1. Introduction
Active pharmaceutical ingredients (APIs) are commonly formulated and delivered to patients in the solid dosage forms (tablets, capsules, powders) for reasons of economy, stability, and convenience of intake [
1]. One of the major problems faced during the formulation of drug is its low bioavailability which is mainly reliant on the solubility and permeability of API [
2,
3], and one of the approaches to enhance the physicochemical and pharmacological properties of API without modifying its intrinsic chemical structure is to develop novel solid forms such as cocrystals [
4,
5,
6,
7]. There are extensive reports on cocrystals for the purpose of improving the pharmaceutical properties including dissolution, permeability, bioavailability, stability, photostability, hygroscopicity, and compressibility [
8,
9].
Cocrystals are much of interest in industrial application as well as academic research because they offer various opportunities for intellectual property rights in respect of the development of new solid forms [
10]. Furthermore, the latest Food and Drug Administration (FDA) guidance on pharmaceutical cocrystals, which recognizes cocrystals as drug substances, provides an excellent opportunity for the pharmaceutical industry to develop commercial products of cocrystals [
11,
12]. According to FDA, cocrystals are “Crystalline materials composed of two or more molecules within the same crystal lattice” [
13]. Pharmaceutical cocrystals, a subclass of cocrystals, are stoichiometric molecular complexes composed of APIs and pharmaceutically acceptable coformers held together by non-covalent interactions such as hydrogen bonding within the same crystal lattice [
14]. Coformers for pharmaceutical cocrystallization should be from the FDA’s list of Everything Added to Food in the United States (EAFUS) or from the Generally Recognized as Safe list (GRAS), as they should have no adverse or pharmacological toxic effects [
1]. The list of acceptable co-formers, in principle, is likely to at least extend into the hundreds, which means that screening of suitable coformers for an API is the most crucial and challenging step in cocrystal development [
15]. Since experimental determination of cocrystals is time-consuming, costly, and labor-intensive, it is valuable to develop complementary tools that can reduce the list of coformers by predicting which coformers are likely to form cocrystals [
16].
Various knowledge-based and computational approaches have been used in the literature to predict cocrystal formation. Supramolecular synthesis introduced by Desiraju is a well-known approach to rationalize the possibility of cocrystal formation [
17,
18,
19]. A common strategy in this method is to first identify the crystal structure of the target molecule and investigate coformers with a desired functional group which can form intermolecular interactions (mainly hydrogen bonding) between the target molecule and coformers [
15]. Knowledge of synthons allows the selection of potential coformers and predicts the interaction outcomes, but there is no guarantee that cocrystals with predicted structures would form. Statistical analysis of cocrystal data from the Cambridge Structural Database (CSD) where more than one million crystal structures of small molecules are available, allows researchers to apply virtual screening techniques to find suitable cocrystal-forming pairs [
20]. Galek et al. introduced hydrogen-bond propensity as a predictive tool and determined the likelihood of co-crystal formation [
15,
21]. Fábián analyzed the possibility of cocrystal formation by correlating the different descriptors such as polarity and molecular shape [
22]. Cocrystal design can also be based on computational approaches, including the use of the
pK value [
23,
24], lattice energy calculation [
25,
26,
27,
28], molecular electrostatic potential surfaces (MEPS) calculation [
29,
30,
31,
32], Hansen solubility parameters calculation [
33,
34] and Conductor like screening Model for Real solvents (COSMO-RS) based enthalpy of mixing calculation [
35,
36,
37,
38].
In recent years, machine-learning (ML) has emerged as promising tool for data-driven predictions in pharmaceutical research, such as quantitative structure-activity/property relationships (QSAR/QSPR), drug-drug interactions, drug repurposing and pharmacogenomics [
39]. In the area of pharmaceutical cocrystal research, Rama Krishna et al. applied artificial neural network to predict three solid-state properties of cocrystals, including melting temperature, lattice energy, and crystal density [
40]. Przybylek et al. developed cocrystal screening models based on simple classification regression and Multivariate Adaptive Regression Splines (MARSplines) algorithm using molecular descriptors for phenolic acid coformers and dicarboxylic acid coformers, respectively [
41]. Wicker et al. created a predictive model, that can classify a pair of coformers as a possible cocrystal or not, using a support vector machine (SVM) and simple descriptors of coformer molecules to guide the selection of coformers in the discovery of new cocrystals [
16]. Devogelaer and co-workers introduced a comprehensive approach to study cocrystallization using network science and linkage prediction algorithms and constructed a data-driven co-crystal prediction tool with co-crystal data extracted from the CSD [
42]. Wang et al. also used a data set with co-crystal data available in the CSD and ultimately developed a machine learning model using different model types and molecular fingerprints that can be used to select appropriate coformers for a target molecule [
43]. The above existing studies have shown successful results, but they have a common limitation that they only compared model performance (e.g., accuracy) without investigating features (i.e., descriptors) importance.
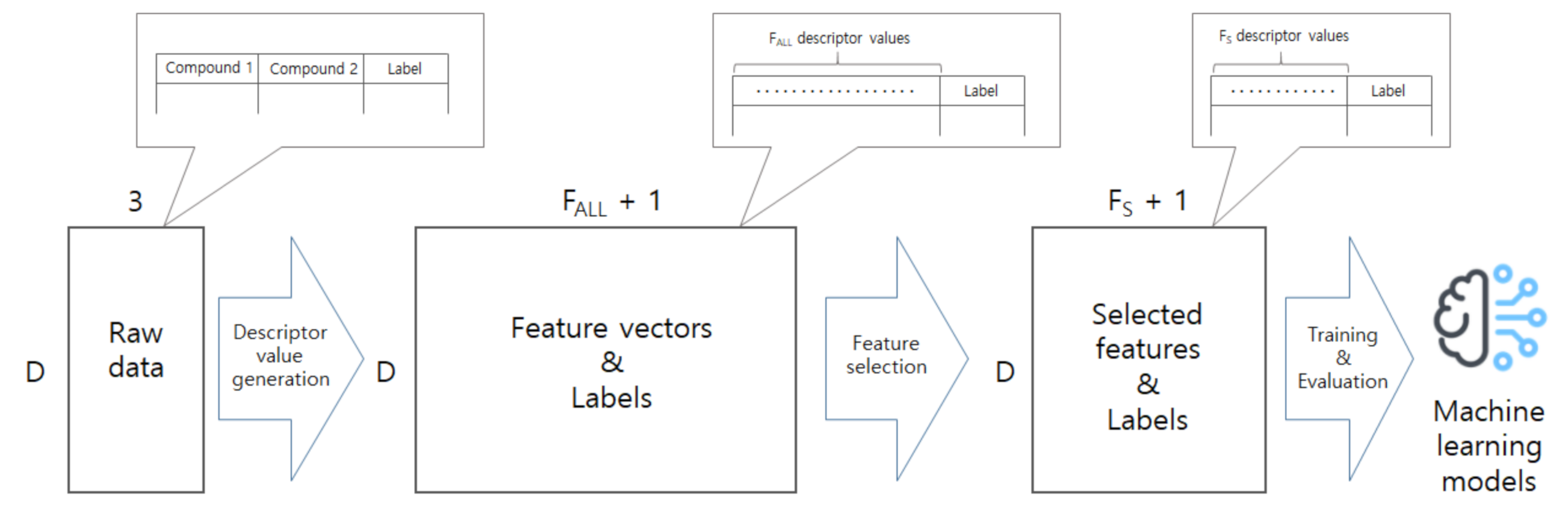
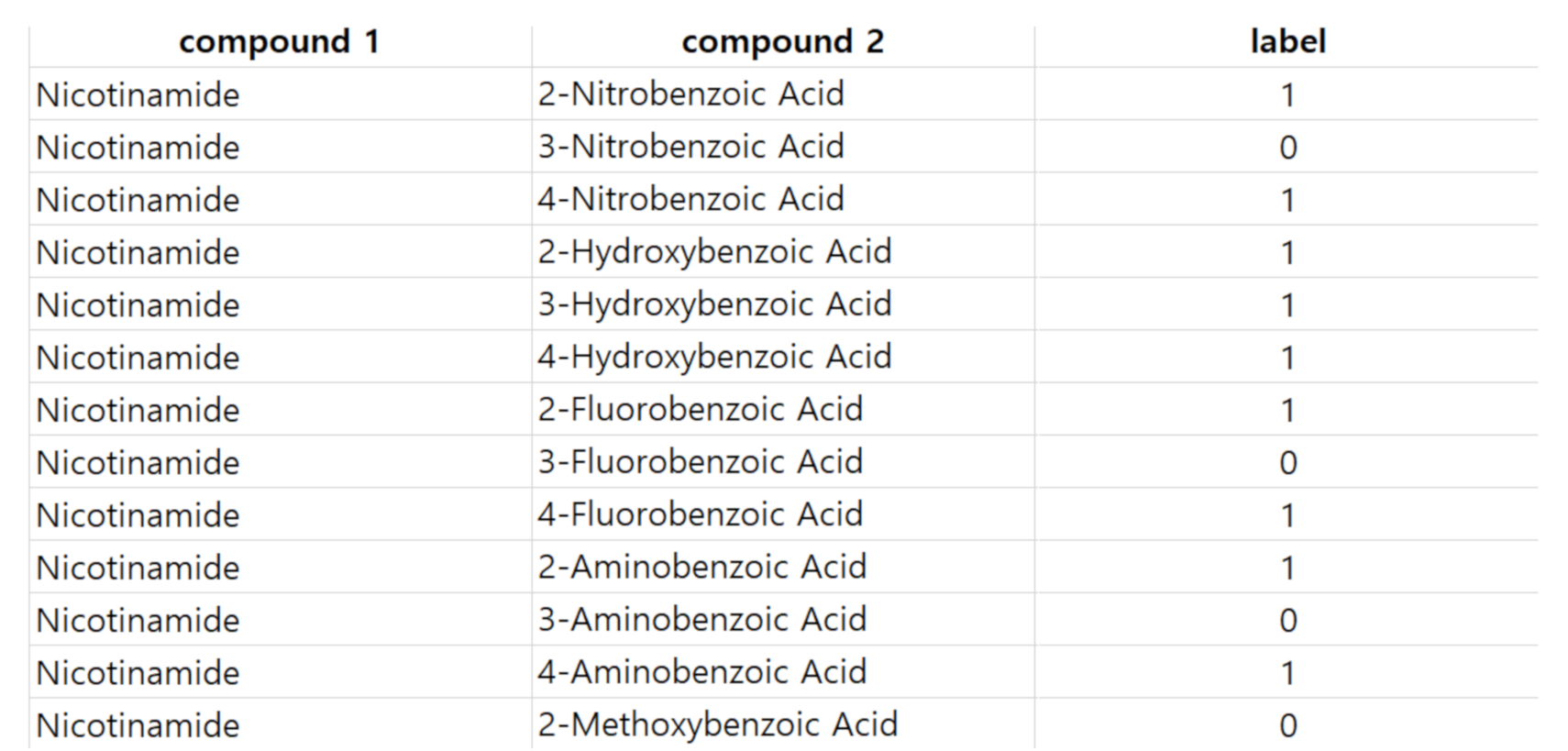
In this work, we develop a model to predict co-crystal formation of API molecules. We use Mordred [
44], one of widely-used descriptor calculators, to extract descriptor values from simplified molecular-input line-entry system (SMILES) strings of API and coformers compounds. There are several other tools or molecular descriptor-calculators used in cheminformatics such as PaDEL [
45], PyDPI [
46], Rcpi [
47], Dragon [
48], BlueDesc (
http://www.ra.cs.uni-tuebingen.de/software/bluedesc) and cinfony [
49]; PaDEL descriptor-calculator is the most well-known tool and provides 1875 descriptors, and cinfony is a collection or a wrapper of other libraries such as Open Babel [
50], RDKit (
http://www.rdkit.org), and Chemistry Development Kit (CDK) [
51]. We chose to use Mordred and used the descriptor values as features and compared different machine learning models through experimental results using our collected data. Our contributions can be summarized as follows. First, we not only extract descriptor values of compound pairs, but also investigate which descriptors are more important, and show that we can achieve good performance even if we use only a small subset of the descriptors. Second, we compare machine learning models through experiments and find that artificial neural networks (ANN) achieve the best performance. Third, we make our dataset available for free through the online website (
http://ant.sch.ac.kr/) so that many other researchers can use the dataset as a benchmark. We believe that this study will advance the field of cocrystal formation prediction, and our dataset will help other researchers to easily develop better models.
4. Discussion
Although the models performed best when we use all features, the feature selection algorithms showed their potential using only half of the features (e.g.,
or 900) gave comparable results. One might want to see what features were valuable than others.
Table 8 shows lists of best and worst features obtained by K-best algorithm with
= 900, where the scores are ANOVA F-values; the features with larger scores turn out to be more vital than others. Note that the features are grouped in terms of
Module; for example, the best feature ‘Mp’ came from ‘Constitutional’ module of Mordred. Interestingly, many of the best and worst features commonly came together from the ‘Autocorrelation’ module, which computes the Moreau-Broto autocorrelation of the topological structure. Most of the best features of the ‘Autocorrelation’ module are Geary coefficients (e.g., ‘GATS’ series), implying that the spatial correlation is particularly essential in predict cocrystal formation. Especially, Geary coefficients weighted by intrinsic state (e.g., GATS4s, GATS6s), valence electrons (e.g., GATS6dv), or atomic number (e.g., GATS3Z, GATS6Z, GATS7Z, GATS8Z) turned out to be extremely important. On the other hand, most of the worst features of the ‘Autocorrelation’ module, are the Moran coefficient (e.g., MATS series). The Moran coefficient focuses on deviations from the mean whereas, the Geary coefficient focuses on the deviations of individual observation area [
61], so we might say that the deviations of each observation area are more meaningful information for cocrystal prediction. It is consistent with a recent study by Shiquan Sun et al. [
62] that revealed that the Moran coefficient is not very competent in detecting spatial patterns other than simple autocorrelation due to its asymptotic normality for p-value computation.
Many of the best features came from the ‘EState’ module, which generates atom type e-state descriptor values [
63]. This implies that the electrostatic interaction of atoms and their topological environment (connections) within a molecule has a significant impact on cocrystallization. It is in line with the fact that the electrostatic interaction between atoms has been treated importantly in pharmaceutics [
64,
65]. Meanwhile, many worst features came from the ‘BCUT’ module that generates burden matrix weighted by ionization potential (e.g., BCUTi-1l), pauling EN (e.g., BCUTpe-1h), or sigma electrons (e.g., BCUTd-1h, BCUTd-1l). Note that this does not mean that these worst descriptor values are harmful to the outcome, but they only have a smaller contribution than the others to the performance (e.g., accuracy).
Table 9 describes a comparison our work with recent studies. The best accuracy of this study is definitely highest among them; although the three studies used different datasets, we might say that we proved the potential of the ANN model to predict cocrystal formation by experimental results. It should be noted that the feature sources are different between these studies. That is, Jerome G. P. Wicker et al. [
16] used the molecular descriptors (i.e., features) as the model inputs and Jan-Joris Devogelaer et al. [
66] used fingerprint vectors and molecular graphs whereas our work uses the molecular descriptors (i.e., features). We employed feature selection algorithms to find some valuable features and explained how they are related to results of previous studies.


 ,
, 





{kind=link}
{kind=link}
{kind=link}