

New Strategies for Potential Contrast Agents’ Synthons Highly Active to MRI Based on Gd3+, Eu3+, and Tb3+



Featured Application
Abstract
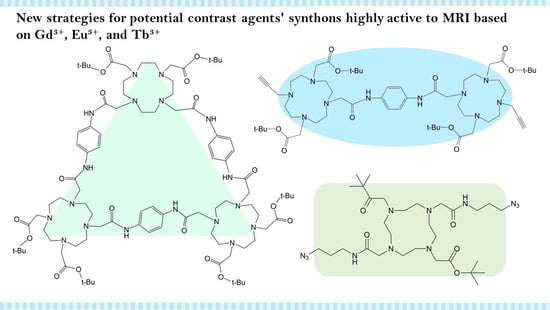
1. Introduction
Designing an Efficient Contrast Agent
2. Materials and Methods
3. Results
4. Discussion
4.1. Click Chemistry Synthetic Pathway
4.2. cisTetraDOTA Synthetic Pathway
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hapuarachchige, S.; Artemov, D. Theranostic Pretargeting Drug Delivery and Imaging Platforms in Cancer Precision Medicine. Front. Oncol. 2020, 10, 1131. [Google Scholar] [CrossRef] [PubMed]
- Yedinak, C. Diagnostic Imaging. In Advanced Practice in Endocrinology Nursing; Llahana, S., Grossman, A., Eds.; Springer International Publishing: Basel, Switzerland, 2019; pp. 305–319. [Google Scholar] [CrossRef]
- McKay, J.A.; Church, A.L.; Rubin, N.; Emory, T.H.; Hoven, N.F.; Kuehn-Hajder, J.E.; Nelson, M.T.; Ramanna, S.; Auerbach, E.J.; Moeller, S.; et al. A Comparison of Methods for High-Spatial-Resolution Diffusion-weighted Imaging in Breast MRI. Radiology 2020, 297, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.S.; Tóth, É. Metal-based environment-sensitive MRI contrast agents. Curr. Opin. Chem. Biol. 2021, 61, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, R.; Yang, Y.; Peng, S.; Xiao, D.; Kong, T.; Cai, X.; Zhu, B. Aptamer-mediated synthesis of multifunctional nano-hydroxyapatite for active tumour bioimaging and treatment. Cell Prolif. 2021, 54, e13105. [Google Scholar] [CrossRef] [PubMed]
- Mnasri, W.; Parvizian, M.; Ammar-Merah, S. Design and Synthesis of Luminescent Lanthanide-Based Bimodal Nanoprobes for Dual Magnetic Resonance (Mr) and Optical Imaging. Nanomaterials 2021, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, L.; Nordberg, G.F.; Nurchi, V.M.; Aaseth, J.O. Gadolinium in Medical Imaging—Usefulness, Toxic Reactions and Possible Countermeasures—A Review. Biomolecules 2022, 12, 742. [Google Scholar] [CrossRef] [PubMed]
- Broch, L.; Wester, S.; Haslund, A.; Edland, A. Anaphylactic Reaction to Mri Contrast Agent. Tidsskr. Den Nor. Legeforening 2020, 140, 8. [Google Scholar] [CrossRef]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of Mri Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2018, 119, 957–1057. [Google Scholar] [CrossRef]
- Merbach, A.S.; Helm, L.; Tóth, E. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging; John Wiley & Sons: Hoboken, NJ, USA, 2013; p. 512. Available online: https://books.google.com.co/books?id=7q0VNrcdD28C&source=gbs_navlinks_s (accessed on 8 June 2022).
- Hsu, J.C.; Naha, P.C.; Lau, K.C.; Chhour, P.; Hastings, R.; Moon, B.F.; Stein, J.M.; Witschey, W.R.T.; McDonald, E.S.; Maidment, A.D.A.; et al. An All-in-One Nanoparticle (Aion) Contrast Agent for Breast Cancer Screening with Dem-Ct-Mri-Nirf Imaging. Nanoscale 2018, 10, 17236–17248. [Google Scholar] [CrossRef]
- Lux, J.; Sherry, A.D. Advances in gadolinium-based Mri contrast agent designs for monitoring biological processes in vivo. Curr. Opin. Chem. Biol. 2018, 45, 121–130. [Google Scholar] [CrossRef]
- Gupta, A.; Caravan, P.; Price, W.S.; Platas-Iglesias, C.; Gale, E.M. Applications for Transition-Metal Chemistry in Contrast-Enhanced Magnetic Resonance Imaging. Inorg. Chem. 2020, 59, 6648–6678. [Google Scholar] [CrossRef] [PubMed]
- Pellico, J.; Ellis, C.M.; Davis, J.J. Nanoparticle-Based Paramagnetic Contrast Agents for Magnetic Resonance Imaging. Contrast Media Mol. Imaging 2019, 2019, 1845637. [Google Scholar] [CrossRef] [PubMed]
- Shuvaev, S.; Akam, E.; Caravan, P. Molecular MR Contrast Agents. Investig. Radiol. 2020, 56, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-K.; Lee, G.H.; Chang, Y. Gadolinium as an Mri contrast agent. Future Med. Chem. 2018, 10, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Hornak, J.P. The Basis of Mri; Muntaabski, Calvar, Eds.; Universidad de San Martín, Rochester Institute of Technology: New York, NY, USA, 2018. Available online: https://www.cis.rit.edu/htbooks/mri/inside.htm (accessed on 23 June 2022).
- De León-Rodríguez, L.M.; Martins, A.F.; Pinho, M.C.; Rofsky, N.M.; Sherry, A.D. Basic MR relaxation mechanisms and contrast agent design. J. Magn. Reson. Imaging 2015, 42, 545–565. [Google Scholar] [CrossRef]
- Zhou, Z.; Yang, L.; Gao, J.; Chen, X. Structure-relaxivity relationships of magnetic nanoparticles for magnetic resonance imaging. Adv. Mater. 2019, 31, e1804567. [Google Scholar] [CrossRef]
- Helm, L. Relaxivity in paramagnetic systems: Theory and mechanisms. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 45–64. [Google Scholar] [CrossRef]
- Rast, S.; Fries, P.H.; Belorizky, E.; Borel, A.; Helm, L.; Merbach, A.E. A general approach to the electronic spin relaxation of Gd(III) complexes in solutions. Monte Carlo simulations beyond the Redfield limit. J. Chem. Phys. 2001, 115, 7554–7563. [Google Scholar] [CrossRef]
- Yang, J.J.; Yang, J.; Wei, L.; Zurkiya, O.; Yang, W.; Li, S.; Zou, J.; Zhou, Y.; Maniccia, A.L.W.; Mao, H.; et al. Rational Design of Protein-Based MRI Contrast Agents. J. Am. Chem. Soc. 2008, 130, 9260–9267. [Google Scholar] [CrossRef]
- Woźniak-Braszak, A.; Jurga, K.; Baranowski, M. The Lipari-Szabo Model-Free Analysis as a Method for Study of Molecular Motion in Solid State Heteronuclear Systems Using NMR Off-Resonance. Appl. Magn. Reson. 2016, 47, 567–574. [Google Scholar] [CrossRef]
- Jacques, V.; Dumas, S.; Sun, W.-C.; Troughton, J.S.; Greenfield, M.T.; Caravan, P. High-Relaxivity Magnetic Resonance Imaging Contrast Agents. Part 2. Optimization of Inner- and Second-Sphere Relaxivity. Investig. Radiol. 2010, 45, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Babič, A.; Vorobiev, V.; Xayaphoummine, C.; Lapicorey, G.; Chauvin, A.-S.; Helm, L.; Allémann, E. Self-Assembled Nanomicelles as Mri Blood-Pool Contrast Agent. Chem. Eur. J. 2017, 24, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Granato, L.; Elst, L.V.; Henoumont, C.; Muller, R.N.; Laurent, S. Optimizing Water Exchange Rates and Rotational Mobility for High-Relaxivity of a Novel Gd-Do3a Derivative Complex Conjugated to Inulin as Macromolecular Contrast Agents for Mri. Chem. Biodivers. 2018, 15, e1700487. [Google Scholar] [CrossRef]
- Yu, K.-C.; Wang, X.-B.; Ye, C.-H.; U, U.-Y.; Liu, M.-L.; Zhuo, R.-X. Synthesis, Relaxivity and Mri Enhancement of Linear Oligo-Gd(III) Complexes with Poly (Dtpa-Ester) Ligands Derived from Amino Acids. Chin. J. Chem. 2010, 19, 788–793. [Google Scholar] [CrossRef]
- Moore, E.G.; Samuel, A.P.S.; Raymond, K.N. From Antenna to Assay: Lessons Learned in Lanthanide Luminescence. Acc. Chem. Res. 2009, 42, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Del Giorgio, E.; Sørensen, T.J. HOCl Responsive Lanthanide Complexes Using Hydroquinone Caging Units. Molecules 2020, 25, 1959. [Google Scholar] [CrossRef]
- Mahawaththa, M.C.; Lee, M.D.; Giannoulis, A.; Adams, L.A.; Feintuch, A.; Swarbrick, J.D.; Graham, B.; Nitsche, C.; Goldfarb, D.; Otting, G. Small neutral Gd(III) tags for distance measurements in proteins by double electron–electron resonance experiments. Phys. Chem. Chem. Phys. 2018, 20, 23535–23545. [Google Scholar] [CrossRef]
- Swetha, M.; Ramana, P.V.; Shirodkar, S.G. Simple and Efficient Method for the Synthesis of Azides in Water-THF Solvent System. Org. Prep. Proced. Int. 2011, 43, 348–353. [Google Scholar] [CrossRef]
- Harris, M.; Carron, S.; Elst, L.V.; Laurent, S.; Muller, R.N.; Parac-Vogt, T.N. Magnetofluorescent micellar complexes of terbium(iii) as potential bimodal contrast agents for magnetic resonance and optical imaging. Chem. Commun. 2015, 51, 2984–2986. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Ding, L.; Liu, J.; Zhao, B.; Deng, Q.; Yan, T. Synthesis and physiochemical properties of novel gemini surfactants with phenyl-1,4-bis(carbamoylmethyl) spacer. RSC Adv. 2015, 5, 74764–74773. [Google Scholar] [CrossRef]
- Suchý, M.; Bartha, R.; Hudson, R.H.E. “Click” chemistry toward bis(DOTA-derived) heterometallic complexes: Potential bimodal Mri/Pet(Spect) molecular imaging probes. RSC Adv. 2013, 3, 3249–3259. [Google Scholar] [CrossRef]
- Stasiuk, G.J.; Smith, H.; Wylezinska-Arridge, M.; Tremoleda, J.L.; Trigg, W.; Luthra, S.K.; Iveson, V.M.; Gavins, F.N.E.; Long, N.J. Gd3+ Cflflfk conjugate for Mri: A targeted contrast agent for FPR1 in inflammation. Chem. Commun. 2012, 49, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; He, X.; Shi, J.; Cui, C.; Xu, Y. A Series of New Eu/Tb Mixed MOFs with Tunable Color Luminescence. Z. Für Anorg. Und Allg. Chem. 2017, 644, 43–49. [Google Scholar] [CrossRef]
- Freslon, S.; Luo, Y.; Daiguebonne, C.; Calvez, G.; Bernot, K.; Guillou, O. Brightness and Color Tuning in a Series of Lanthanide-Based Coordination Polymers with Benzene-1,2,4,5-tetracarboxylic Acid as a Ligand. Inorg. Chem. 2015, 55, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Feng, Y.; Guo, N.; Sun, Y.; Zhang, T.; Ma, L.; Wang, L. Series d–f Heteronuclear Metal–Organic Frameworks: Color Tunability and Luminescent Probe with Switchable Properties. Inorg. Chem. 2017, 56, 1713–1721. [Google Scholar] [CrossRef]
- Cheng, R.-H.; Chen, J.-M.; Chen, Y.-W.; Cai, H.; Cui, X.; Hwang, D.W.; Chen, Z.; Ding, S. Macromolecular Crowding May Significantly Affect the Performance of an Mri Contrast Agent: A 1H NMR Spectroscopy, Microimaging, and Fast-Field-Cycling NMR Relaxometry Study. ChemistryOpen 2018, 7, 288–296. [Google Scholar] [CrossRef]
- Lauffer, R.B. Paramagnetic metal complexes as water proton relaxation agents for NMR imaging: Theory and design. Chem. Rev. 1987, 87, 901–927. [Google Scholar] [CrossRef]
- Diaferia, C.; Gianolio, E.; Accardo, A. Peptide-based building blocks as structural elements for supramolecular Gd-containing Mri contrast agents. J. Pept. Sci. 2019, 25, e3157. [Google Scholar] [CrossRef]
- Accardo, A.; Tesauro, D.; Aloj, L.; Pedone, C.; Morelli, G. Supramolecular aggregates containing lipophilic Gd(III) complexes as contrast agents in Mri. Co-Ord. Chem. Rev. 2009, 253, 2193–2213. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.2; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ditchfield, R. Molecular Orbital Theory of Magnetic Shielding and Magnetic Susceptibility. J. Chem. Phys. 1972, 56, 5688–5691. [Google Scholar] [CrossRef]
- De Bie, M.J.A. Carbon-13 NMR Spectroscopy. Hans-Otto Kalinowski, Stefan Berger and Siegmar Braun; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1988; Volume 107, p. 652. ISBN 0-471-913065. [Google Scholar]
- Pihlaja, K.; Kleinpeter, E. Carbon-13 NMR Chemical Shifts in Structural and Stereochemical Analysis; VCH: New York, NY, USA, 1994. [Google Scholar]
- Soria-Martínez, R.; Mendoza-Meroño, R.; García-Granda, S. Synthesis, crystal structure, spectroscopic characterization and theoretical study of (2E)-N-phenyl-2-(pyridin-3-ylmethylidene)hydrazinecarboxamide. J. Mol. Struct. 2016, 1105, 322–331. [Google Scholar] [CrossRef]
- Goodreid, J.D.; Duspara, P.A.; Bosch, C.; Batey, R.A. Amidation Reactions from the Direct Coupling of Metal Carboxylate Salts with Amines. J. Org. Chem. 2013, 79, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, H.; Sumana, G.; Marquette, C.A. A label-free ultrasensitive microfluidic surface Plasmon resonance biosensor for Aflatoxin B1 detection using nanoparticles integrated gold chip. Food Chem. 2019, 307, 125530. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.; Kimura, T.; Kishida, A. Controlling Coupling Reaction of EDC and NHS for Preparation of Collagen Gels Using Ethanol/Water Co-Solvents. Macromol. Biosci. 2008, 8, 32–37. [Google Scholar] [CrossRef]
- Sureshbabu, V.V.; Lalithamba, H.S.; Narendra, N.; Hemantha, H.P. New and simple synthesis of acid azides, ureas and carbamates from carboxylic acids: Application of peptide coupling agents EDC and HBTU. Org. Biomol. Chem. 2009, 8, 835–840. [Google Scholar] [CrossRef]
- Lin, M.; Liang, W.; Ma, J.; Wu, Y.; Xiao, X.; Ying, J.; Yan, Y.; Zhao, L.; Jin, H.; Liang, H.; et al. Efficient and Chemoselective Amidation of β-Carboline Carboxylic Acids. ChemistrySelect 2019, 4, 12978–12982. [Google Scholar] [CrossRef]
- Mi, X.; Sheng, D.; Yu, Y.; Wang, Y.; Zhao, L.; Lu, J.; Li, Y.; Li, D.; Dou, J.; Duan, J.; et al. Tunable Light Emission and Multiresponsive Luminescent Sensitivities in Aqueous Solutions of Two Series of Lanthanide Metal–Organic Frameworks Based on Structurally Related Ligands. ACS Appl. Mater. Interfaces 2019, 11, 7914–7926. [Google Scholar] [CrossRef]
- Wang, J.-X.; Yin, J.; Shekhah, O.; Bakr, O.M.; Eddaoudi, M.; Mohammed, O.F. Energy Transfer in Metal–Organic Frameworks for Fluorescence Sensing. ACS Appl. Mater. Interfaces 2022, 14, 9970–9986. [Google Scholar] [CrossRef]
- Ji, H.; Wang, X.; Wang, P.; Gong, Y.; Wang, Y.; Liu, C.; Ji, G.; Wang, X.; Wang, M. Lanthanide-based metal–organic frameworks solidified by gelatin-methacryloyl hydrogels for improving the accuracy of localization and excision of small pulmonary nodules. J. Nanobiotechnol. 2022, 20, 60. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Base | Base Eq. | T (°C) | Time (h) | Eq. C17 | Eq. C12 | Result | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| Chloroform | TEA | 3 | r.t. | 3 | 1 | 5 | - | - |
| Chloroform | Pyridine | 3 | 0 | 12 | 1 | 1 | - | - |
| Chloroform | Pyridine | 5 | −78 | 12 | 1 | 1 | - | - |
| DMF | TEA | 10 | r.t | 12 | 1 | 5 | - | - |
| DMF | TEA | 10 | r.t | 12 | 1 | 2 | - | - |
| DMF | TEA | 10 | 50 | 12 | 1 | 1 | - | - |
| DMF | Cs2CO3 | 5 | 50 | 12 | 1 | 2.2 | - | - |
| DMF | Cs2CO3 | 10 | 60 | 12 | 1 | 2.2 | - | - |
| Toluene | Cs2CO3 | 10 | 110 | 12 | 1 | 2.2 | - | - |
| Toluene | Cs2CO3/1% NaI | 10 | 110 | 12 | 1 | 2.2 | - | - |
| Toluene | Cs2CO3/5% NaI | 10 | 110 | 24 | 1 | 2.2 | - | - |
| Toluene | Cs2CO3/5% NaI | 10 | 110 | 48 | 1 | 2.2 | - | - |
| Acetonitrile:0.1 DMF | Cs2CO3/5% NaI | 10 | 110 | 48 | 1 | 2.2 | * | * |
| Acetonitrile | Cs2CO3/5% NaI | 10 | 82 | 24 | 1 | 2.2 | - | - |
| Acetonitrile | NaHCO3 | 2 | 82 | 48 | 1 | 2.2 | - | - |
| Acetonitrile | NaHCO3 | 2 | 82 | 56 | 1 | 2.2 | - | - |
| DMF | TEA | 5 | 80 | 48 | 1 | 2.2 | - | - |
| DMF | NaHCO3 | 5 | 80 | 48 | 1 | 2.2 | - | - |
| Toluene:0.1 DMF | Cs2CO3/5% NaI | 10 | 110 | 48 | 1 | 2.2 | * | * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzmán, C.; Soria-Martínez, R.; Urresta, J. New Strategies for Potential Contrast Agents’ Synthons Highly Active to MRI Based on Gd3+, Eu3+, and Tb3+. Appl. Sci. 2022, 12, 9969. https://doi.org/10.3390/app12199969
Guzmán C, Soria-Martínez R, Urresta J. New Strategies for Potential Contrast Agents’ Synthons Highly Active to MRI Based on Gd3+, Eu3+, and Tb3+. Applied Sciences. 2022; 12(19):9969. https://doi.org/10.3390/app12199969
Chicago/Turabian StyleGuzmán, Carlos, Rubén Soria-Martínez, and Julián Urresta. 2022. "New Strategies for Potential Contrast Agents’ Synthons Highly Active to MRI Based on Gd3+, Eu3+, and Tb3+" Applied Sciences 12, no. 19: 9969. https://doi.org/10.3390/app12199969
APA StyleGuzmán, C., Soria-Martínez, R., & Urresta, J. (2022). New Strategies for Potential Contrast Agents’ Synthons Highly Active to MRI Based on Gd3+, Eu3+, and Tb3+. Applied Sciences, 12(19), 9969. https://doi.org/10.3390/app12199969