
Pan-Cancer Analysis for Immune Cell Infiltration and Mutational Signatures Using Non-Negative Canonical Correlation Analysis


Abstract
:Simple Summary
Abstract
1. Introduction
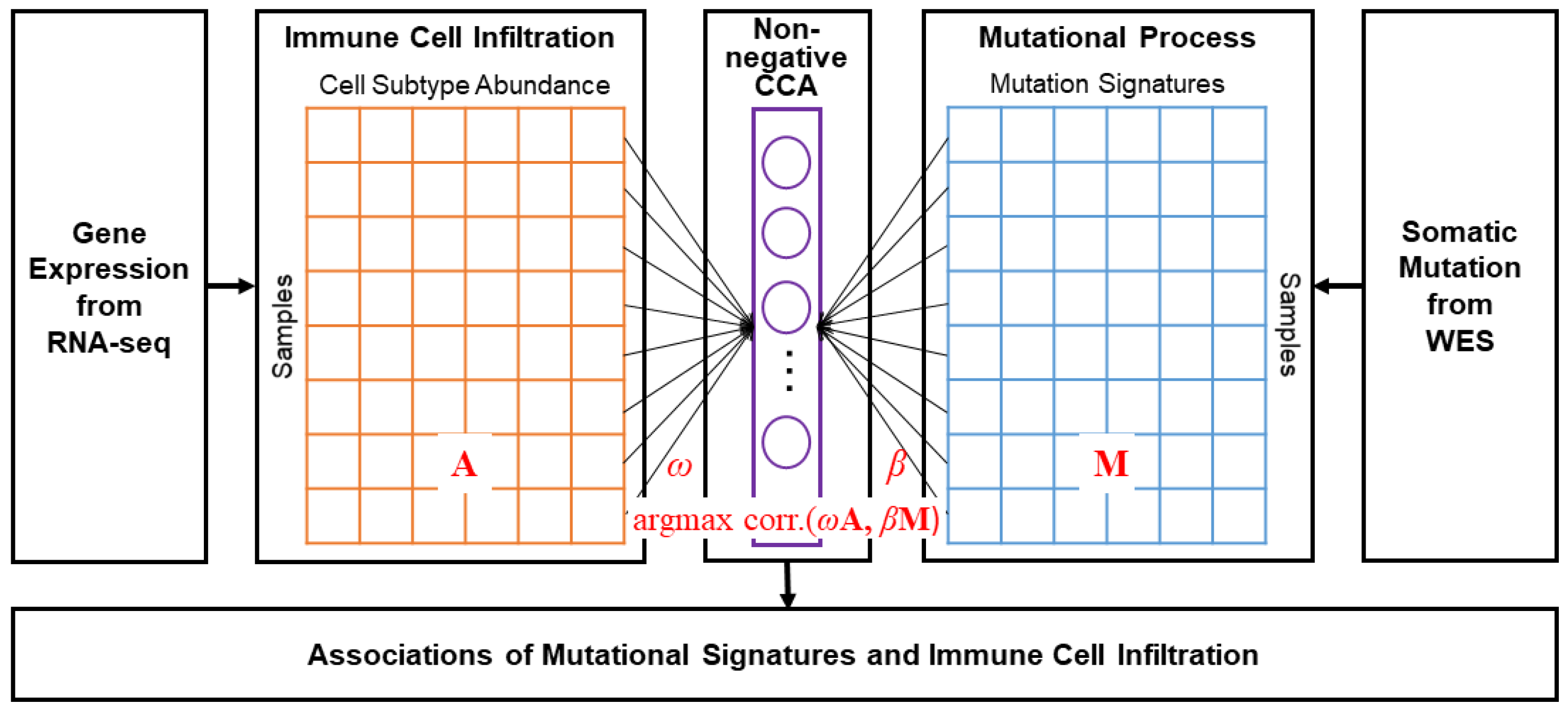
2. Materials and Methods
2.1. Data Preparation
2.2. Estimation of Mutational Signatures and Immune Cell Infiltration
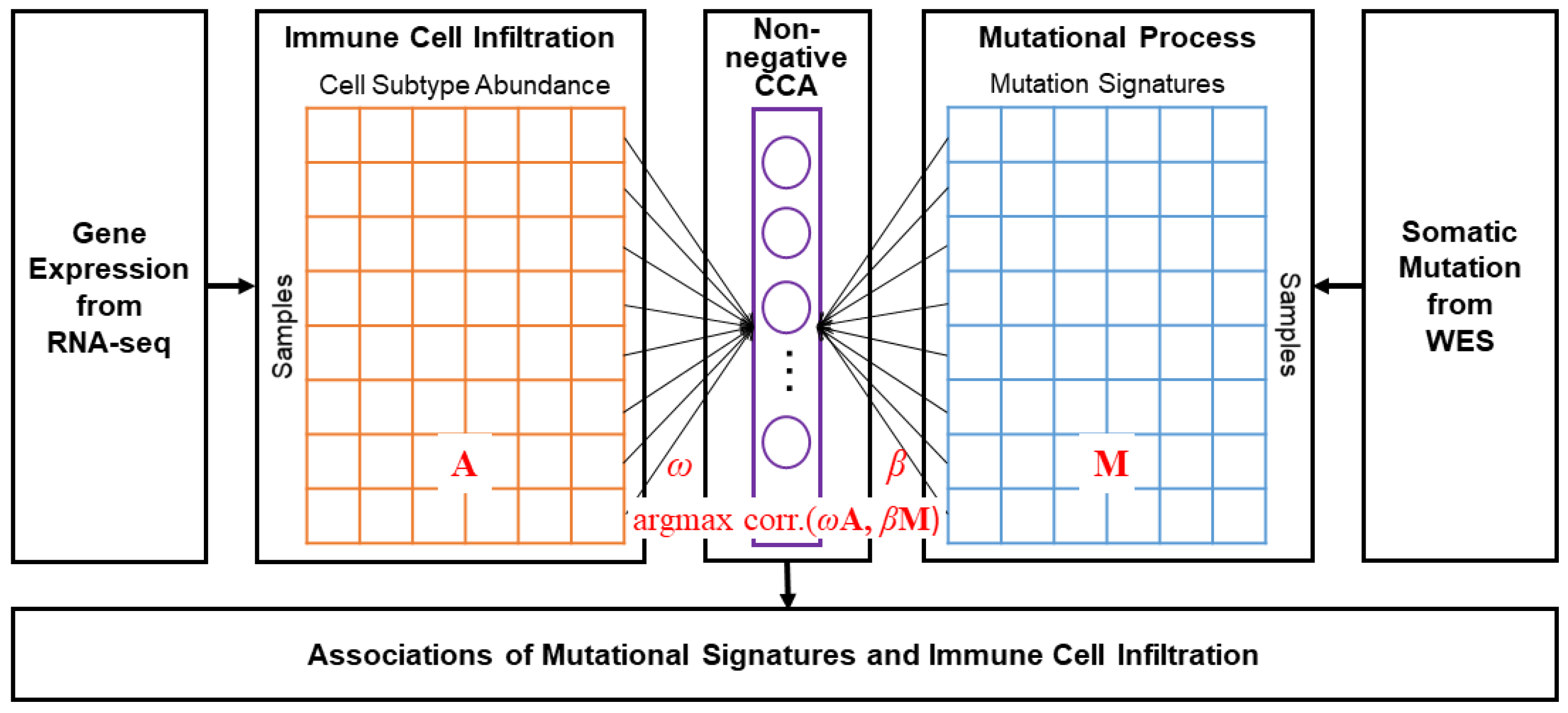
2.3. Non-Negative CCA and Estimation of Coefficients
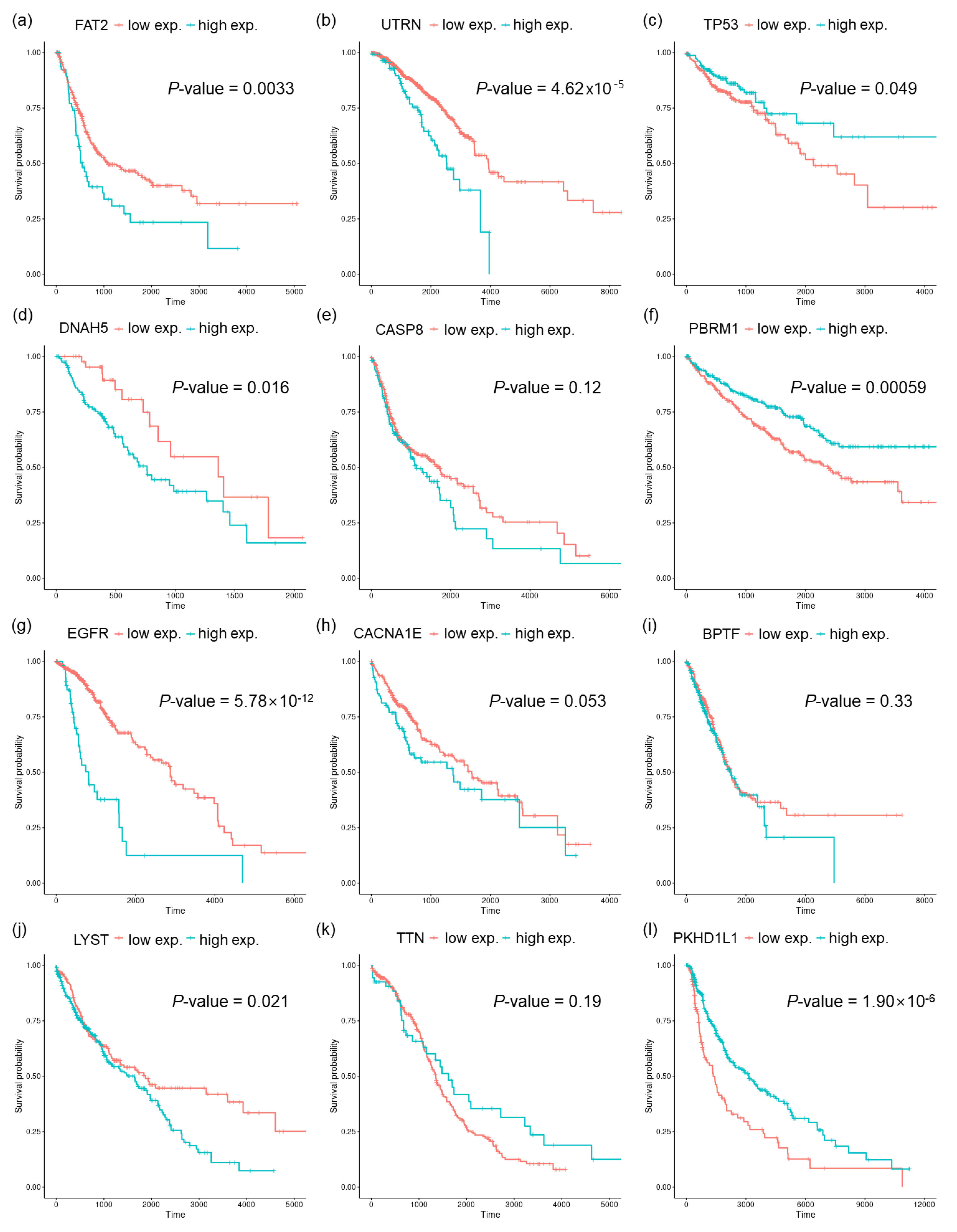
2.4. Finding Genes Correlated to Mutational Signatures and Survival Analysis
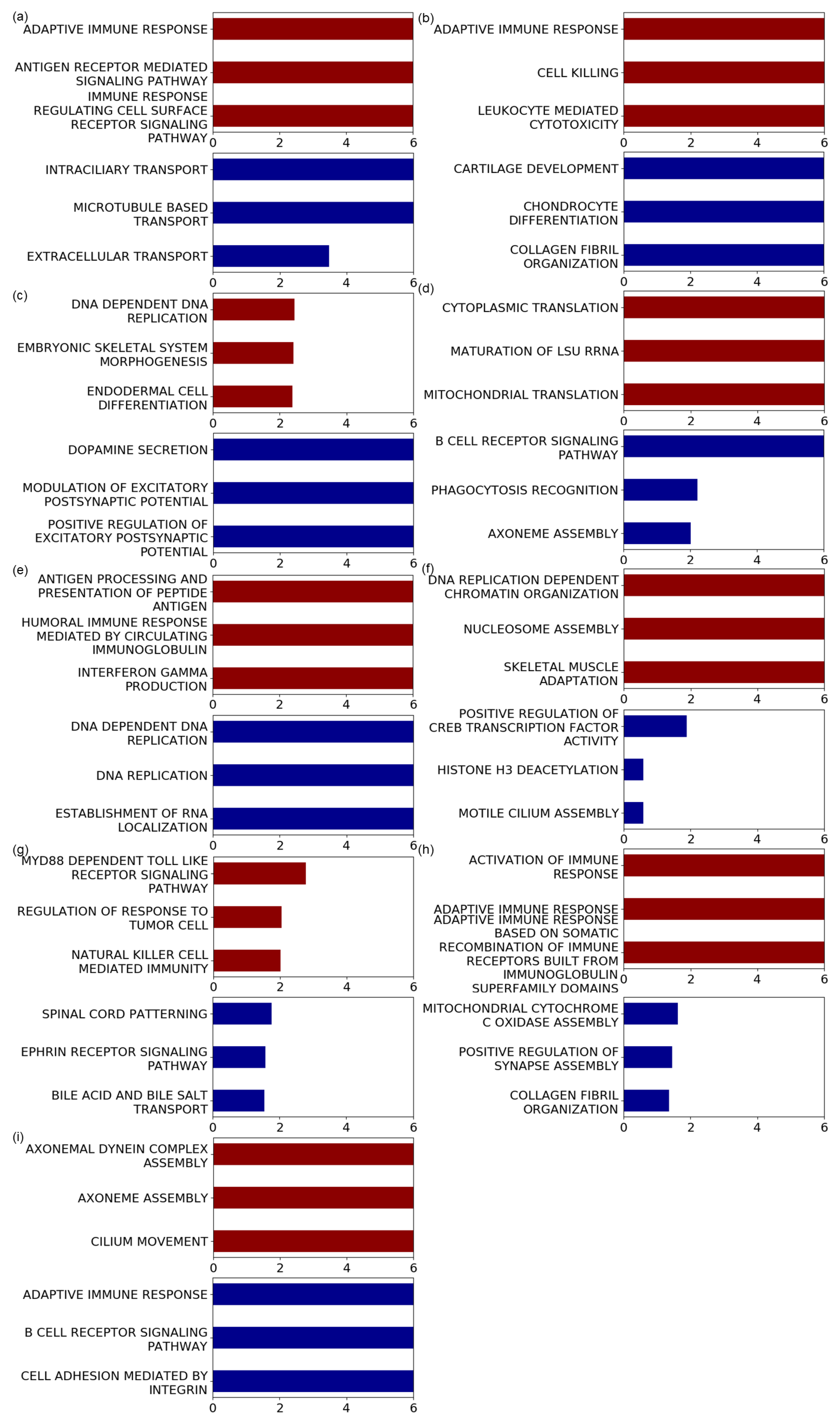
2.5. Geneset Enrichment Analysis Using a Pre-Ranked Gene List
3. Results
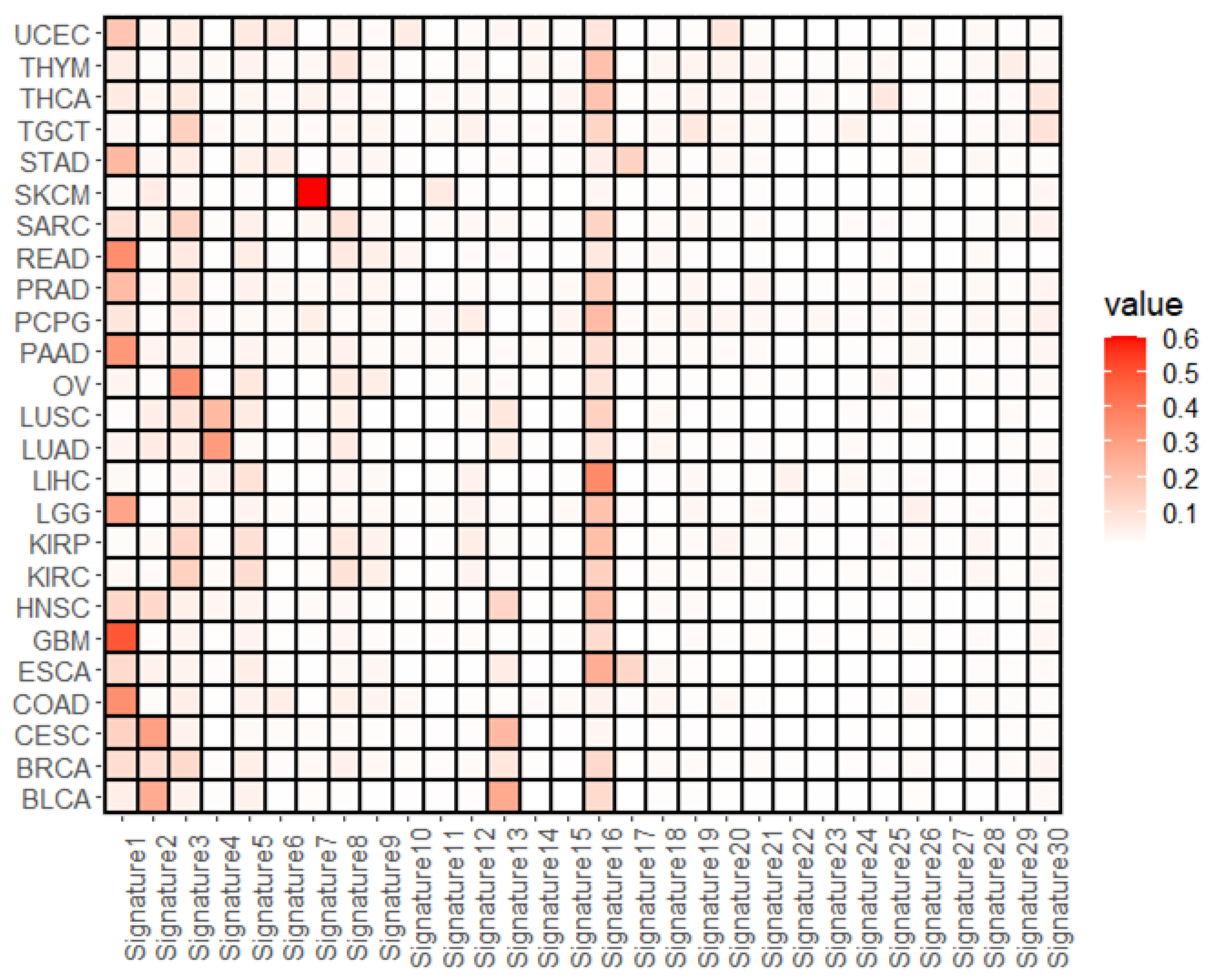
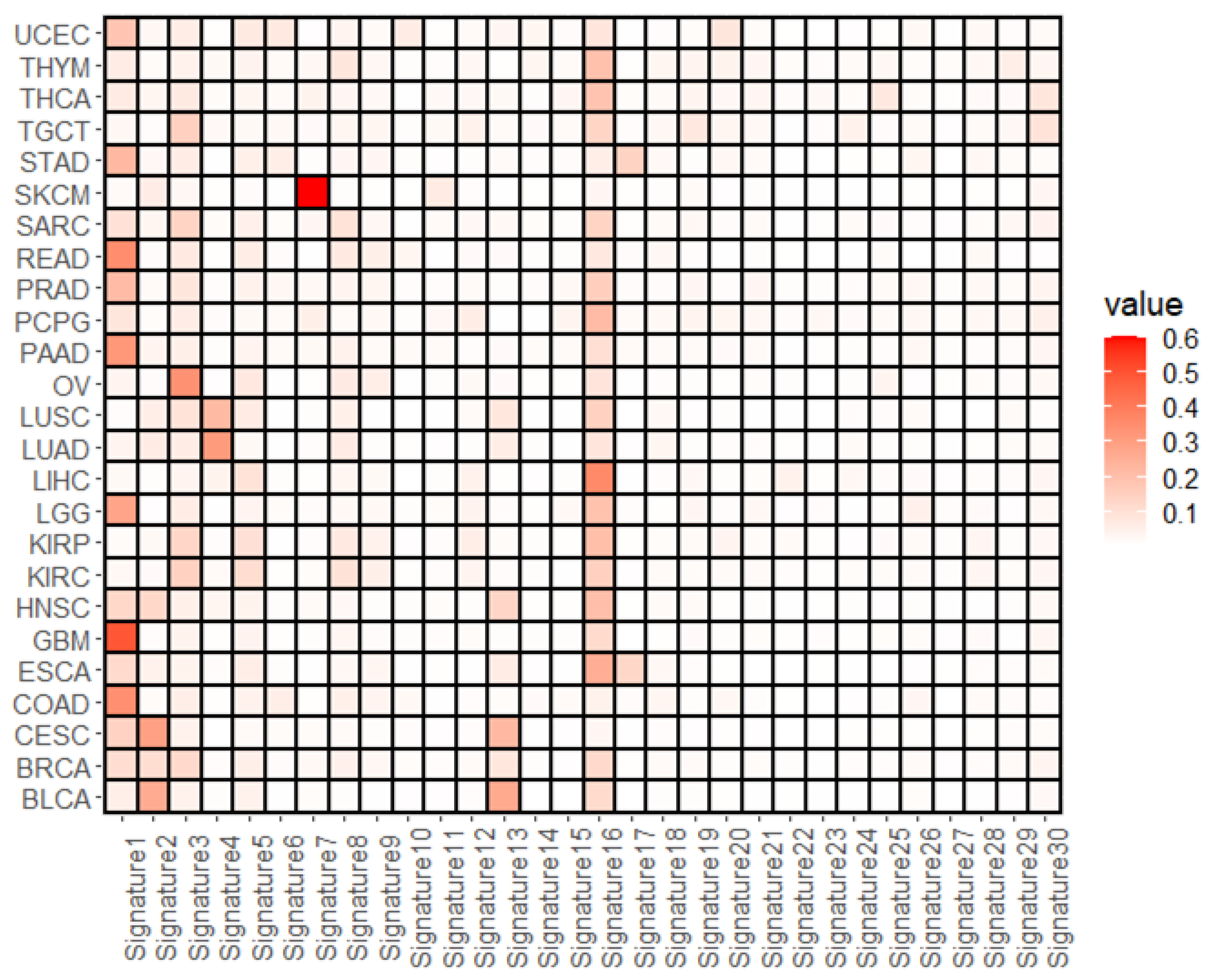
3.1. Overall Representation of Mutational Signatures
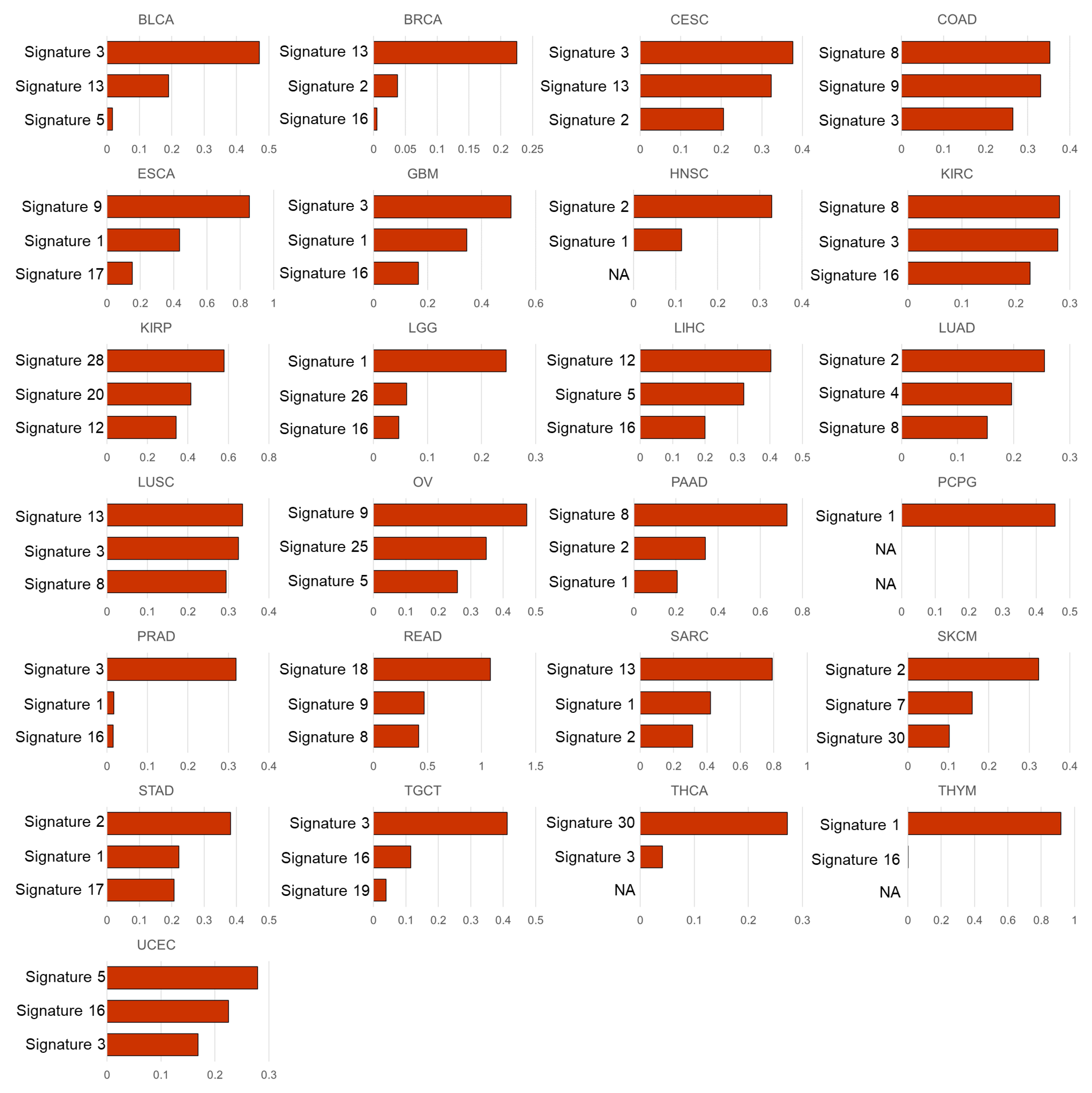
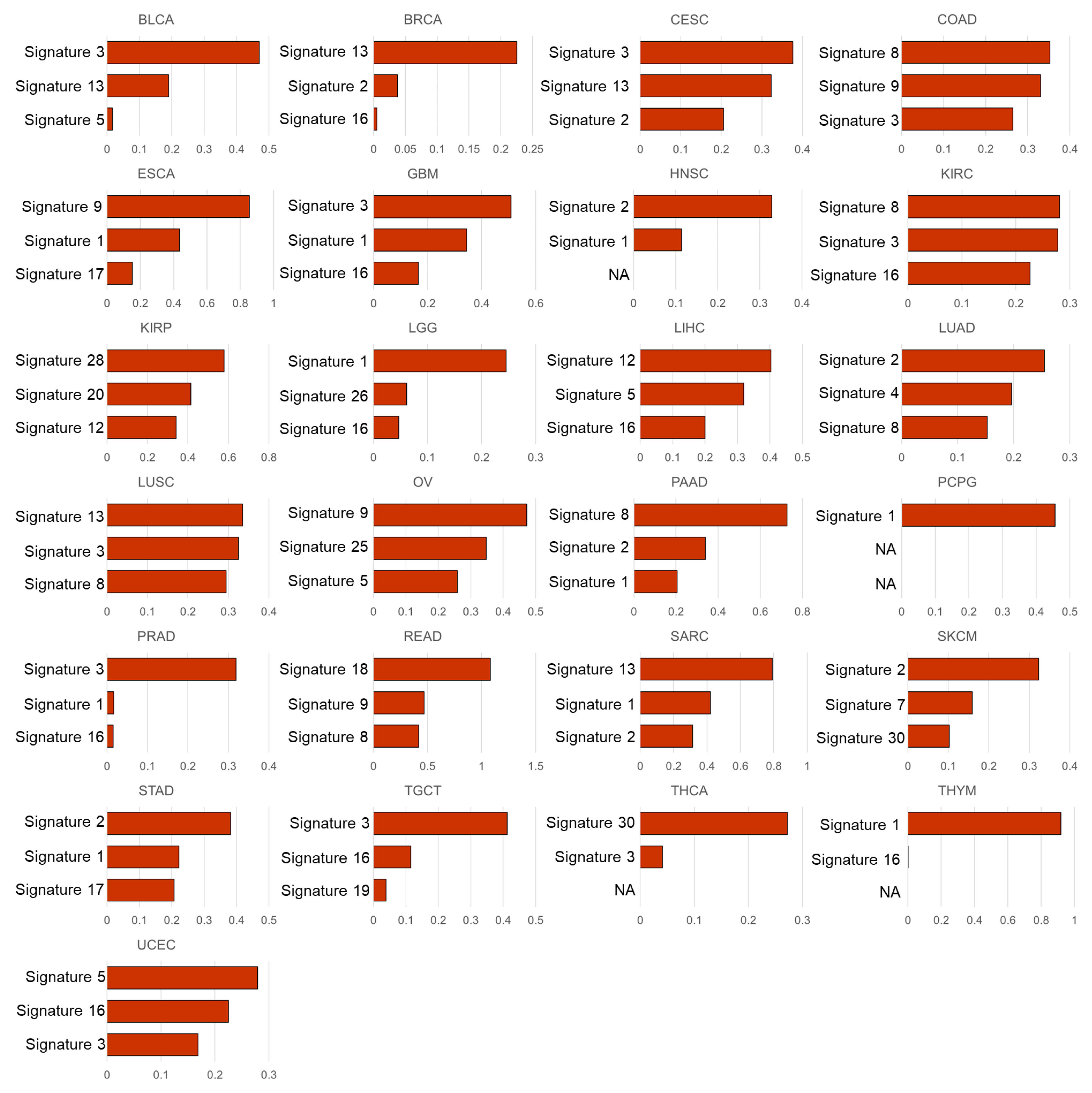
3.2. Mutational Signatures Associated with the Cell Subtype Composition
3.3. Genesets Associated with the Identified Mutational Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poon, S.L.; McPherson, J.R.; Tan, P.; Teh, B.T.; Rozen, S.G. Mutation signatures of carcinogen exposure: Genome-wide detection and new opportunities for cancer prevention. Genome Med. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rideout, W.M., III; Coetzee, G.A.; Olumi, A.F.; Jones, P.A. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990, 249, 1288–1290. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Cervantes-Gracia, K.; Gramalla-Schmitz, A.; Weischedel, J.; Chahwan, R. APOBECs orchestrate genomic and epigenomic editing across health and disease. Trends Genet. 2021, 37, 1028–1043. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.A.; Leiserson, M.D.; Moorjani, P.; Sharan, R.; Wojtowicz, D.; Przytycka, T.M. Mutational signatures: From methods to mechanisms. Annu. Rev. Biomed. Data Sci. 2021, 4, 189–206. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Van Hoeck, A.; Tjoonk, N.H.; van Boxtel, R.; Cuppen, E. Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer 2019, 19, 457. [Google Scholar] [CrossRef]
- Koh, G.; Degasperi, A.; Zou, X.; Momen, S.; Nik-Zainal, S. Mutational signatures: Emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 2021, 21, 619–637. [Google Scholar] [CrossRef]
- Brady, S.W.; Gout, A.M.; Zhang, J. Therapeutic and prognostic insights from the analysis of cancer mutational signatures. Trends Genet. 2021, 38, 194–208. [Google Scholar] [CrossRef]
- Chen, H.; Chong, W.; Teng, C.; Yao, Y.; Wang, X.; Li, X. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci. 2019, 110, 2348. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jia, M.; He, Z.; Liu, X.S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 2018, 37, 3924–3936. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Middlebrooks, C.D.; Banday, A.R.; Matsuda, K.; Udquim, K.I.; Onabajo, O.O.; Paquin, A.; Figueroa, J.D.; Zhu, B.; Koutros, S.; Kubo, M.; et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat. Genet. 2016, 48, 1330–1338. [Google Scholar] [CrossRef]
- Härdle, W.K.; Simar, L. Canonical correlation analysis. In Applied Multivariate Statistical Analysis; Springer: Berlin/Heidelberg, Germany, 2015; pp. 443–454. [Google Scholar]
- Luo, Y.; Tao, D.; Ramamohanarao, K.; Xu, C.; Wen, Y. Tensor canonical correlation analysis for multi-view dimension reduction. IEEE Trans. Knowl. Data Eng. 2015, 27, 3111–3124. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Liu, W.; Liu, W.; Tao, D. A survey on canonical correlation analysis. IEEE Trans. Knowl. Data Eng. 2019, 33, 2349–2368. [Google Scholar] [CrossRef]
- Sun, L.; Ji, S.; Ye, J. Canonical correlation analysis for multilabel classification: A least-squares formulation, extensions, and analysis. IEEE Trans. Pattern Anal. Mach. Intell. 2010, 33, 194–200. [Google Scholar]
- Shen, C.; Sun, M.; Tang, M.; Priebe, C.E. Generalized canonical correlation analysis for classification. J. Multivar. Anal. 2014, 130, 310–322. [Google Scholar] [CrossRef]
- Chaudhuri, K.; Kakade, S.M.; Livescu, K.; Sridharan, K. Multi-view clustering via canonical correlation analysis. In Proceedings of the 26th Annual International Conference on Machine Learning, Montreal, BC, Canada, 14–18 June 2009; pp. 129–136. [Google Scholar]
- Rhee, J.K.; Joung, J.G.; Chang, J.H.; Fei, Z.; Zhang, B.T. Identification of cell cycle-related regulatory motifs using a kernel canonical correlation analysis. BMC Genom. Biomed. Cent. 2009, 10, S29. [Google Scholar] [CrossRef] [Green Version]
- Soneson, C.; Lilljebjörn, H.; Fioretos, T.; Fontes, M. Integrative analysis of gene expression and copy number alterations using canonical correlation analysis. BMC Bioinform. 2010, 11, 191. [Google Scholar] [CrossRef] [Green Version]
- Rodosthenous, T.; Shahrezaei, V.; Evangelou, M. Integrating multi-OMICS data through sparse canonical correlation analysis for the prediction of complex traits: A comparison study. Bioinformatics 2020, 36, 4616–4625. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Zhang, X.; Lan, L.; Huang, X.; Luo, Z. Nonnegative constrained graph based canonical correlation analysis for multi-view feature learning. Neural Process. Lett. 2019, 50, 1215–1240. [Google Scholar] [CrossRef]
- Sigg, C.; Fischer, B.; Ommer, B.; Roth, V.; Buhmann, J. Nonnegative CCA for audiovisual source separation. In Proceedings of the 2007 IEEE Workshop on Machine Learning for Signal Processing, Thessaloniki, Greece, 27–29 August 2007; pp. 253–258. [Google Scholar]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Pachter, L. Models for transcript quantification from RNA-Seq. arXiv 2011, arXiv:1104.3889. [Google Scholar]
- Zhao, Y.; Li, M.C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M. TPM, FPKM, or normalized counts? A comparative study of quantification measures for the analysis of RNA-seq data from the NCI patient-derived models repository. J. Transl. Med. 2021, 19, 1–15. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mutational Signatures v2. Available online: https://cancer.sanger.ac.uk/signatures/signatures_v2/ (accessed on 31 March 2015).
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Mackey, L. Deflation methods for sparse PCA. In Proceedings of the 21st International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 8–10 December 2008; pp. 1017–1024. [Google Scholar]
- Uurtio, V.; Monteiro, J.M.; Kandola, J.; Shawe-Taylor, J.; Fernandez-Reyes, D.; Rousu, J. A tutorial on canonical correlation methods. ACM Comput. Surv. 2017, 50, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Vía, J.; Santamaría, I.; Pérez, J. A robust RLS algorithm for adaptive canonical correlation analysis. In Proceedings of the (ICASSP’05) IEEE International Conference on Acoustics, Speech, and Signal Processing, Philadelphia, PA, USA, 23 March 2005; Volume 4, pp. iv365–iv368. [Google Scholar]
- Fischer, B.; Roth, V.; Buhmann, J.M. Time-series alignment by non-negative multiple generalized canonical correlation analysis. BMC Bioinform. Biomed. Cent. 2007, 8, S4. [Google Scholar]
- Sun, L.; Ji, S.; Ye, J. A least squares formulation for canonical correlation analysis. In Proceedings of the 25th International Conference on Machine Learning, Helsinki, Finland, 5–9 July 2008; pp. 1024–1031. [Google Scholar]
- Hothorn, T.; Lausen, B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Póti, Á.; Gyergyák, H.; Németh, E.; Rusz, O.; Tóth, S.; Kovácsházi, C.; Chen, D.; Szikriszt, B.; Spisák, S.; Takeda, S.; et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Devarajan, K. Nonnegative matrix factorization: An analytical and interpretive tool in computational biology. PLoS Comput. Biol. 2008, 4, e1000029. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Michailidis, G. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics 2016, 32, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Boutros, P.C. Optimization and expansion of non-negative matrix factorization. BMC Bioinform. 2020, 21, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, i436–i445. [Google Scholar] [CrossRef] [PubMed]
- DiMarco, A.V.; Qin, X.; McKinney, B.J.; Garcia, N.M.G.; van Alsten, S.C.; Mendes, E.A.; Force, J.; Hanks, B.A.; Troester, M.A.; Owzar, K.; et al. APOBEC Mutagenesis Inhibits Breast Cancer Growth through Induction of T cell–Mediated Antitumor Immune Responses. Cancer Immunol. Res. 2022, 10, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Rosenthal, R.; Kanu, N.; McGranahan, N.; Bartek, J.; Quezada, S.; Hare, J.; Harris, R.; Swanton, C. Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann. Oncol. 2018, 29, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Faden, D.L.; Ding, F.; Lin, Y.; Zhai, S.; Kuo, F.; Chan, T.A.; Morris, L.G.; Ferris, R.L. APOBEC mutagenesis is tightly linked to the immune landscape and immunotherapy biomarkers in head and neck squamous cell carcinoma. Oral Oncol. 2019, 96, 140–147. [Google Scholar] [CrossRef]
- Li, C.; Egloff, A.M.; Sen, M.; Grandis, J.R.; Johnson, D.E. Caspase-8 mutations in head and neck cancer confer resistance to death receptor-mediated apoptosis and enhance migration, invasion, and tumor growth. Mol. Oncol. 2014, 8, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Ghanekar, Y.; Sadasivam, S. In silico analysis reveals a shared immune signature in CASP8-mutated carcinomas with varying correlations to prognosis. PeerJ 2019, 7, e6402. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Lu, J.J.; Guo, W.; Yu, W.; Wang, Q.; Tang, R.; Tang, Z.; Xiao, Y.; Li, Z.; Sun, W.; et al. BPTF promotes tumor growth and predicts poor prognosis in lung adenocarcinomas. Oncotarget 2015, 6, 33878. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Liu, D.; Li, X.; Dai, S. BPTF biomarker correlates with poor survival in human NSCLC. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 102–107. [Google Scholar]
- Mayes, K.; Alkhatib, S.G.; Peterson, K.; Alhazmi, A.; Song, C.; Chan, V.; Blevins, T.; Roberts, M.; Dumur, C.I.; Wang, X.Y.; et al. BPTF Depletion Enhances T-cell–Mediated Antitumor Immunity. Cancer Res. 2016, 76, 6183–6192. [Google Scholar] [CrossRef] [Green Version]
- Miao, Z.; Ali, A.; Hu, L.; Zhao, F.; Yin, C.; Chen, C.; Yang, T.; Qian, A. Microtubule actin cross-linking factor 1, a novel potential target in cancer. Cancer Sci. 2017, 108, 1953–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Hu, K.; Zeng, Z.; Xu, C.; Lv, J.; Lin, Z.; Wen, B. Expression and Clinical Significance of Microtubule-Actin Cross-Linking Factor 1 in Serous Ovarian Cancer. Recent Patents-Anti-Cancer Drug Discov. 2021, 16, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Warrell, J.; Li, S.; McGillivray, P.D.; Meyerson, W.; Salichos, L.; Harmanci, A.; Martinez-Fundichely, A.; Chan, C.W.; Nielsen, M.M.; et al. Passenger mutations in more than 2,500 cancer genomes: Overall molecular functional impact and consequences. Cell 2020, 180, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Temko, D.; Tomlinson, I.P.; Severini, S.; Schuster-Böckler, B.; Graham, T.A. The effects of mutational processes and selection on driver mutations across cancer types. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.K.; Aichmüller, C.; Schulze, M.; Hlevnjak, M.; Elgaafary, S.; Lichter, P.; Zapatka, M. Association of mutation signature effectuating processes with mutation hotspots in driver genes and non-coding regions. Nat. Commun. 2022, 13, 1–13. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
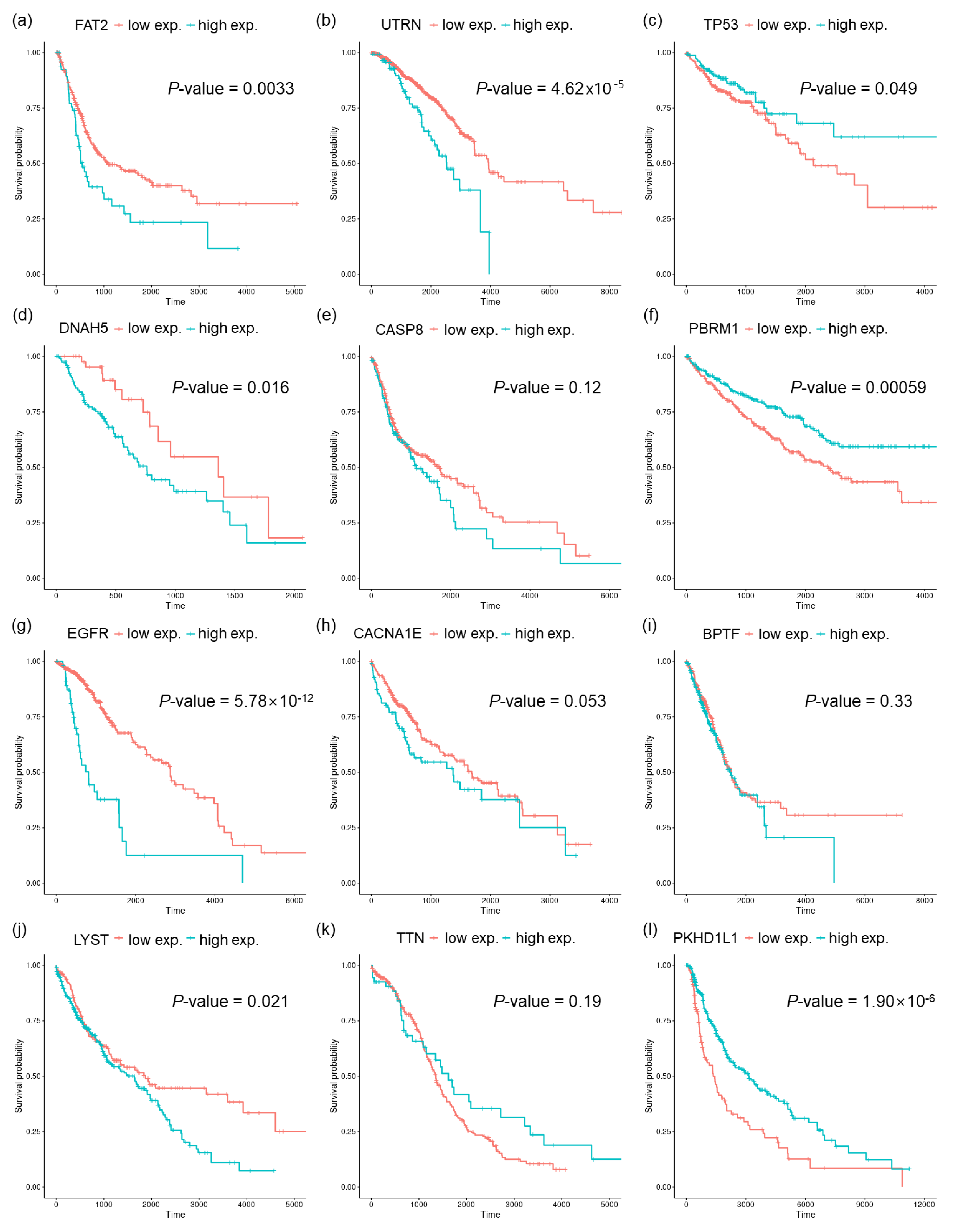{kind=link}
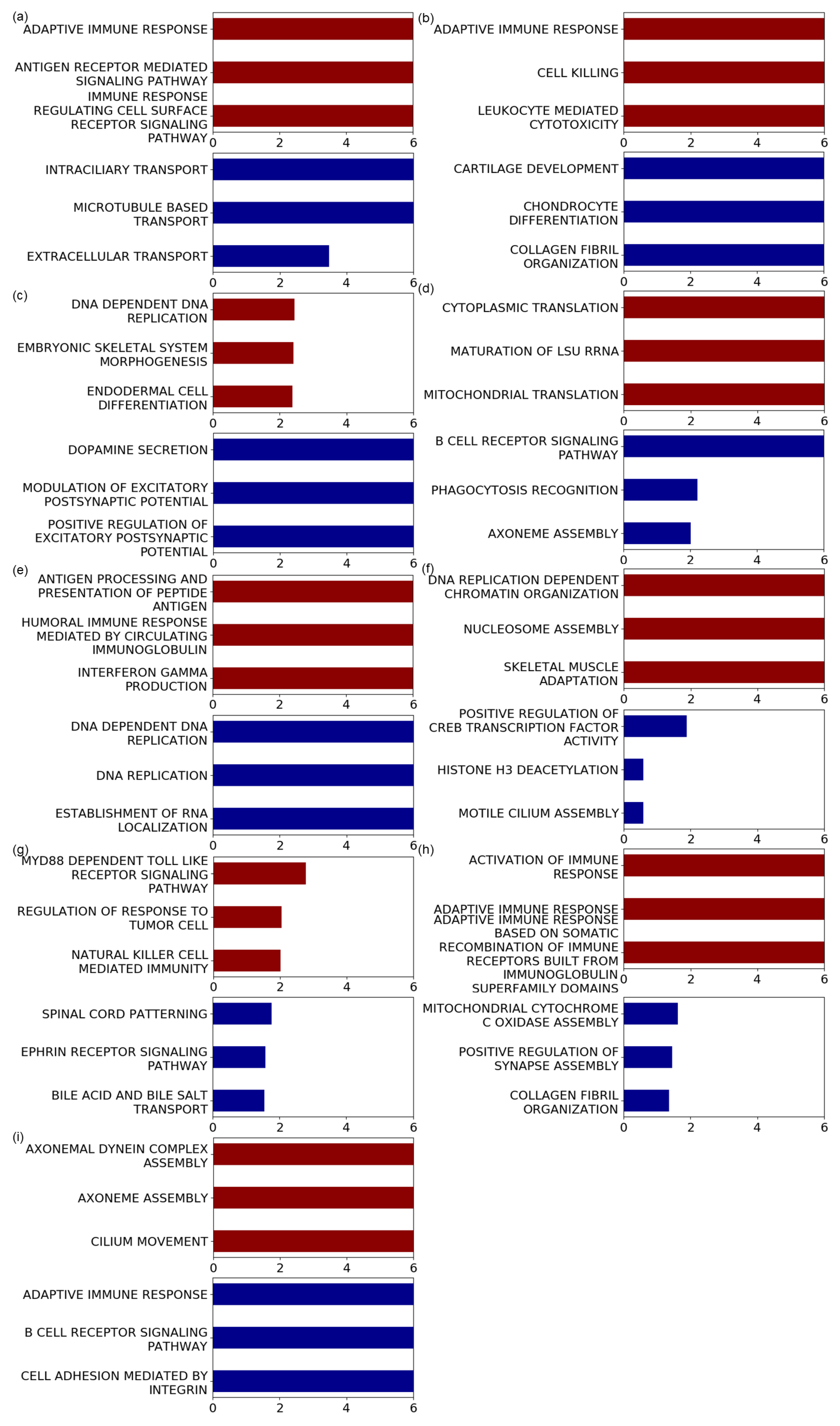{kind=link}
| Tumor Type | Samples |
|---|---|
| Bladder urothelial carcinoma (BLCA) | 409 |
| Breast invasive carcinoma (BRCA) | 974 |
| Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) | 287 |
| Colon adenocarcinoma (COAD) | 397 |
| Esophageal carcinoma (ESCA) | 161 |
| Glioblastoma multiforme (GBM) | 150 |
| Head and neck squamous cell carcinoma (HNSC) | 494 |
| Kidney renal clear cell carcinoma (KIRC) | 333 |
| Kidney renal papillary cell carcinoma (KIRP) | 279 |
| Brain lower grade glioma (LGG) | 504 |
| Liver hepatocellular carcinoma (LIHC) | 360 |
| Lung adenocarcinoma (LUAD) | 510 |
| Lung squamous cell carcinoma (LUSC) | 490 |
| Ovarian serous cystadenocarcinoma (OV) | 273 |
| Pancreatic adenocarcinoma (PAAD) | 170 |
| Pheochromocytoma and paraganglioma (PCPG) | 179 |
| Prostate adenocarcinoma (PRAD) | 493 |
| Rectum adenocarcinoma (READ) | 134 |
| Sarcoma (SARC) | 236 |
| Skin cutaneous melanoma (SKCM) | 466 |
| Stomach adenocarcinoma (STAD) | 373 |
| Testicular germ cell tumors (TGCT) | 145 |
| Thyroid carcinoma (THCA) | 488 |
| Thymoma (THYM) | 119 |
| Uterine corpus endometrial carcinoma (UCEC) | 527 |
| Mutational Signature | Average |
|---|---|
| Signature 1 | 0.1250 |
| Signature 2 | 0.0605 |
| Signature 3 | 0.0801 |
| Signature 4 | 0.0392 |
| Signature 5 | 0.0472 |
| Signature 6 | 0.0174 |
| Signature 7 | 0.0458 |
| Signature 8 | 0.0403 |
| Signature 9 | 0.0218 |
| Signature 10 | 0.0106 |
| Signature 11 | 0.0120 |
| Signature 12 | 0.0203 |
| Signature 13 | 0.0519 |
| Signature 14 | 0.0083 |
| Signature 15 | 0.0102 |
| Signature 16 | 0.1335 |
| Signature 17 | 0.0135 |
| Signature 18 | 0.0169 |
| Signature 19 | 0.0176 |
| Signature 20 | 0.0167 |
| Signature 21 | 0.0146 |
| Signature 22 | 0.0069 |
| Signature 23 | 0.0050 |
| Signature 24 | 0.0095 |
| Signature 25 | 0.0134 |
| Signature 26 | 0.0175 |
| Signature 27 | 0.0042 |
| Signature 28 | 0.0136 |
| Signature 29 | 0.0125 |
| Signature 30 | 0.0282 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhee, J.-K. Pan-Cancer Analysis for Immune Cell Infiltration and Mutational Signatures Using Non-Negative Canonical Correlation Analysis. Appl. Sci. 2022, 12, 6596. https://doi.org/10.3390/app12136596
Rhee J-K. Pan-Cancer Analysis for Immune Cell Infiltration and Mutational Signatures Using Non-Negative Canonical Correlation Analysis. Applied Sciences. 2022; 12(13):6596. https://doi.org/10.3390/app12136596
Chicago/Turabian StyleRhee, Je-Keun. 2022. "Pan-Cancer Analysis for Immune Cell Infiltration and Mutational Signatures Using Non-Negative Canonical Correlation Analysis" Applied Sciences 12, no. 13: 6596. https://doi.org/10.3390/app12136596
APA StyleRhee, J.-K. (2022). Pan-Cancer Analysis for Immune Cell Infiltration and Mutational Signatures Using Non-Negative Canonical Correlation Analysis. Applied Sciences, 12(13), 6596. https://doi.org/10.3390/app12136596