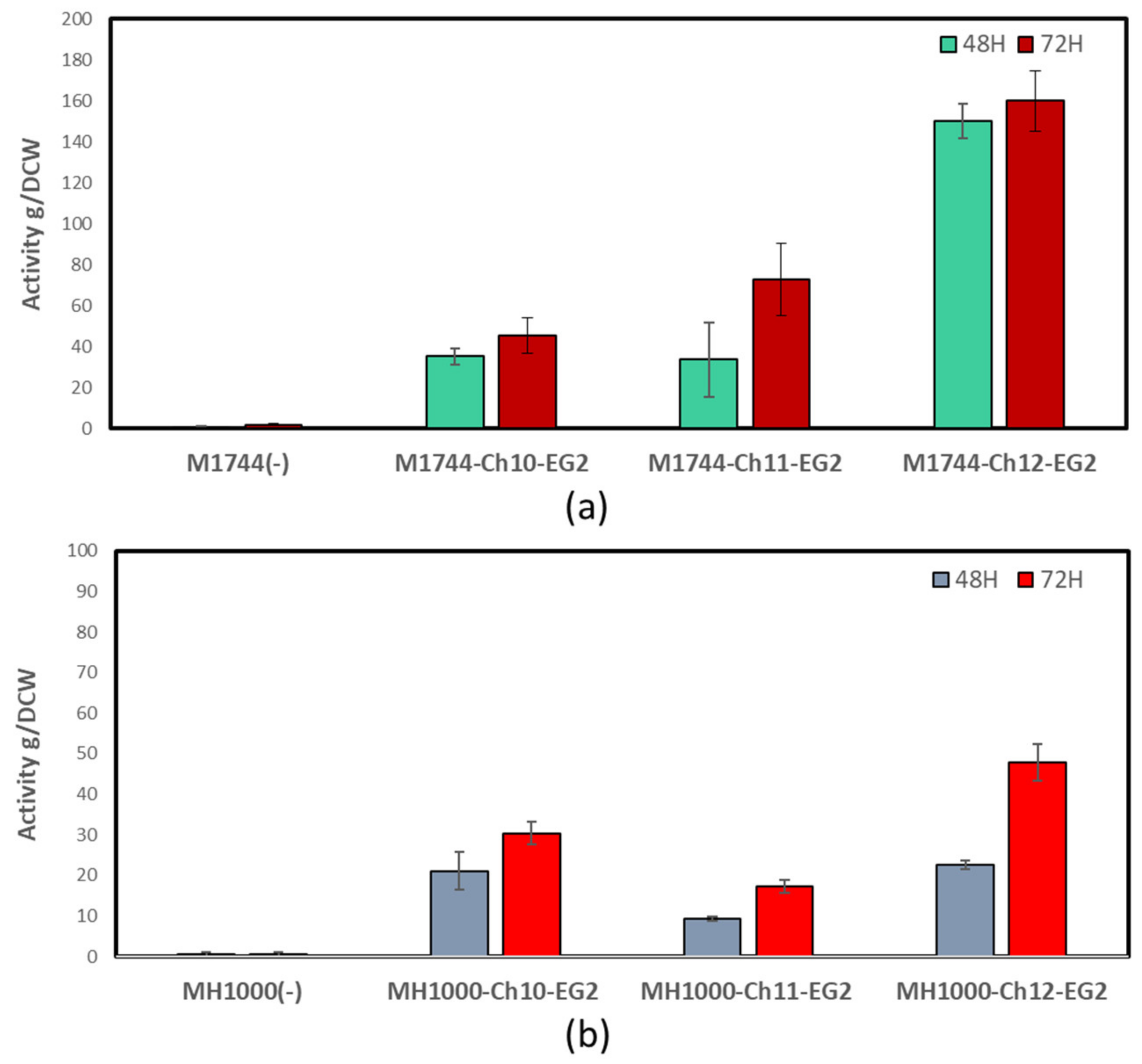
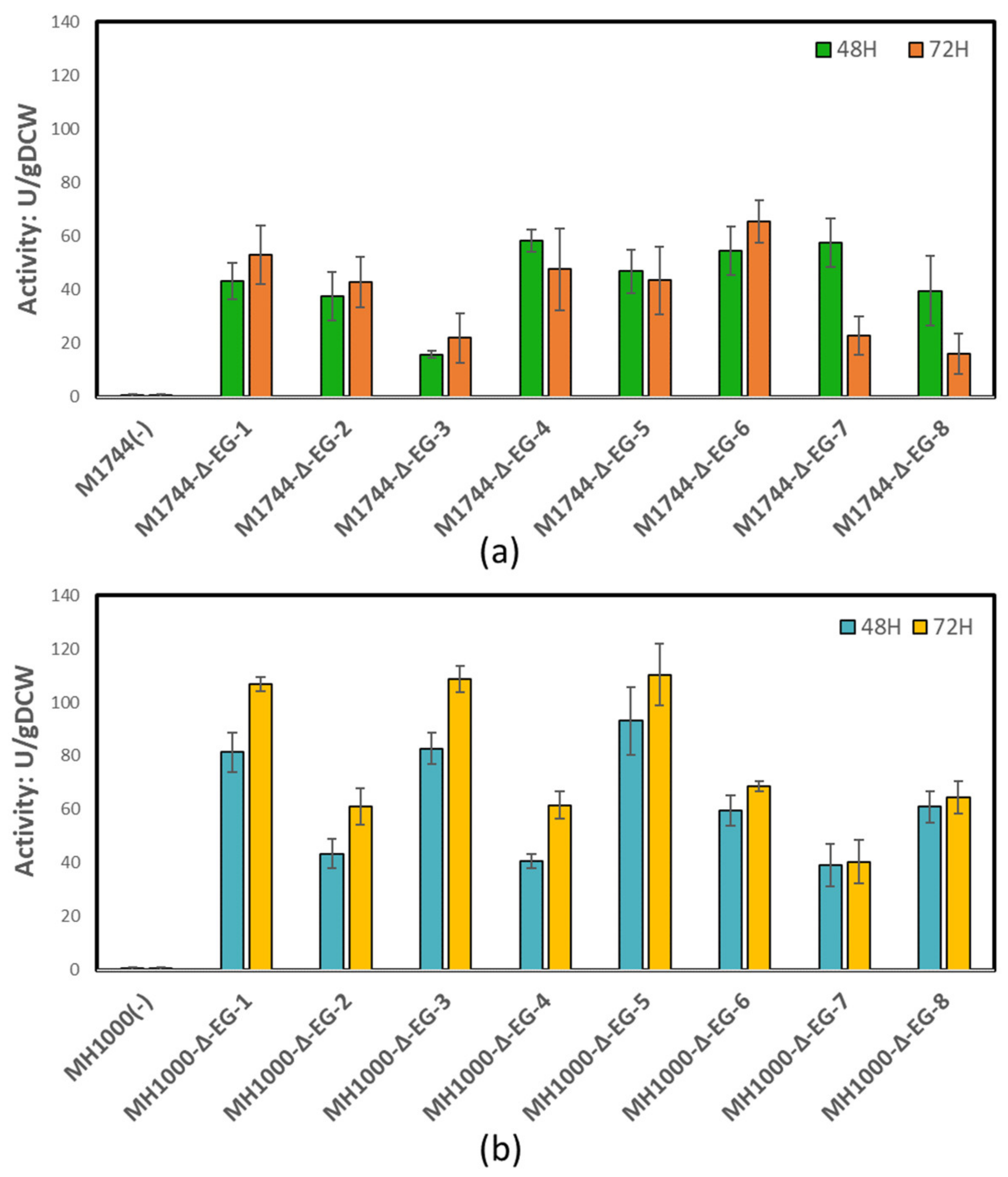
3.2. Endoglucanase Integration and Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T.r.eg2 Transformed Stains | T.r.eg2 Copy Number |
|---|---|
| M1744-Ch10-EG2 (Haploid) | 1 |
| M1744-Ch11-EG2 | 1 |
| M1744-Ch12-EG2 | 3 |
| M1744-Δ-EG2-1 | 1 |
| M1744-Δ-EG2-2 | 1 |
| MH1000-Ch10-EG2 (Diploid) | 2 |
| MH1000-Ch11-EG2 | 2 |
| MH1000-Ch12-EG2 | N.D. |
| MH1000-Δ-EG2-1 | 3 |
| MH1000-Δ-EG2-1 | 1 |
| T.e.cbh1 transformed stains | T.e.cbh1 copy number |
| M1744-Ch10-CBH1 | 1 |
| M1744-Ch11-CBH1 | 1 |
| M1744-Ch12-CBH1 | 4 |
| M1744-Δ-CBH1-1 | 2 |
| M1744-Δ-CBH1-2 | 1 |
| M1744-Δ-CBH1-3 | 2 |
| M1744-Δ-CBH1-4 | 2 |
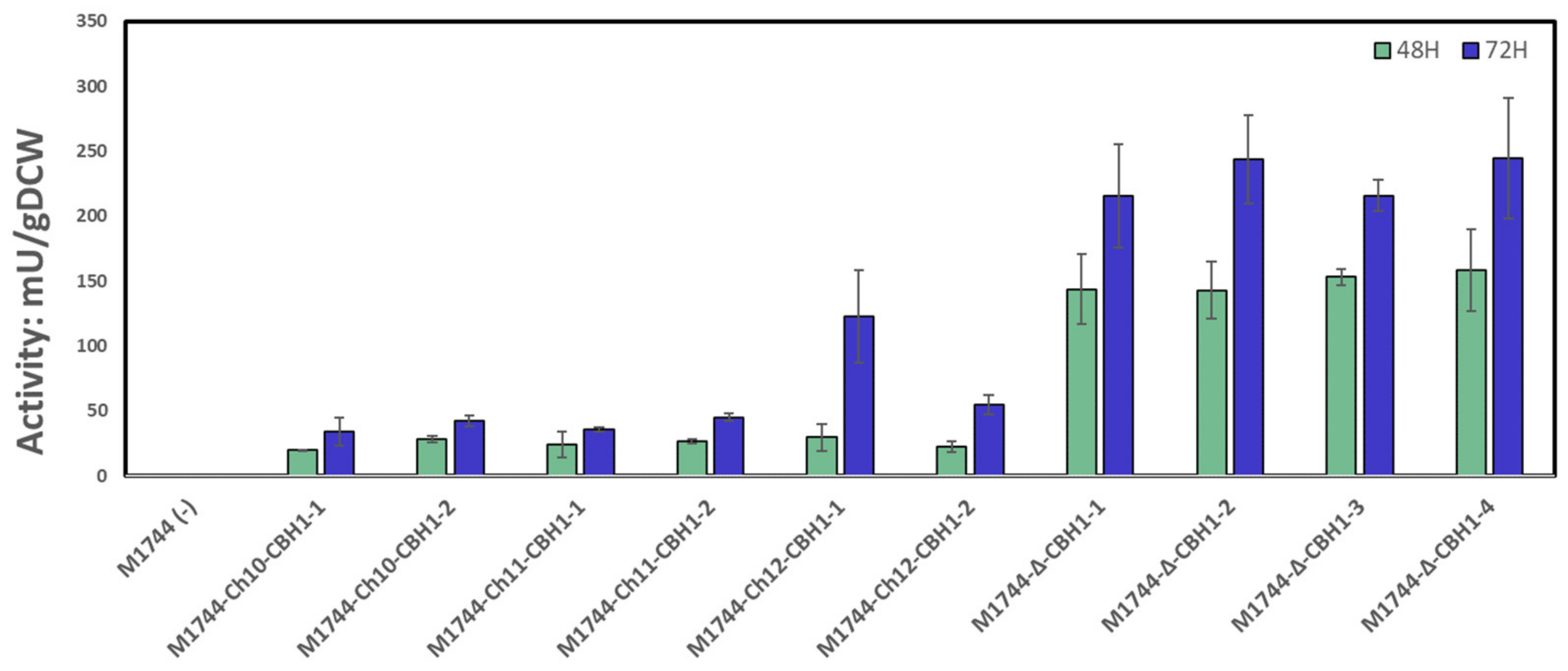
3.3. Cellobiohydrolase Integration and Activity
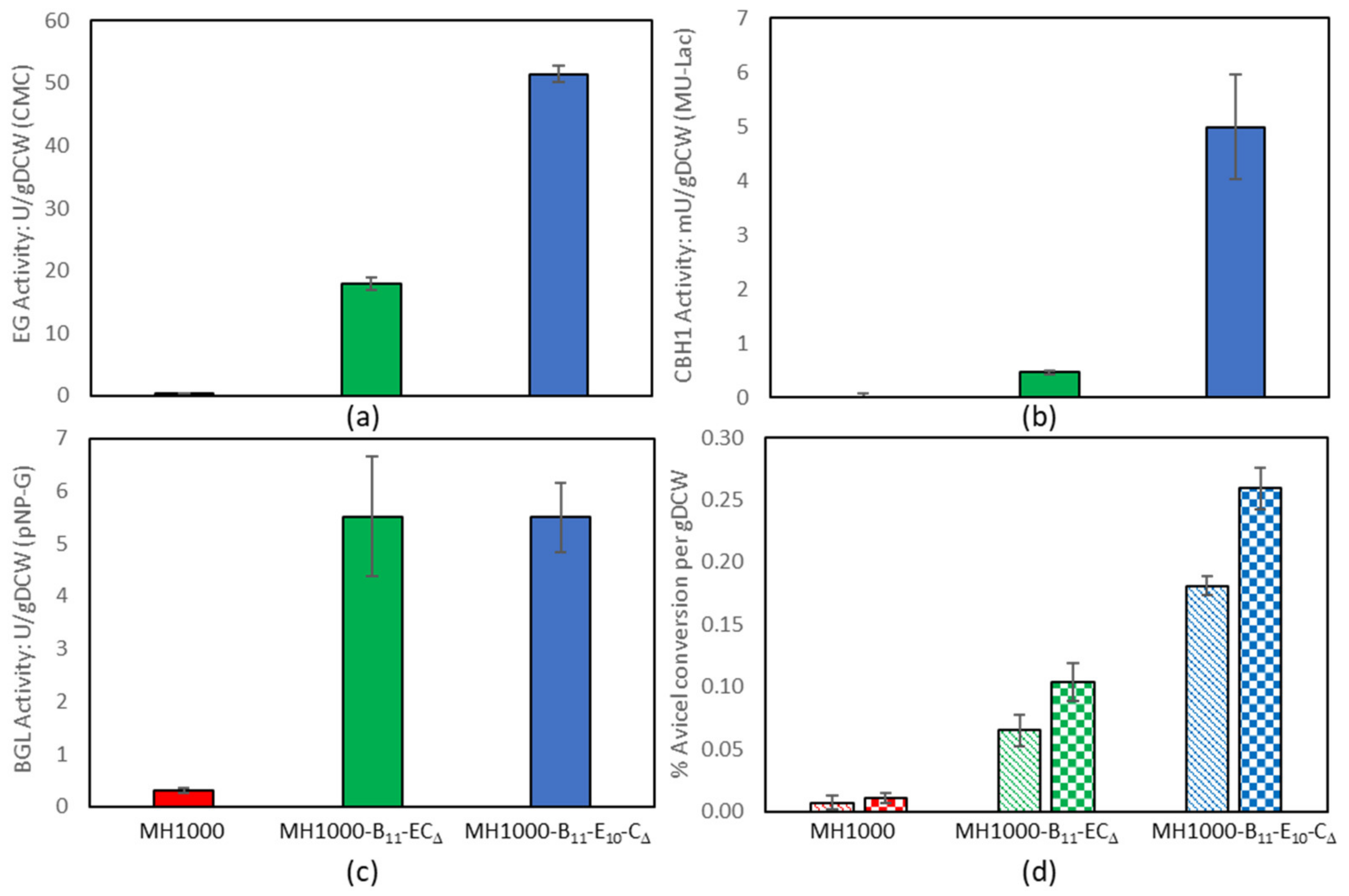
3.4. Constructing Yeast Strains for Cellulose CBP
4. Discussion
4.1. Endoglucanase Integration and Activity
4.2. Cellobiohydrolase Integration and Activity
4.3. Constructing Yeast Strains for Cellulose CBP
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding yeast for budding geneticists: A primer on the Saccharomyces cerevisiae model system. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Borodina, I.; Nielsen, J. Advances in metabolic engineering of yeast Saccharomyces cerevisiae for production of chemicals. Biotechnol. J. 2014, 9, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Demeke, M.M.; Dumortier, F.; Li, Y.; Broeckx, T.; Foulquie-Moreno, M.R.; Thevelein, J.M. Combining inhibitor tolerance and D-xylose fermentation in industrial Saccharomyces cerevisiae for efficient lignocellulose-based bioethanol production. Biotechnol. Biofuels 2013, 6, 120. [Google Scholar] [CrossRef] [PubMed]
- Lugani, Y.; Rai, R.; Prabhu, A.A.; Maan, P.; Meenu, H.; Kumar, V.; Kumar, S.; Chandel, A.K.; Sengar, R.S. Recent advances in bioethanol production from lignocelluloses: A comprehensive review with a focus on enzyme engineering and designer biocatalysts. Biofuel Res. J. 2020, 7, 1267–1295. [Google Scholar] [CrossRef]
- Hasunuma, T.; Kondo, A. Development of yeast cell factories for consolidated bioprocessing of lignocellulose to bioethanol through cell surface engineering. Biotechnol. Adv. 2012, 30, 1207–1218. [Google Scholar] [CrossRef]
- Oh, E.J.; Jin, Y.S. Engineering of Saccharomyces cerevisiae for efficient fermentation of cellulose. FEMS Yeast Res. 2020, 20, foz089. [Google Scholar] [CrossRef]
- Lynd, L.R.; Van Zyl, W.H.; McBride, J.E.; Laser, M. Consolidated bioprocessing of cellulosic biomass: An update. Curr. Opin. Biotechnol. 2005, 16, 577–583. [Google Scholar] [CrossRef]
- Van Zyl, W.H.; Lynd, L.R.; Den Haan, R.; McBride, J.E. Consolidated Bioprocessing for Bioethanol Production Using Saccharomyces cerevisiae. Adv. Biochem. Eng. Biotechnol. 2007, 108, 205–235. [Google Scholar]
- Yamada, R.; Hasunuma, T.; Kondo, A. Endowing non-cellulolytic microorganisms with cellulolytic activity aiming for consolidated bioprocessing. Biotechnol. Adv. 2013, 31, 754–763. [Google Scholar] [CrossRef]
- Den Haan, R.; van Rensburg, E.; Rose, S.H.; Gorgens, J.F.; Van Zyl, W.H. Progress and challenges in the engineering of non-cellulolytic microorganisms for consolidated bioprocessing. Curr. Opin. Biotechnol. 2015, 33, 32–38. [Google Scholar] [CrossRef]
- Lane, S.; Dong, J.; Jin, Y.S. Value-added biotransformation of cellulosic sugars by engineered Saccharomyces cerevisiae. Bioresour. Technol. 2018, 260, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Den Haan, R.; Rose, S.H.; Cripwell, R.A.; Trollope, K.M.; Myburgh, M.W.; Viljoen-Bloom, M.; van Zyl, W.H. Heterologous production of cellulose- and starch-degrading hydrolases to expand Saccharomyces cerevisiae substrate utilization: Lessons learnt. Biotechnol. Adv. 2021, 53, 107859. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Borodina, I. Application of synthetic biology for production of chemicals in yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2015, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Stovicek, V.; Borodina, I.; Forster, J. CRISPR−Cas system enables fast and simple genome editing of industrial Saccharomyces cerevisiae strains. Metab. Eng. Commun. 2015, 2, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ronda, C.; Maury, J.; Jakočiunas, T.; Jacobsen, S.A.; Germann, S.M.; Harrison, S.J.; Borodina, I.; Keasling, J.D.; Jensen, M.K.; Nielsen, A.T. CrEdit: CRISPR mediated multi-loci gene integration in Saccharomyces cerevisiae. Microb. Cell Fact. 2015, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Brevnova, E.; McBride, J.; Wiswall, E.; Wenger, K.; Caiazza, N.; Hau, H.; Argyros, A.; Agbogbo, F.; Rice, C.; Barret, T.; et al. Yeast Expressing Saccharolytic Enzymes for Consolidated Bioprocessing Using Starch and Cellulose. WO/2011/153516, 6 March 2011. [Google Scholar]
- Ilmén, M.; Den Haan, R.; Brevnova, E.; McBride, J.; Wiswall, E.; Froehlich, A.; Koivula, A.; Voutilainen, S.P.; Siika-aho, M.; Lagrange, D.C.; et al. High level secretion of cellobiohydrolases by Saccharomyces cerevisiae. Biotechnol. Biofuels 2011, 4, 30. [Google Scholar] [CrossRef]
- Inokuma, K.; Kitada, Y.; Bamba, T.; Kobayashi, Y.; Yukawa, T.; den Haan, R.; van Zyl, W.H.; Kondo, A.; Hasunuma, T. Improving the functionality of surface-engineered yeast cells by altering the cell wall morphology of the host strain. Appl. Microbiol. Biotechnol. 2021, 105, 5895–5904. [Google Scholar] [CrossRef]
- Kruger, F.; den Haan, R. Surface tethered xylosidase activity improved xylan conversion in engineered strains of Saccharomyces cerevisiae. J. Chem. Technol. Biotechnol. 2022, 97, 1099–1111. [Google Scholar] [CrossRef]
- Davison, S.A.; Den Haan, R.; Van Zyl, W.H. Heterologous expression of cellulase genes in natural Saccharomyces cerevisiae strains. Appl. Microbiol. Biotechnol. 2016, 100, 8241–8254. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.M.; Yoo, Y.J.; Kang, H.S. δ-Integration of endo/exo-glucanase and β-glucosidase genes into the yeast chromosomes for direct conversion of cellulose to ethanol. Enzym. Microb. Technol. 1999, 25, 23–30. [Google Scholar] [CrossRef]
- Moriguchi, K.; Yamamoto, S.; Ohmine, Y.; Suzuki, K. A fast and practical yeast transformation method mediated by Escherichia coli based on a trans-kingdom conjugal transfer system: Just mix two cultures and wait one hour. PLoS ONE 2016, 11, e0148989. [Google Scholar] [CrossRef]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, C.S.; Winston, F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 1987, 57, 267–272. [Google Scholar] [CrossRef]
- Chetty, B.J.; Inokuma, K.; Hasunuma, T.; van Zyl, W.H.; den Haan, R. Improvement of cell-tethered cellulase activity in recombinant strains of Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2022, 106, 6347–6361. [Google Scholar] [CrossRef]
- La Grange, D.C.; Pretorius, I.S.; Claeyssens, M.; Van Zyl, W.H. Degradation of xylan to D-xylose by recombinant Saccharomyces cerevisiae coexpressing the Aspergillus niger beta-xylosidase (xlnD) and the Trichoderma reesei xylanase II (xyn2) genes. Appl. Environ. Microbiol. 2001, 67, 5512–5519. [Google Scholar] [CrossRef]
- Den Haan, R.; van Zyl, J.M.; Harms, T.M.; Van Zyl, W.H. Modeling the minimum enzymatic requirements for optimal cellulose conversion. Environ. Res. Lett 2013, 8, 025013. [Google Scholar] [CrossRef]
- Teste, M.A.; Duquenne, M.; Francois, J.M.; Parrou, J.L. Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol. Biol. 2009, 10, 99. [Google Scholar] [CrossRef]
- Zhang, G.C.; Kong, I.I.; Kim, H.; Liu, J.J.; Cate, J.H.; Jin, Y.S. Construction of a quadruple auxotrophic mutant of an industrial polyploid Saccharomyces cerevisiae strain by using RNA-guided Cas9 nuclease. Appl. Env. Microbiol. 2014, 80, 7694–7701. [Google Scholar] [CrossRef]
- Van Wyk, N.; Kroukamp, H.; Espinosa, M.I.; von Wallbrunn, C.; Wendland, J.; Pretorius, I.S. Blending wine yeast phenotypes with the aid of CRISPR DNA editing technologies. Int. J. Food Microbiol. 2020, 324, 108615. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.D.; Buron, L.D.; Salomonsen, B.; Olsen, C.E.; Hansen, B.G.; Mortensen, U.H.; Halkier, B.A. Microbial production of indolylglucosinolate through engineering of a multi-gene pathway in a versatile yeast expression platform. Metab. Eng. 2012, 14, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Flagfeldt, D.B.; Siewers, V.; Huang, L.; Nielsen, J. Characterization of chromosomal integration sites for heterologous gene expression in Saccharomyces cerevisiae. Yeast 2009, 26, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, R.; Yamada, R.; Ogino, H. CRISPR system in the yeast Saccharomyces cerevisiae and its application in the bioproduction of useful chemicals. World J. Microbiol. Biotechnol. 2019, 35, 111. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Taniguchi, N.; Tanaka, T.; Ogino, C.; Fukuda, H.; Kondo, A. Cocktail δ-integration: A novel method to construct cellulolytic enzyme expression ratio-optimized yeast strains. Microb. Cell Fact. 2010, 9, 32. [Google Scholar] [CrossRef]
- Sasaki, Y.; Mitsui, R.; Yamada, R.; Ogino, H. Secretory overexpression of the endoglucanase by Saccharomyces cerevisiae via CRISPR-δ-integration and multiple promoter shuffling. Enzym. Microb. Technol. 2019, 121, 17–22. [Google Scholar] [CrossRef]
- Shi, S.; Liang, Y.; Zhang, M.M.; Ang, E.L.; Zhao, H. A highly efficient single-step, markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae. Metab. Eng. 2016, 33, 19–27. [Google Scholar] [CrossRef]
- Fleiss, A.; O’Donnell, S.; Fournier, T.; Lu, W.; Agier, N.; Delmas, S.; Schacherer, J.; Fischer, G. Reshuffling yeast chromosomes with CRISPR/Cas9. PLoS Genet. 2019, 15, e1008332. [Google Scholar] [CrossRef]
- Jensen, N.B.; Strucko, T.; Kildegaard, K.R.; David, F.; Maury, J.; Mortensen, U.H.; Forster, J.; Nielsen, J.; Borodina, I. EasyClone: Method for iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae. FEMS Yeast Res. 2014, 14, 238–248. [Google Scholar] [CrossRef]
- Wu, X.-L.; Li, B.-Z.; Zhang, W.-Z.; Song, K.; Qi, H.; Dai, J.-B.; Yuan, Y.-J. Genome-wide landscape of position effects on heterogeneous gene expression in Saccharomyces cerevisiae. Biotechnol. Biofuels 2017, 10, 189. [Google Scholar] [CrossRef]
- Xu, Q.; Singh, A.; Himmel, M.E. Perspectives and new directions for the production of bioethanol using consolidated bioprocessing of lignocellulose. Curr. Opin. Biotechnol. 2009, 20, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Van Dyk, J.S.; Pletschke, B.I. A review of lignocellulose bioconversion using enzymatic hydrolysis and synergistic cooperation between enzymes--factors affecting enzymes, conversion and synergy. Biotechnol. Adv. 2012, 30, 1458–1480. [Google Scholar] [CrossRef] [PubMed]
- Adiego-Pérez, B.; Randazzo, P.; Daran, J.M.; Verwaal, R.; Roubos, J.A.; Daran-Lapujade, P.; van der Oost, J. Multiplex genome editing of microorganisms using CRISPR-Cas. FEMS Microbiol. Lett. 2019, 366, fnz086. [Google Scholar] [CrossRef]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Labuhn, M.; Adams, F.F.; Ng, M.; Knoess, S.; Schambach, A.; Charpentier, E.M.; Schwarzer, A.; Mateo, J.L.; Klusmann, J.H.; Heckl, D. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 2018, 46, 1375–1385. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef]
- Liao, C.; Ttofali, F.; Slotkowski, R.A.; Denny, S.R.; Cecil, T.D.; Leenay, R.T.; Keung, A.J.; Beisel, C.L. Modular one-pot assembly of CRISPR arrays enables library generation and reveals factors influencing crRNA biogenesis. Nat. Commun. 2019, 10, 2948. [Google Scholar] [CrossRef]
- Bourgeois, L.; Pyne, M.E.; Martin, V.J.J. A highly characterized synthetic landing pad system for precise multicopy gene integration in yeast. ACS Synth. Biol. 2018, 7, 2675–2685. [Google Scholar] [CrossRef]
- Baek, S.; Utomo, J.C.; Lee, J.Y.; Dalal, K.; Yoon, Y.J.; Ro, D.K. The yeast platform engineered for synthetic gRNA-landing pads enables multiple gene integrations by a single gRNA/Cas9 system. Metab. Eng. 2021, 64, 111–121. [Google Scholar] [CrossRef]
- Mans, R.; van Rossum, H.M.; Wijsman, M.; Backx, A.; Kuijpers, N.G.; van den Broek, M.; Daran-Lapujade, P.; Pronk, J.T.; van Maris, A.J.; Daran, J.M. CRISPR/Cas9: A molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 2015, 15, fov004. [Google Scholar] [CrossRef]
- Verkuijl, S.A.; Rots, M.G. The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr. Opin. Biotechnol. 2019, 55, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Reider Apel, A.; d’Espaux, L.; Wehrs, M.; Sachs, D.; Li, R.A.; Tong, G.J.; Garber, M.; Nnadi, O.; Zhuang, W.; Hillson, N.J.; et al. A Cas9-based toolkit to program gene expression in Saccharomyces cerevisiae. Nucleic Acids Res. 2017, 45, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Kroukamp, H.; den Haan, R.; van Zyl, J.-H.; van Zyl, W.H. Rational strain engineering interventions to enhance cellulase secretion by Saccharomyces cerevisiae. Biofuels Bioprod. Biorefining 2018, 12, 108–124. [Google Scholar] [CrossRef]
- Davison, S.A.; den Haan, R.; van Zyl, W.H. Exploiting strain diversity and rational engineering strategies to enhance recombinant cellulase secretion by Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2020, 104, 5163–5184. [Google Scholar] [CrossRef]





| Plasmid | Description | Reference |
|---|---|---|
| pRDH180 | eg2 plasmid, carrying the ENO1 promoter, terminator and T.r.eg2. Used to produce PCR products carrying the eg2 gene cassette. | [18] |
| pMI529 | cbh1 plasmid, carrying the ENO1 promoter, terminator, and T.e.cbh1. Used to produce the PCR product carrying the cbh1 gene cassette. | [19] |
| pIBG-SSAD | bgl1 plasmid, carrying the SED1 promoter, DIT1 terminator, and A.a.bgl1. Used to produce the PCR product carrying the bgl1 gene cassette. | [20] |
| pCas9NAT | CEN6/ARS4 plasmid, TEF1 promoter, CYC1 terminator, SV40 Nuclear Localization Sequence, human codon optimized S.p.Cas9; CloNAT resistance. Low copy plasmid carrying the cas9 encoding gene for the 2-plasmid CRISPR system. | Addgene |
| pRS42-G_ChX | Guide RNA expression plasmid for the 2-plasmid system targeting Chromosome X (Ch10) intergenic region; G418 resistance; contains the SNR52 promoter and SUP4 terminator for gRNA expression | [21] |
| pRS42-G-DELTA | Similar to pRS42G_ChX, but targeting the yeast DELTA sequences | This study |
| pRSCG_ChXI | pRS423-cas9-gRNA-G418 targeting Chromosome XI (Ch11) intergenic region protospacer; G418 resistance; this plasmid also contains the S.p.Cas9 under TEF1 promoter and CYC1 terminator – 1-plasmid system. | [21] |
| pRSCG_ChXII | Similar to pRSCG_ChXI but targets a Chromosome XII (Ch12) intergenic region protospacer. | This study |
| Strain | Abbreviation | Description | Reference |
|---|---|---|---|
| S. cerevisiae MH1000 | MH1000 | Industrial yeast strain, diploid, no auxotrophy | [22] |
| S. cerevisiae M1744 | M1744 | Haploid yeast strain with uracil auxotrophy (Δura3) | [18] |
| S. cerevisiae M1744 + pCas9 + pRS42-G_ChX + T.r.eg2 | M1744-Ch10-EG2 | S. cerevisiae M1744 with the T.r.eg2 integrated at the chromosome X intergenic site using pCas9NAT and pRS42H_ChX | This study |
| S. cerevisiae M1744 + pRSCG_ChXI + T.r.eg2 | M1744-Ch11-EG2 | S. cerevisiae M1744 with the T.r.eg2 integrated at the chromosome XI intergenic site using pRSCG_ChXI | This study |
| S. cerevisiae M1744 + pRSCG_ChXII + T.r.eg2 | M1744-Ch12-EG2 | S. cerevisiae M1744 with the T.r.eg2 integrated at the chromosome XII intergenic site using pRSCG_ChXII | This study |
| S. cerevisiae MH1000 + pCas9 + pRS42-G_ChX + T.r.eg2 | MH1000-Ch10-EG2 | S. cerevisiae MH1000 with the T.r.eg2 integrated at the chromosome X intergenic site using pCas9NAT and pRS42H_ChX | This study |
| S. cerevisiae MH1000 + pRSCG_ChXI + T.r.eg2 | MH1000-Ch11-EG2 | S. cerevisiae MH1000 with the T.r.eg2 integrated at the chromosome XI intergenic site using pRSCG_ChXI | This study |
| S. cerevisiae MH1000 + pRSCG_ChXII + T.r.eg2 | MH1000-Ch12-EG2 | S. cerevisiae MH1000 with the T.r.eg2 integrated at the chromosome XII intergenic site using pRSCG_ChXII | This study |
| S. cerevisiae M1744 + pCas9 + pRS42-G_ ChX + T.e.cbh1 | M1744-Ch10-CBH1 | S. cerevisiae M1744 with the T.e.cbh1 integrated at the chromosome X intergenic site using pCas9NAT and pRS42H_ChX | This study |
| S. cerevisiae M1744 + pRSCG_ChXI + T.e.cbh1 | M1744-Ch11-CBH1 | S. cerevisiae M1744 with the T.e.cbh1 integrated at the chromosome XI intergenic site using pRSCG_ChXI | This study |
| S. cerevisiae M1744 + pRSCG_ChXII + T.e.cbh1 | M1744-Ch12-CBH1 | S. cerevisiae M1744 with the T.e.cbh1 integrated at the chromosome XII intergenic site using pRSCG_ChXII | This study |
| S. cerevisiae M1744 + pRS42-G-DELTA + T.r.eg2 | M1744-Δ-EG2 | S. cerevisiae M1744 with the T.r.eg2 integrated at delta sites in the genome using pCas9NAT and pRS42G-DELTA | This study |
| S. cerevisiae M1744 + pRS42-G-DELTA + T.e.cbh1 | M1744-Δ-CBH1 | S. cerevisiae M1744 with the T.e.cbh1 integrated at delta sites in the genome using pCas9NAT and pRS42-G-DELTA | This study |
| S. cerevisiae MH1000 + pRS42-G-DELTA + T.e.cbh1 | MH1000-Δ-EG2 | S. cerevisiae MH1000 with the T.e.cbh1 integrated at delta sites in the genome using pCas9NAT and pRS42-G-DELTA | This study |
| S. cerevisiae MH1000 + pRS42-G-DELTA + T.e.cbh1 | MH1000-Δ-CBH1 | S. cerevisiae MH1000 with the T.e.cbh1 integrated at delta sites in the genome using pCas9NAT and pRS42-G-DELTA | This study |
| S. cerevisiae MH1000 + (A.a.bgl1 + T.r.eg2 + T.e.cbh1) | MH1000-B11-E10-CΔ | S. cerevisiae MH1000 with A.a.bgl1 targeted to Ch11, T.r.eg2 targeted to Ch10 and T.e.cbh1 targeted to the delta sequences | This study |
| S. cerevisiae MH1000 + (A.a.bgl1 + T.r.eg2 + T.e.cbh1) | MH1000-B11-ECΔ | S. cerevisiae MH1000 with A.a.bgl1 targeted to Ch11, and, T.r.eg2 and T.e.cbh1 targeted to the delta sequences. | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacob, O.; van Lill, G.R.; den Haan, R. CRISPR-Based Multi-Gene Integration Strategies to Create Saccharomyces cerevisiae Strains for Consolidated Bioprocessing. Appl. Sci. 2022, 12, 12317. https://doi.org/10.3390/app122312317
Jacob O, van Lill GR, den Haan R. CRISPR-Based Multi-Gene Integration Strategies to Create Saccharomyces cerevisiae Strains for Consolidated Bioprocessing. Applied Sciences. 2022; 12(23):12317. https://doi.org/10.3390/app122312317
Chicago/Turabian StyleJacob, Odwa, Gert Rutger van Lill, and Riaan den Haan. 2022. "CRISPR-Based Multi-Gene Integration Strategies to Create Saccharomyces cerevisiae Strains for Consolidated Bioprocessing" Applied Sciences 12, no. 23: 12317. https://doi.org/10.3390/app122312317
APA StyleJacob, O., van Lill, G. R., & den Haan, R. (2022). CRISPR-Based Multi-Gene Integration Strategies to Create Saccharomyces cerevisiae Strains for Consolidated Bioprocessing. Applied Sciences, 12(23), 12317. https://doi.org/10.3390/app122312317