
Salt-Induced Homogeneous Liquid–Liquid Microextraction of Piroxicam and Meloxicam from Human Urine Prior to Their Determination by HPLC-DAD




Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Instrumentation and HPLC Conditions
2.3. Sample Collection
2.4. SI-HLLME Procedure
3. Results and Discussion
3.1. Influence of Extraction Solvent and Salt Type
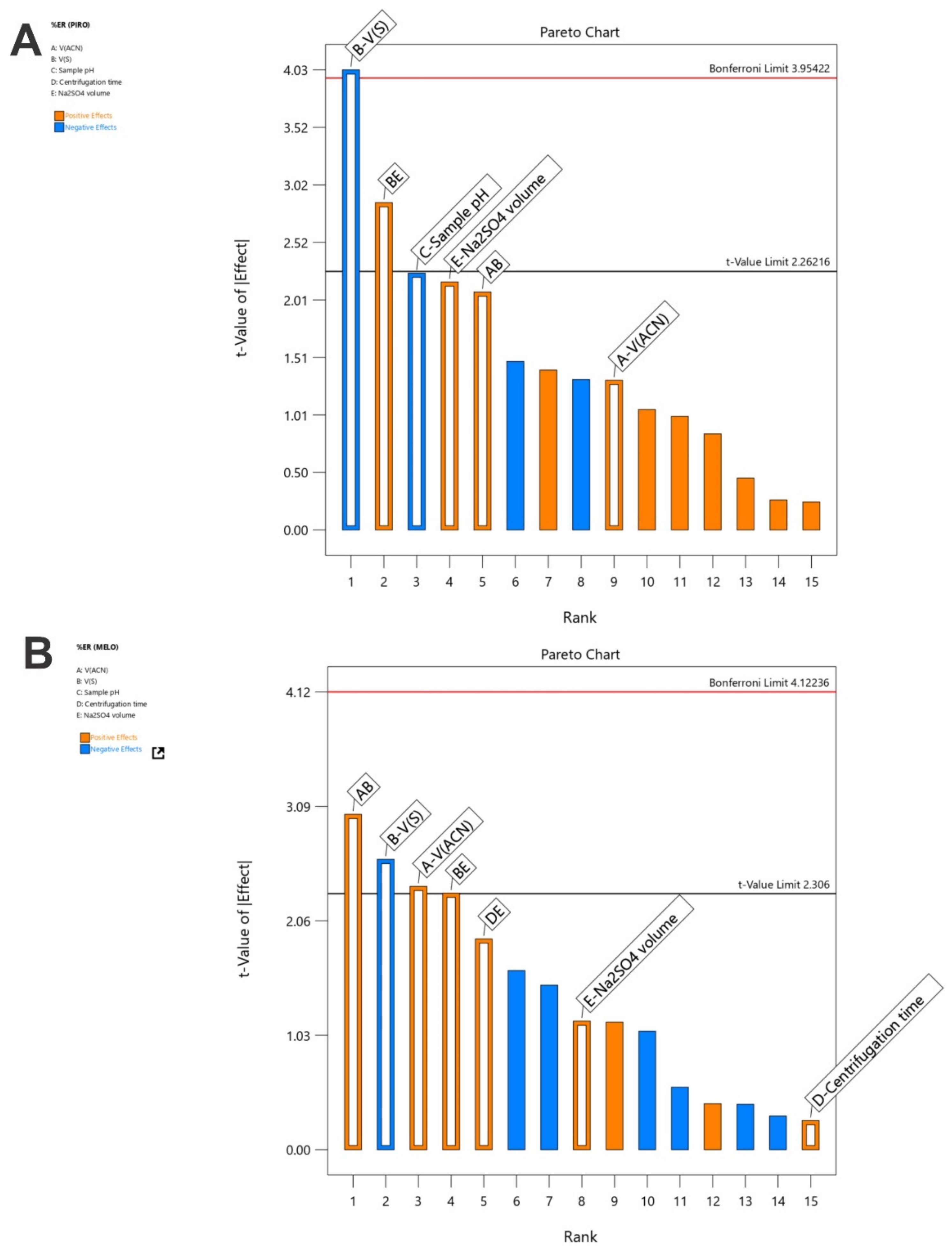
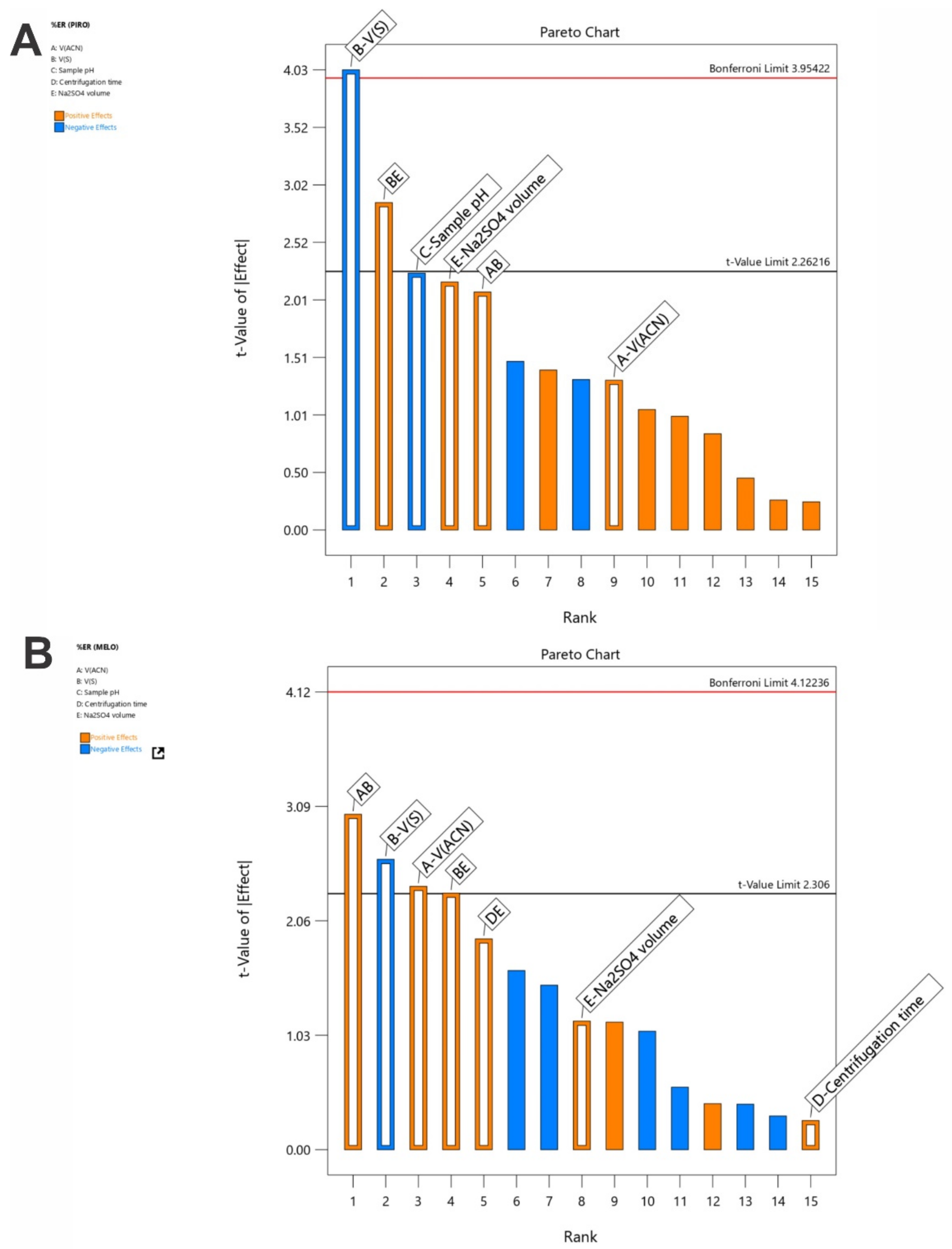
3.2. Examination of the SI-HLLME Parameters Using 2-Level FFD
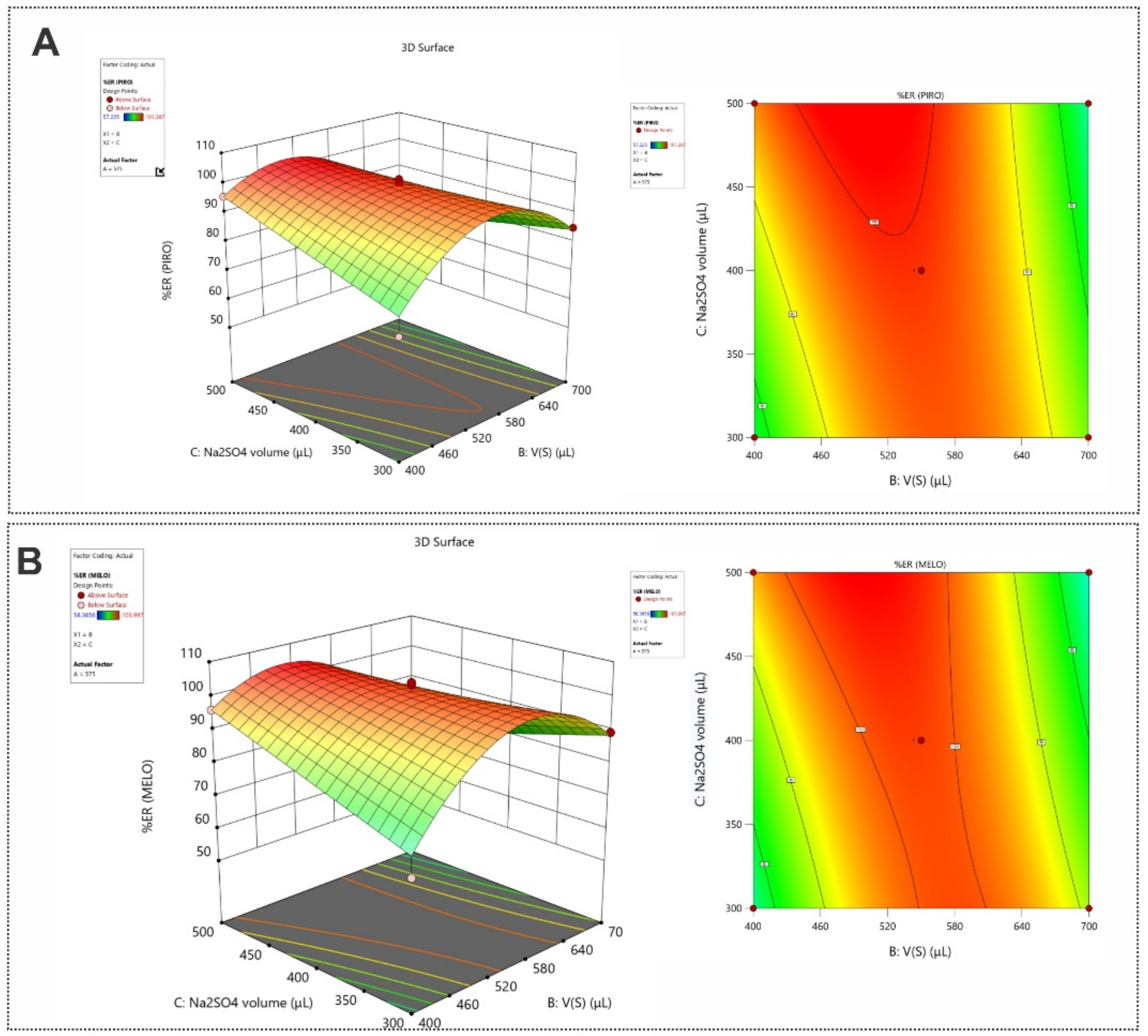
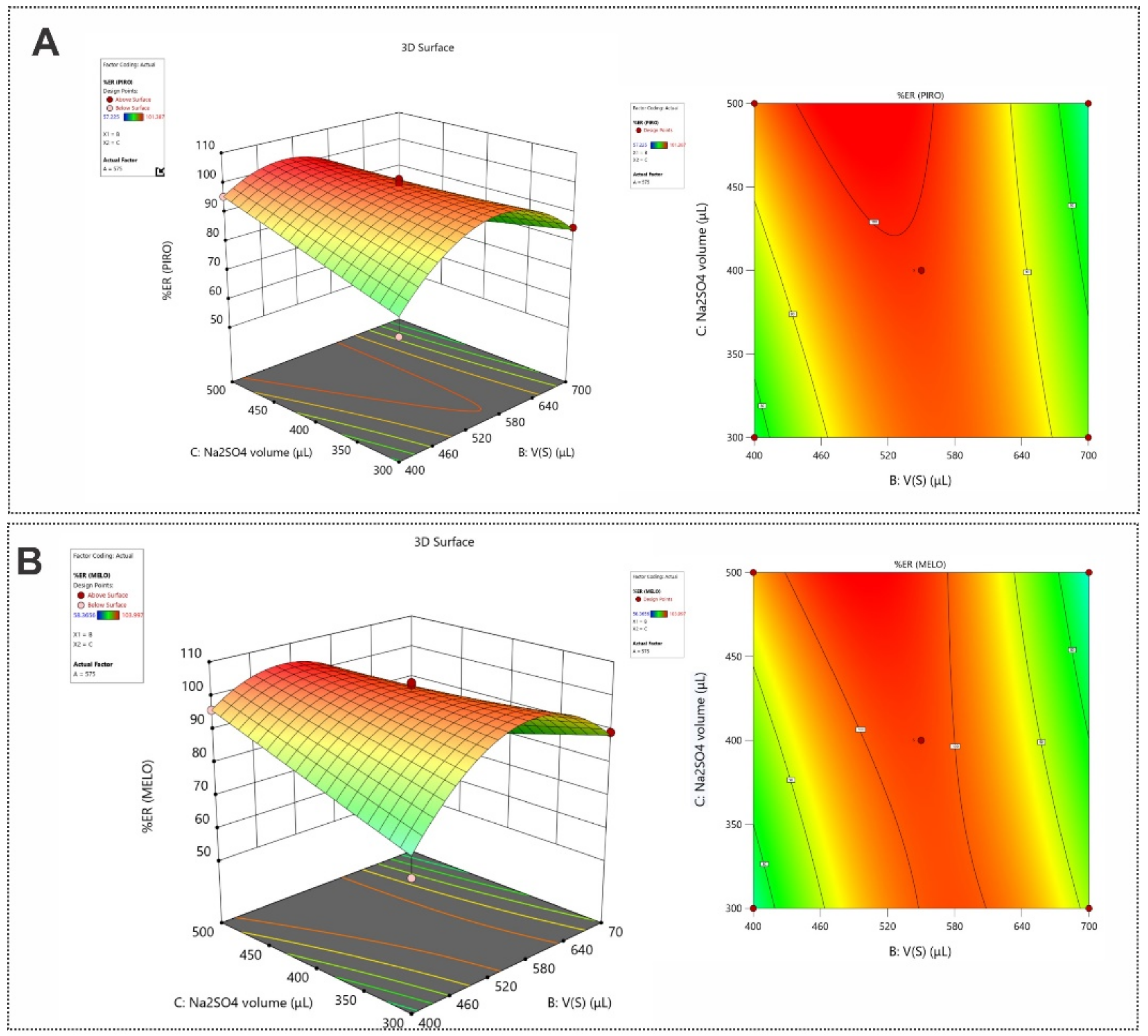
3.3. Optimization of the SI-HLLME Parameters Using Box–Behnken Design
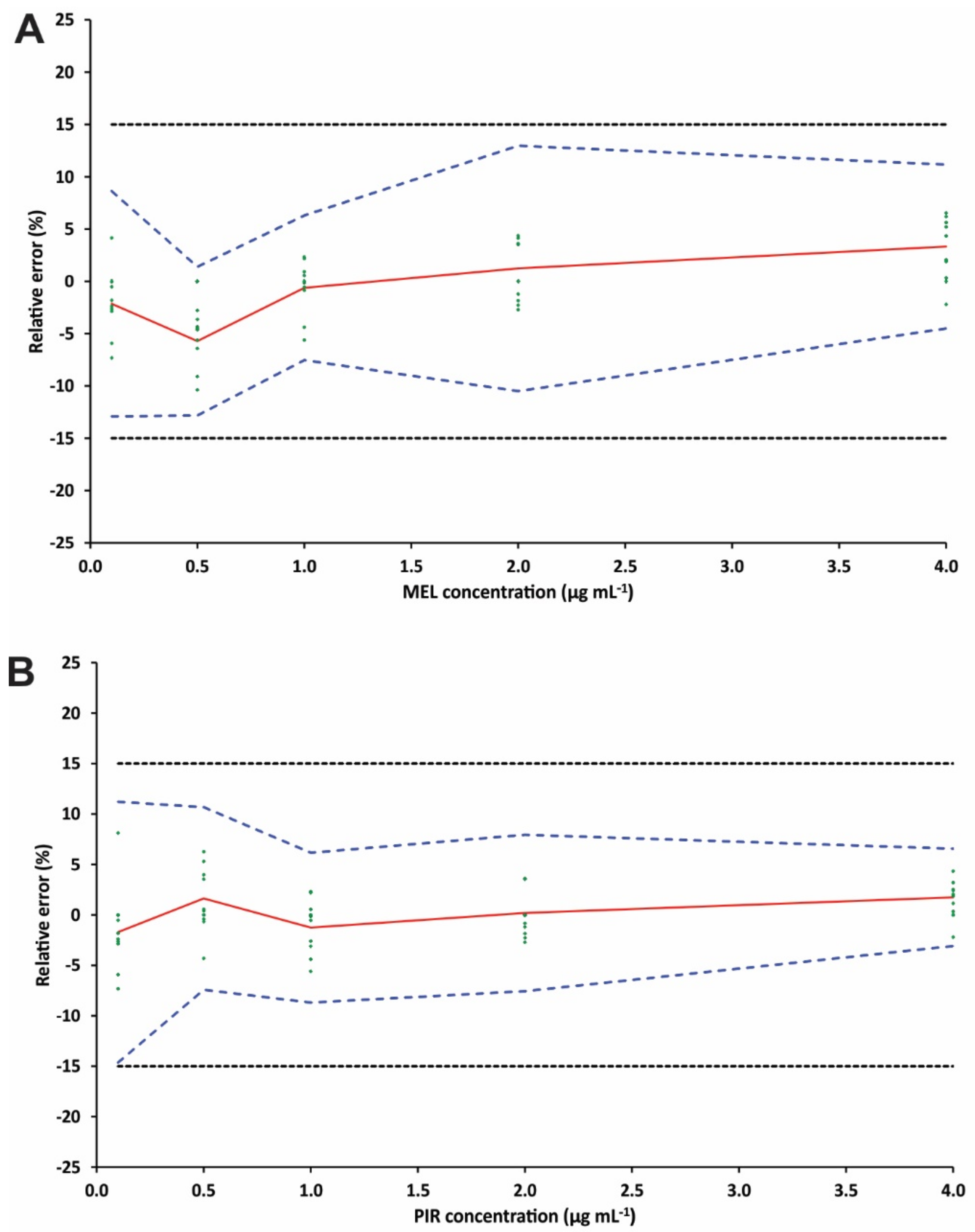
3.4. Method Validation
3.5. Stability Studies
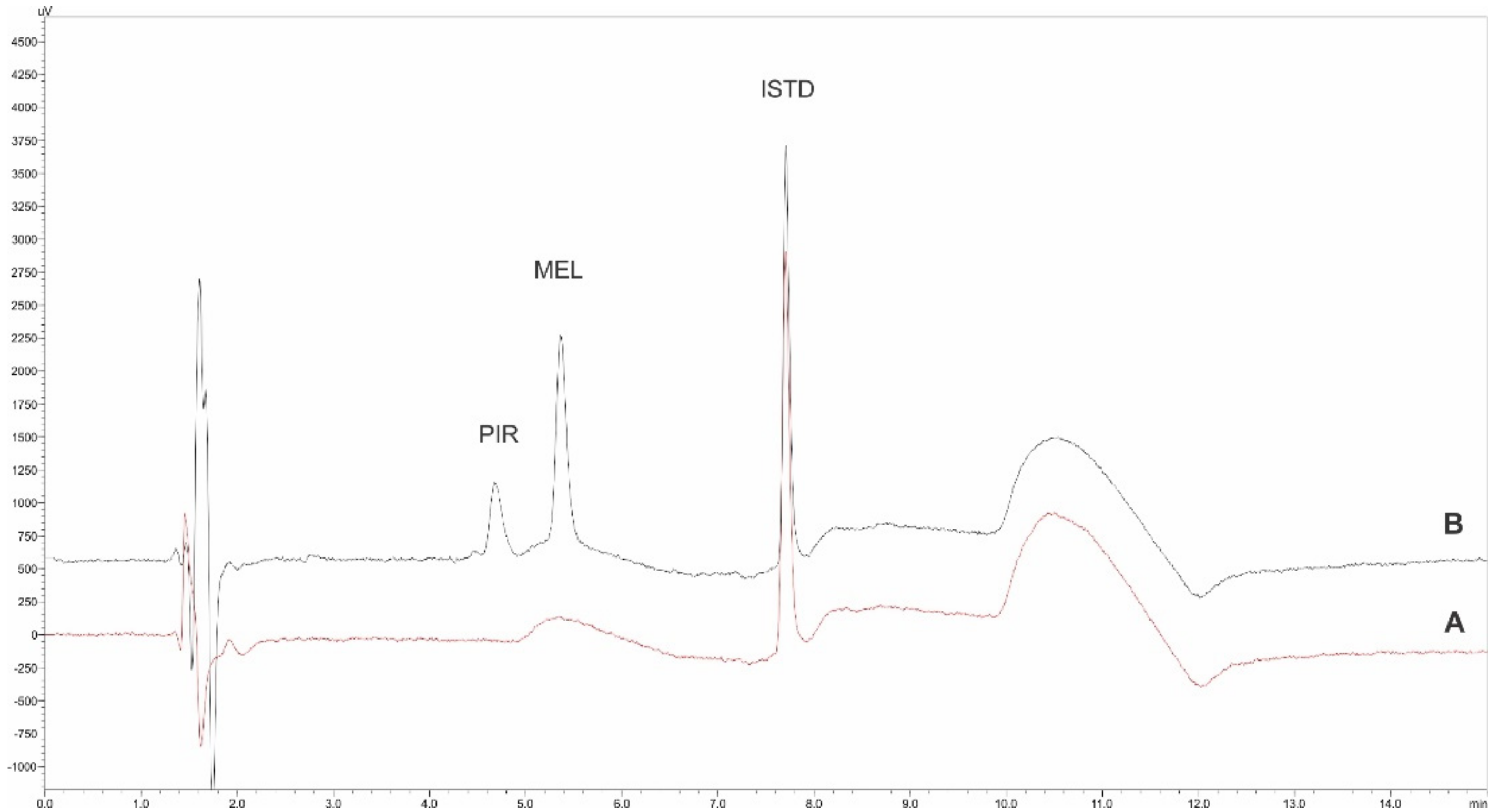
3.6. Application to Real Urine Analysis
3.7. Comparison of the Proposed Method with Other Studies
3.8. Investigation of the Green Potential of the Developed SI-HLLME-HPLC-DAD Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cox, S.; Hayes, J.; Yarbrough, J.; Veiga-Parga, T.; Greenacre, C. High-Performance Liquid Chromatography Determination of Meloxicam and Piroxicam with Ultraviolet Detection. Chromatogr. Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Tabrizi, A.B.; Tutunchi, N.S. Analysis of piroxicam in pharmaceutical formulation and human urine by dispersive liquid-liquid microextraction combined with spectrophotometry. Adv. Pharm. Bull. 2013, 3, 37–44. [Google Scholar]
- Xu, S.; Rouzer, C.A.; Marnett, L.J. Oxicams, a class of nonsteroidal anti-inflammatory drugs and beyond. IUBMB Life 2014, 66, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.W.; Kim, M.J.; Jang, C.G.; Lee, S.Y. Determination of meloxicam in human plasma using a HPLC method with UV detection and its application to a pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 859, 69–73. [Google Scholar] [CrossRef]
- Milligan, P.A. Determination of piroxicam and its major metabolites in the plasma, urine and bile of humans by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1992, 576, 121–128. [Google Scholar] [CrossRef]
- Martinez-Sena, T.; Armenta, S.; de la Guardia, M.; Esteve-Turrillas, F.A. Determination of non-steroidal anti-inflammatory drugs in water and urine using selective molecular imprinted polymer extraction and liquid chromatography. J. Pharm. Biomed. Anal. 2016, 131, 48–53. [Google Scholar] [CrossRef]
- Andruch, V.; Varfalvyová, A.; Halko, R.; Jatkowska, N.; Płotka-Wasylka, J. Application of deep eutectic solvents in bioanalysis. TrAC Trends Anal. Chem. 2022, 154, 116660. [Google Scholar] [CrossRef]
- Martin, P.D.; Jones, G.R.; Stringer, F.; Wilson, I.D. Comparison of extraction of a β-blocker from plasma onto a molecularly imprinted polymer with liquid-liquid extraction and solid phase extraction methods. J. Pharm. Biomed. Anal. 2004, 35, 1231–1239. [Google Scholar] [CrossRef]
- Câmara, J.S.; Perestrelo, R.; Berenguer, C.V.; Andrade, C.F.P.; Gomes, T.M.; Olayanju, B.; Kabir, A.; Rocha, C.M.R.; Teixeira, J.A.; Pereira, J.A.M. Green Extraction Techniques as Advanced Sample Preparation Approaches in Biological, Food, and Environmental Matrices: A Review. Molecules 2022, 27, 2953. [Google Scholar] [CrossRef]
- Armenta, S.; Garrigues, S.; de la Guardia, M. Green Analytical Chemistry. TrAC-Trends Anal. Chem. 2008, 27, 497–511. [Google Scholar] [CrossRef]
- Queiroz, M.E.C.; de Souza, I.D.; de Oliveira, I.G.; Grecco, C.F. In vivo solid phase microextraction for bioanalysis. TrAC Trends Anal. Chem. 2022, 153, 116656. [Google Scholar] [CrossRef]
- Rigkos, G.; Alampanos, V.; Kabir, A.; Furton, K.G.; Roje, Ž.; Vrček, I.V.; Panderi, I.; Samanidou, V. An improved fabric-phase sorptive extraction protocol for the determination of seven parabens in human urine by HPLC–DAD. Biomed. Chromatogr. 2021, 35, e4974. [Google Scholar] [CrossRef] [PubMed]
- Manousi, N.; Kabir, A.; Furton, K.G.; Samanidou, V.F.; Zacharis, C.K. Exploiting the capsule phase microextraction features in bioanalysis: Extraction of ibuprofen from urine samples. Microchem. J. 2021, 172, 106934. [Google Scholar] [CrossRef]
- Giakisikli, G.; Anthemidis, A.N. Magnetic materials as sorbents for metal/metalloid preconcentration and/or separation. A review. Anal. Chim. Acta 2013, 789, 1–16. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, Y.; Zhang, Q.; Zang, L.; Chen, B.; Hu, B. Stir bar sorptive extraction and its application. J. Chromatogr. A 2021, 1637, 461810. [Google Scholar] [CrossRef]
- Turoňová, D.; Kujovská Krčmová, L.; Švec, F. Application of microextraction in pipette tips in clinical and forensic toxicology. TrAC Trends Anal. Chem. 2021, 143, 116404. [Google Scholar] [CrossRef]
- Mansour, F.R.; Khairy, M.A. Pharmaceutical and biomedical applications of dispersive liquid–liquid microextraction. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1061, 382–391. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, S.; Huang, C.; Gjelstad, A. Electromembrane extraction–Recent trends and where to go. J. Pharm. Anal. 2017, 7, 141–147. [Google Scholar] [CrossRef]
- Manousi, N.; Vlachaki, A.; Kika, F.S.; Markopoulou, C.K.; Tzanavaras, P.D.; Zacharis, C.K. Salting-out homogeneous liquid-liquid microextraction for the determination of azole drugs in human urine: Validation using total error concept. J. Sep. Sci. 2022, 45, 1240–1251. [Google Scholar] [CrossRef]
- Li, N.; Zhang, T.; Chen, G.; Xu, J.; Ouyang, G.; Zhu, F. Recent advances in sample preparation techniques for quantitative detection of pharmaceuticals in biological samples. TrAC Trends Anal. Chem. 2021, 142, 116318. [Google Scholar] [CrossRef]
- Dmitrienko, S.G.; Apyari, V.V.; Gorbunova, M.V.; Tolmacheva, V.V.; Zolotov, Y.A. Homogeneous Liquid–Liquid Microextraction of Organic Compounds. J. Anal. Chem. 2020, 75, 1371–1383. [Google Scholar] [CrossRef]
- Zacharis, C.K. Extraction In Aqueous Two-Phase Extraction; Worsfold, P., Poole, C., Townshend, A., Miró, M.B.T.-E., Third, E., Eds.; Academic Press: Oxford, UK, 2019; pp. 47–51. ISBN 978-0-08-101984-9. [Google Scholar]
- Hammad, S.F.; Abdallah, I.A.; Bedair, A.; Mansour, F.R. Homogeneous liquid–liquid extraction as an alternative sample preparation technique for biomedical analysis. J. Sep. Sci. 2021, 45, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, R.R.; Gurrani, S.; Tsai, P.-C.; Ponnusamy, V.K. Novel Salt-Assisted Liquid-Liquid Microextraction Technique for Environmental, Food, and Biological Samples Analysis Applications: A Review. Curr. Anal. Chem. 2022, 18, 577–587. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Y.; Wang, J.; Li, X.; Wang, W.; Huang, Z. Sugaring-out assisted liquid-liquid extraction coupled with high performance liquid chromatography-electrochemical detection for the determination of 17 phenolic compounds in honey. J. Chromatogr. A 2019, 1601, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Akramipour, R.; Fattahi, N.; Pirsaheb, M.; Gheini, S. Combination of counter current salting-out homogenous liquid-liquid extraction and dispersive liquid-liquid microextraction as a novel microextraction of drugs in urine samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1012, 162–168. [Google Scholar] [CrossRef]
- Tsanaktsidou, E.; Markopoulou, C.K.; Tzanavaras, P.D.; Zacharis, C.K. Homogeneous liquid phase microextraction using hydrophilic media for the determination of fluoroquinolones in human urine using HPLC-FLD. Microchem. J. 2022, 172, 106906. [Google Scholar] [CrossRef]
- Feriduni, B.; Farajzadeh, M.A.; Jouyban, A. Determination of two antiepileptic drugs in urine by homogenous liquid-liquid extraction performed in a narrow tube combined with dispersive liquid-liquid microextraction followed by gas chromatography-flame ionization detection. Iran. J. Pharm. Res. 2019, 18, 620–630. [Google Scholar]
- Sorouraddin, S.M.; Farajzadeh, M.A.; Hassanyani, A.; Afshar Mogaddam, M.R. Combination of homogenous liquid-liquid extraction and dispersive liquid-liquid microextraction for extraction and preconcentration of amantadine from biological samples followed by its indirect determination by flame atomic absorption spectrometry. RSC Adv. 2016, 6, 108603–108610. [Google Scholar] [CrossRef]
- López-Lorente, Á.I.; Pena-Pereira, F.; Pedersen-Bjergaard, S.; Zuin, V.G.; Ozkan, S.A.; Psillakis, E. The ten principles of green sample preparation. TrAC-Trends Anal. Chem. 2022, 148, 11653. [Google Scholar] [CrossRef]
- Brooks, T.; Keevil, C.W. A simple artificial urine for the growth of urinary pathogens. Lett. Appl. Microbiol. 1997, 24, 203–206. [Google Scholar] [CrossRef]
- Ferreira, S.L.C.; Bruns, R.E.; Ferreira, H.S.; Matos, G.D.; David, J.M.; Brandão, G.C.; da Silva, E.G.P.; Portugal, L.A.; dos Reis, P.S.; Souza, A.S.; et al. Box-Behnken design: An alternative for the optimization of analytical methods. Anal. Chim. Acta 2007, 597, 179–186. [Google Scholar] [CrossRef] [PubMed]
- FDA. Bioanalytical Method Validation Guidance. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 11 January 2022).
- Stege, P.W.; Lapierre, A.V.; Martinez, L.D.; Messina, G.A.; Sombra, L.L. A combination of single-drop microextraction and open tubular capillary electrochromatography with carbon nanotubes as stationary phase for the determination of low concentration of illicit drugs in horse urine. Talanta 2011, 86, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Zeeb, M.; Farahani, H.; Mirza, B.; Papan, M.K. Quantification of Meloxicam in Human Plasma Using Ionic Liquid-Based Ultrasound-Assisted In Situ Solvent Formation Microextraction Followed by High-Performance Liquid Chromatography. J. Chromatogr. Sci. 2018, 56, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.Y.; Shi, Y.P.; Chen, J. A novel extraction technique based on carbon nanotubes reinforced hollow fiber solid/liquid microextraction for the measurement of piroxicam and diclofenac combined with high performance liquid chromatography. Talanta 2012, 100, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Choi, H.K. Analysis of meloxicam by high-performance liquid chromatography with cloud-point extraction. Anal. Bioanal. Chem. 2008, 392, 947–953. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef]
- Płotka-Wasylka, J.; Wojnowski, W. Complementary green analytical procedure index (ComplexGAPI) and software. Green Chem. 2021, 23, 8657–8665. [Google Scholar] [CrossRef]
- Plastiras, O.; Andreasidou, E.; Samanidou, V.F. Microextraction Techniques with Deep Eutectic Solvents. Molecules 2020, 25, 6026. [Google Scholar] [CrossRef]
- Cielecka-Piontek, J.; Zalewski, P.; Jelińska, A.; Garbacki, P. UHPLC: The greening face of liquid chromatography. Chromatographia 2013, 76, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.A.; Dewe, W.; Feinberg, M.; Lallier, M.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal—Part I. J. Pharm. Biomed. Anal. 2004, 36, 579–586. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standard Run | VACN (μL) | VS (μL) | Sample pH | Centrifugation Time | Vsalt (μL) |
|---|---|---|---|---|---|
| 14 | 700 | 400 | 6 | 10 | 300 |
| 10 | 700 | 400 | 3 | 10 | 500 |
| 3 | 450 | 700 | 3 | 1 | 300 |
| 5 | 450 | 400 | 6 | 1 | 300 |
| 15 | 450 | 700 | 6 | 10 | 300 |
| 2 | 700 | 400 | 3 | 1 | 300 |
| 11 | 450 | 700 | 3 | 10 | 500 |
| 8 | 700 | 700 | 6 | 1 | 300 |
| 6 | 700 | 400 | 6 | 1 | 500 |
| 7 | 450 | 700 | 6 | 1 | 500 |
| 13 | 450 | 400 | 6 | 10 | 500 |
| 1 | 450 | 400 | 3 | 1 | 500 |
| 12 | 700 | 700 | 3 | 10 | 300 |
| 16 | 700 | 700 | 6 | 10 | 500 |
| 4 | 700 | 700 | 3 | 1 | 500 |
| 9 | 450 | 400 | 3 | 10 | 300 |
| Standard Run | VACN (μL) | VS (μL) | Vsalt (μL) | % ER (PIR) | % ER (MEL) |
|---|---|---|---|---|---|
| 11 | 575 | 400 | 500 | 95.45 | 95.93 |
| 7 | 450 | 550 | 500 | 82.27 | 84.33 |
| 14 | 575 | 550 | 400 | 100.38 | 103.25 |
| 15 | 575 | 550 | 400 | 101.38 | 103.99 |
| 4 | 700 | 700 | 400 | 76.77 | 76.55 |
| 1 | 450 | 400 | 400 | 78.26 | 76.83 |
| 3 | 450 | 700 | 400 | 57.22 | 58.36 |
| 6 | 700 | 550 | 300 | 100.88 | 101.43 |
| 13 | 575 | 550 | 400 | 98.16 | 99.66 |
| 16 | 575 | 550 | 400 | 99.92 | 100.29 |
| 5 | 450 | 550 | 300 | 85.78 | 88.05 |
| 9 | 575 | 400 | 300 | 70.42 | 68.56 |
| 8 | 700 | 550 | 500 | 98.84 | 97.62 |
| 17 | 575 | 550 | 400 | 94.81 | 97.41 |
| 12 | 575 | 700 | 500 | 79.42 | 78.63 |
| 2 | 700 | 400 | 400 | 85.04 | 82.28 |
| 10 | 575 | 700 | 300 | 84.96 | 89.31 |
| Validation Criteria | |||
|---|---|---|---|
| Response function (linear regression) | Slope | Intercept (×103) | r2 |
| (k a = 3; m = 5; n = 3) (0.1–4.0 μg mL−1) | |||
| Day 1 | 0.6971 | −0.0375 | 0.9998 |
| Day 2 | 0.7015 | −0.0367 | 0.9996 |
| Day 3 | 0.7066 | −0.0399 | 0.9995 |
| Precision (k = 3; n = 3) | |||
| C (μg mL−1) | sr (%) b | sR (%) c | |
| 0.1 | 2.5 | 2.6 | |
| 0.5 | 2.9 | 4.4 | |
| 1 | 2.8 | 3.9 | |
| 2 | 2.0 | 2.8 | |
| 4 | 2.8 | 2.6 | |
| Trueness (k = 3; n = 3) | |||
| C (μg mL−1) | Relative bias (%) | ||
| 0.1 | −2.2 | ||
| 0.5 | −5.7 | ||
| 1 | −0.62 | ||
| 2 | + 1.2 | ||
| 4 | + 3.4 | ||
| Accuracy (k = 5; n = 3) | |||
| C (%) | Relative β-ΕΤΙ (%) | ||
| 0.1 | [−12.92, 8.61] | ||
| 0.5 | [−12.82, 1.40] | ||
| 1 | [−7.54, 6.30] | ||
| 2 | [−10.49, 12.96] | ||
| 4 | [−4.50, 11.16] | ||
| Linearity (k = 3; n = 3; m = 5) (0.1–4.0 μg mL−1) | |||
| Slope | 1.042 | ||
| Intercept | −0.024 | ||
| r2 | 0.9998 | ||
| LOD (μg mL−1) | 0.02 | ||
| LLOQ (μg mL−1) | 0.1 | ||
| Analyte | Sample | Sample Preparation 1 | Analytical Determination 2 | RR% | RSD% | LOD (μg mL−1) | Ref. |
|---|---|---|---|---|---|---|---|
| PIR | Horse urine | SDME | CEC-DAD | 94.8 | <4.32 | 0.01764 | [34] |
| MEL | Human plasma | IL-UA-ISFME | HPLC-UV | 82.1–93.6 | <5.1 | 0.0001 | [35] |
| PIR | Water samples | HF-SLPME | HPLC-DAD | 70.02–110.98 | <4.27 | 0.00458 | [36] |
| MEL | Human serum | Cloud-point extraction | HPLC-UV | >92 | 6.9 | 0.01 | [37] |
| PIR | Pharmaceutical formulations, human urine | DLLME | Spectrophotometry | 97–110 | 2.83 | 0.058 | [2] |
| PIR MEL | Human urine | SI-HLLME | HPLC-DAD | 94.3–103.4 | <4.4 | 0.02–0.03 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manousi, N.; Tsiona, S.V.; Zacharis, C.K. Salt-Induced Homogeneous Liquid–Liquid Microextraction of Piroxicam and Meloxicam from Human Urine Prior to Their Determination by HPLC-DAD. Appl. Sci. 2022, 12, 6658. https://doi.org/10.3390/app12136658
Manousi N, Tsiona SV, Zacharis CK. Salt-Induced Homogeneous Liquid–Liquid Microextraction of Piroxicam and Meloxicam from Human Urine Prior to Their Determination by HPLC-DAD. Applied Sciences. 2022; 12(13):6658. https://doi.org/10.3390/app12136658
Chicago/Turabian StyleManousi, Natalia, Sotiria V. Tsiona, and Constantinos K. Zacharis. 2022. "Salt-Induced Homogeneous Liquid–Liquid Microextraction of Piroxicam and Meloxicam from Human Urine Prior to Their Determination by HPLC-DAD" Applied Sciences 12, no. 13: 6658. https://doi.org/10.3390/app12136658
APA StyleManousi, N., Tsiona, S. V., & Zacharis, C. K. (2022). Salt-Induced Homogeneous Liquid–Liquid Microextraction of Piroxicam and Meloxicam from Human Urine Prior to Their Determination by HPLC-DAD. Applied Sciences, 12(13), 6658. https://doi.org/10.3390/app12136658