
Structural Analysis, Phase Stability, Electronic Band Structures, and Electric Transport Types of (Bi2)m(Bi2Te3)n by Density Functional Theory Calculations


Abstract
:1. Introduction
2. Methods and Calculations Details
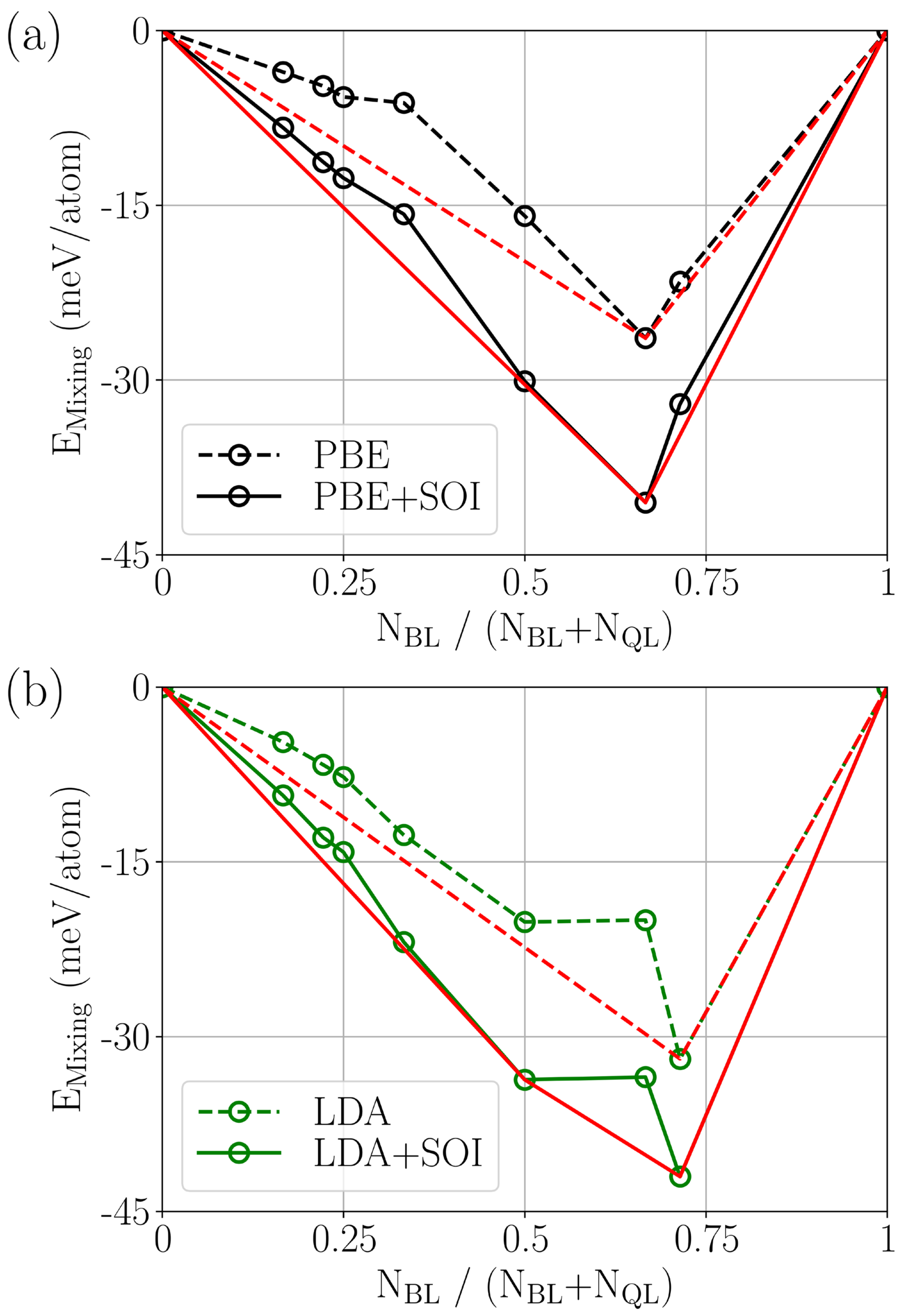
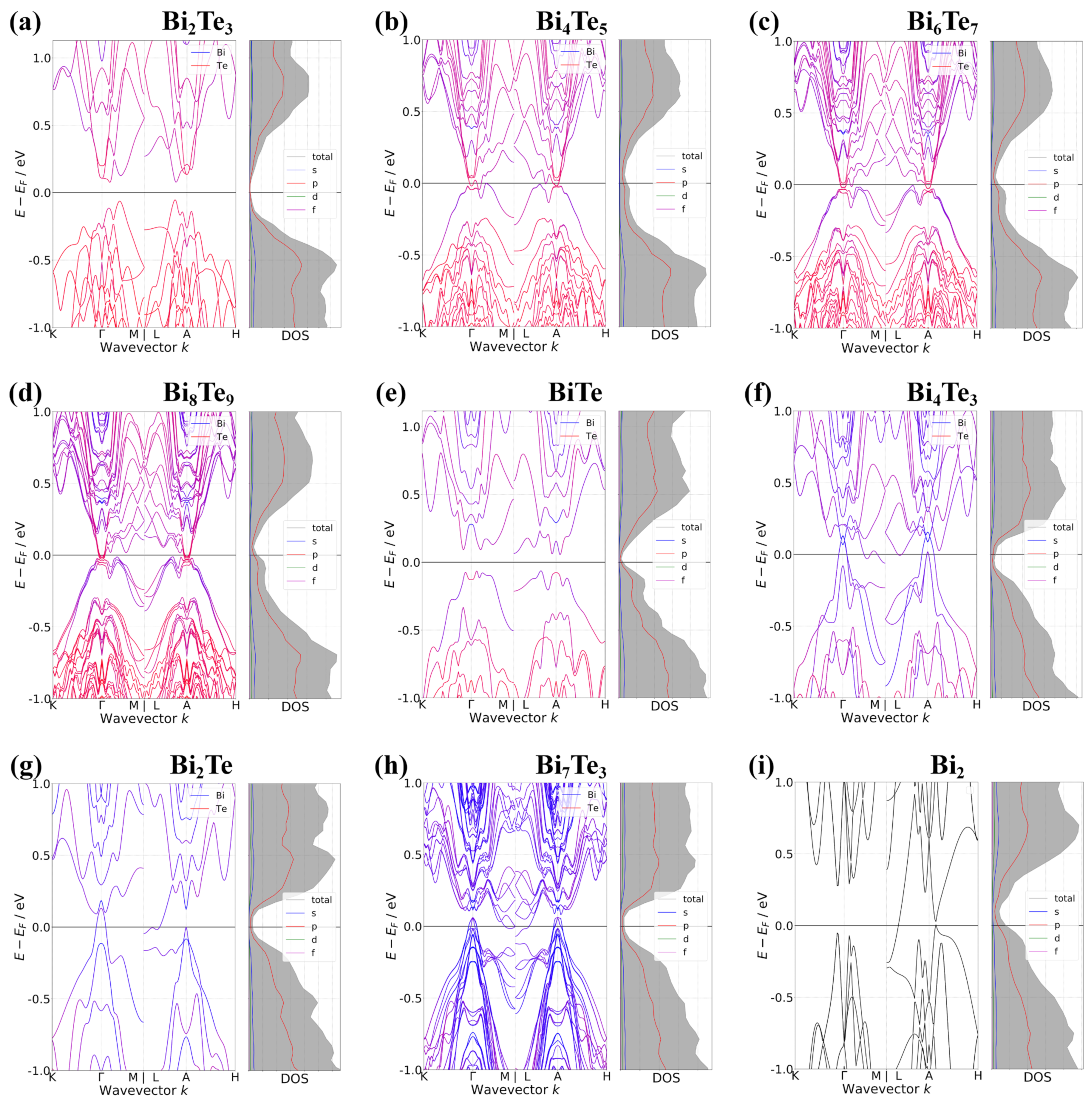
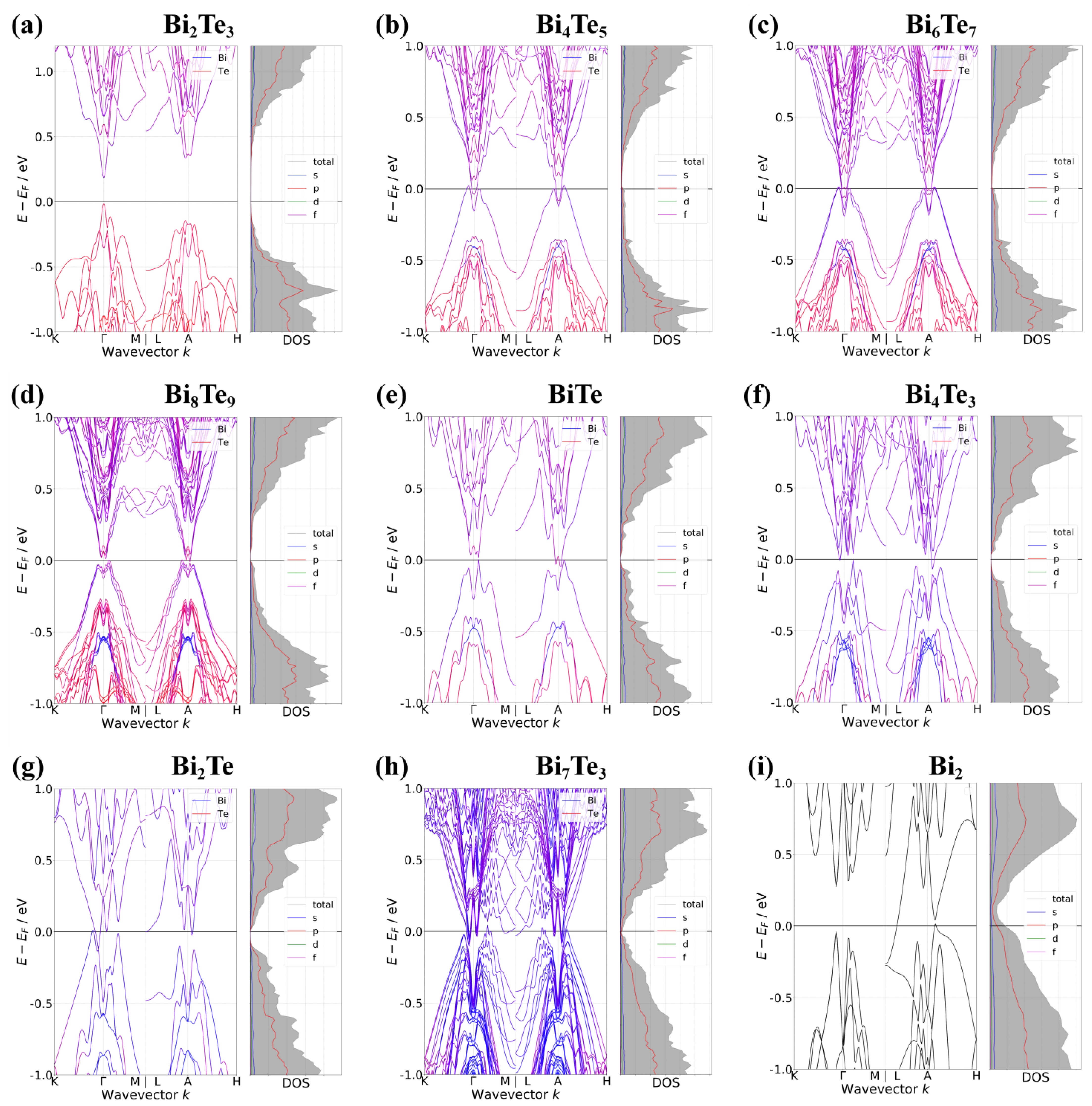
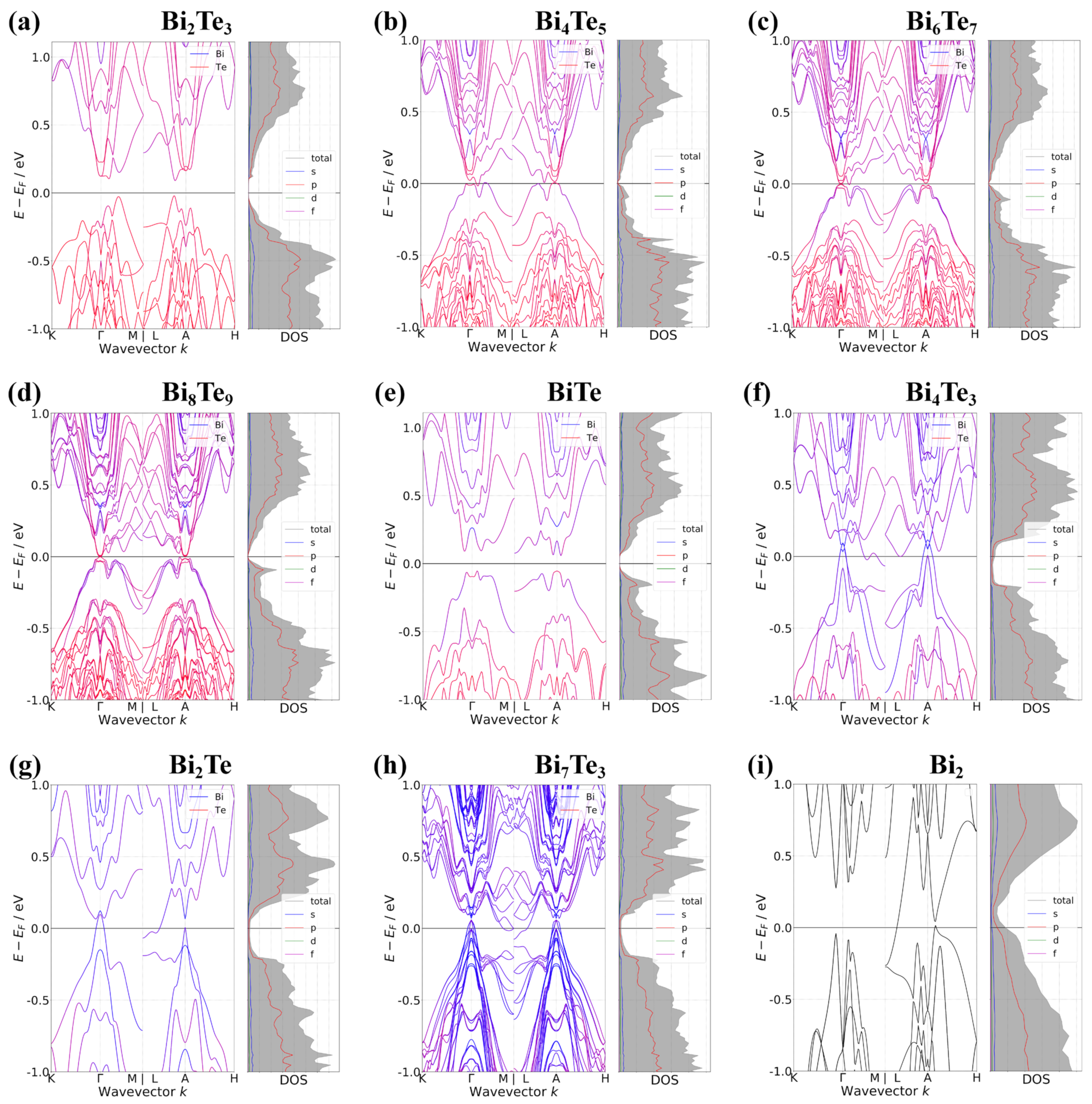
3. Results and Discussions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| Acronym | Meaning |
| BL | bilayer |
| CBM | conduction band minimum |
| DFT | density functional theory |
| DOS | density of states |
| EF | Fermi level |
| EMixing | layer mixing energy |
| direct energy gap (bandgap) | |
| indirect energy gap (bandgap) | |
| EVBM | energy of valence band maximum |
| ECBM | energy of conduction band minimum |
| Exc | exchange–correlation functional |
| reciprocal position of direct energy gap (bandgap) | |
| kVBM | reciprocal position of valence band maximum |
| kCBM | reciprocal position of conduction band minimum |
| LDA | local density approximation |
| LDA + SOI | local density approximation with spin–orbit interaction |
| NAtom | total number of atoms in the supercell |
| NBL | the number of bilayers |
| NQL | the number of quintuple layers |
| PAW | projector augmented wave |
| PBE | Perdew–Burke–Ernzerhof |
| PBE + SOI | Perdew–Burke–Ernzerhof with spin–orbit interaction |
| QL | quintuple layer |
| SC | semiconductor |
| SM | semimetal |
| SOI | spin–orbit interaction |
| VBM | valence band maximum |
References
- Hsu, C.; Yao, D.; Ye, K.; Yu, B. Renewable energy of waste heat recovery system for automobiles. J. Renew. Sustain. Energy 2010, 2, 013105. [Google Scholar] [CrossRef]
- Yu, S.; Du, Q.; Diao, H.; Shu, G.; Jiao, K. Start-up modes of thermoelectric generator based on vehicle exhaust waste heat recovery. Appl. Energy 2015, 138, 276–290. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.; Kim, S.; Rhi, S. A thermoelectric generator using engine coolant for light-duty internal combustion engine-powered vehicles. J. Electron. Mater. 2011, 40, 812–816. [Google Scholar] [CrossRef]
- Elsheikh, M.H.; Shnawah, D.A.; Sabri, M.F.M.; Said, S.B.M.; Hassan, M.H.; Bashir, M.B.A.; Mohamad, M. A review on thermoelectric renewable energy: Principle parameters that affect their performance. Renew. Sustain. Energy Rev. 2014, 30, 337–355. [Google Scholar] [CrossRef]
- Orr, B.; Akbarzadeh, A.; Mochizuki, M.; Singh, R. A review of car waste heat recovery systems utilising thermoelectric generators and heat pipes. Appl. Therm. Eng. 2016, 101, 490–495. [Google Scholar] [CrossRef]
- Enescu, D. Thermoelectric energy harvesting: Basic principles and applications. In Green Energy Advances; Enescu, D., Ed.; Open Access Peer-Reviewed Edited Vol.; IntechOpen Limited: London, UK, 2019. [Google Scholar] [CrossRef]
- Hsiao, Y.Y.; Chang, W.C.; Chen, S.L. A mathematic model of thermoelectric module with applications on waste heat recovery from automobile engine. Energy 2010, 35, 1447–1454. [Google Scholar] [CrossRef]
- Arnaud, L.; Ludovic, G.; Mouad, D.; Hamid, Z.; Vincent, L. Comparison and impact of waste heat recovery technologies on passenger car fuel consumption in a normalized driving cycle. Energies 2014, 7, 5273–5290. [Google Scholar] [CrossRef] [Green Version]
- Ryu, B.; Chung, J.; Choi, E.; Ziolkowski, P.; Müller, E.; Park, S.D. Counterintuitive example on relation between ZT and thermoelectric efficiency. Appl. Phys. Lett. 2020, 116, 193903. [Google Scholar] [CrossRef]
- Ryu, B.; Chung, J.; Park, S.D. Thermoelectric degrees of freedom determining thermoelectric efficiency. iScience 2021, 24, 102934. [Google Scholar] [CrossRef]
- Selvan, K.V.; Ali, M.S.M. Micro-scale energy harvesting devices: Review of methodological performances in the last decade. Renew. Sustain. Energy Rev. 2016, 54, 1035–1047. [Google Scholar] [CrossRef]
- Mamur, H.; Bhuiyan, M.R.A.; Korkmaz, F.; Nil, M. A review on bismuth telluride (Bi2Te3) nanostructure for thermoelectric applications. Renew. Sustain. Energy Rev. 2018, 82, 4159–4169. [Google Scholar] [CrossRef]
- Burkov, A.T. Silicide thermoelectrics: Materials for energy harvesting. Phys. Status Solidi A 2018, 215, 1800105. [Google Scholar] [CrossRef]
- Bos, J.W.G.; Zandbergen, H.W.; Lee, M.-H.; Ong, N.P.; Cava, R.J. Structures and thermoelectric properties of the infinitely adaptive series (Bi2)m(Bi2Te3)n. Phys. Rev. B 2007, 75, 195203. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.W.G.; Faucheux, F.; Downie, R.A.; Marcinkova, A. Phase stability, structures and properties of the (Bi2)m (Bi2Te3)n natural superlattices. J. Solid State Chem. 2012, 193, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, P.I.; Yapaskurt, V.O.; Shchamkhalova, B.S.; Shcherbakov, V.D.; Yakushcheva, G.G.; Luzanov, V.A.; Jitov, V.A. Growth of Bi2Te3 films and other phases of Bi-Te system by MOVPE. J. Cryst. Growth 2016, 455, 122–128. [Google Scholar] [CrossRef]
- Mao, C.; Tan, M.; Zhang, L.; Wu, D.; Bai, W.; Liu, L. Experimental reinvestigation and thermodynamic description of Bi-Te binary system. Calphad 2018, 60, 81–89. [Google Scholar] [CrossRef]
- Zhu, H.Z.; Zhao, C.; Nan, P.; Jiang, X.; Zhao, J.; Ge, B.; Xiao, C.; Xie, Y. Intrinsically low lattice thermal conductivity in natural superlattice (Bi2)m(Bi2Te3)n thermoelectric materials. Chem. Mater. 2021, 33, 1140–1148. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251. [Google Scholar] [CrossRef] [PubMed]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Kaupp, M.; Malkina, O.L.; Malkin, V.G.; Pyykkö, P. How do spin–orbit-induced heavy-atom effects on NMR chemical shifts function? Validation of a simple analogy to spin–spin coupling by density functional theory (DFT) calculations on some iodo compounds. Chem. Eur. J. 1998, 4, 118–126. [Google Scholar] [CrossRef]
- Autschbach, J.; Ziegler, T. Nuclear spin–spin coupling constants from regular approximate relativistic density functional calculations. I. Formalism and scalar relativistic results for heavy metal compounds. J. Chem. Phys. 2000, 113, 936. [Google Scholar] [CrossRef]
- Autschbach, J.; Ziegler, T. Nuclear spin–spin coupling constants from regular approximate relativistic density functional calculations. II. Spin–orbit coupling effects and anisotropies. J. Chem. Phys. 2000, 113, 9410. [Google Scholar] [CrossRef]
- Ong, S.P.; Richards, W.D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V.; Persson, K.A.; Ceder, G. Python materials genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 2013, 68, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Goldsmid, H. Recent studies of bismuth telluride and its alloys. J. Appl. Phys. 1961, 32, 2198. [Google Scholar] [CrossRef]
- Thomas, G.; Rapkine, D.; Van Dover, R.; Mattheiss, L.; Sunder, W.; Schneemeyer, L.; Waszczak, J. Large electronic-density increase on cooling a layered metal: Doped Bi2Te3. Phys. Rev. B 1992, 46, 1553. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ryu, B. Hybrid-density functional theory study on the band structures of tetradymite-Bi2Te3, Sb2Te3, Bi2Se3, and Sb2Se3 thermoelectric materials. J. Korean Phys. Soc. 2016, 69, 1683–1687. [Google Scholar] [CrossRef] [Green Version]
- Lannin, J.; Calleja, J.; Cardona, M. Second-order Raman scattering in the group-Vb semimetals: Bi, Sb, and As. Phys. Rev. B 1975, 12, 585. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | Bi2Te3 | Bi4Te5 | Bi6Te7 | Bi8Te9 | BiTe | Bi4Te3 | Bi2Te | Bi7Te3 | Bi2 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phase properties | Hexagonal compositions | Bi6Te9 | Bi12Te15 | Bi18Te21 | Bi24Te27 | Bi6Te6 | Bi12Te9 | Bi6Te3 | Bi42Te18 | Bi6 | ||
| NBL: NQL | 0:3 | 1:5 | 2:7 | 3:9 | 1:2 | 3:3 | 2:1 | 15:6 | 3:0 | |||
| NBL/(NBL + NQL) | 0 | 0.167 | 0.222 | 0.250 | 0.333 | 0.500 | 0.667 | 0.714 | 1 | |||
| Structural parameters | Volume/NAtom (Å3) | 33.892 | 33.974 | 33.986 | 34.015 | 33.887 | 34.224 | 35.093 | 34.352 | 35.383 | ||
| aHex (Å) | 4.388 | 4.415 | 4.424 | 4.410 | 4.423 | 4.451 | 4.490 | 4.472 | 4.546 | |||
| cHex (Å) | 30.488 | 54.330 | 78.200 | 103.000 | 24.002 | 41.890 | 18.090 | 119.000 | 11.862 | |||
| cHex/NAtom (Å) | 2.033 | 2.012 | 2.005 | 2.020 | 2.000 | 1.995 | 2.010 | 1.983 | 1.977 | |||
| PBE | w/o SOI | Intralayer thickness (Å) | BL | - | 1.697 | 1.693 | 1.701 | 1.692 | 1.681 | 1.671 | 1.674 | 1.977 |
| QL | 7.529 | 7.472 | 7.453 | 7.492 | 7.449 | 7.415 | 7.390 | 7.358 | - | |||
| Interlayer distance (Å) | BL-BL | - | - | - | - | - | - | 2.464 | 2.375 | 1.977 | ||
| BL-QL | - | 2.436 | 2.428 | 2.453 | 2.421 | 2.434 | 2.447 | 2.363 | - | |||
| QL-QL | 2.634 | 2.600 | 2.586 | 2.626 | 2.570 | - | - | - | - | |||
| w/ SOI | Intralayer thickness (Å) | BL | - | 1.758 | 1.753 | 1.762 | 1.748 | 1.729 | 1.704 | 1.702 | 1.977 | |
| QL | 7.623 | 7.560 | 7.538 | 7.579 | 7.525 | 7.480 | 7.454 | 7.409 | - | |||
| Interlayer distance (Å) | BL-BL | - | - | - | - | - | - | 2.468 | 2.375 | 1.977 | ||
| BL-QL | - | 2.412 | 2.400 | 2.423 | 2.386 | 2.377 | 2.380 | 2.303 | - | |||
| QL-QL | 2.539 | 2.488 | 2.465 | 2.494 | 2.433 | - | - | - | - | |||
| LDA | w/o SOI | Intralayer thickness (Å) | BL | - | 1.661 | 1.657 | 1.66 | 1.657 | 1.645 | 1.632 | 1.638 | 1.977 |
| QL | 7.454 | 7.403 | 7.384 | 7.419 | 7.383 | 7.346 | 7.310 | 7.291 | - | |||
| Interlayer distance (Å) | BL-BL | - | - | - | - | - | - | 2.525 | 2.426 | 1.977 | ||
| BL-QL | - | 2.474 | 2.469 | 2.492 | 2.467 | 2.486 | 2.495 | 2.404 | - | |||
| QL-QL | 2.709 | 2.677 | 2.663 | 2.714 | 2.645 | - | - | - | - | |||
| w/ SOI | Intralayer thickness (Å) | BL | - | 1.718 | 1.714 | 1.722 | 1.710 | 1.693 | 1.661 | 1.661 | 1.977 | |
| QL | 7.565 | 7.509 | 7.489 | 7.526 | 7.476 | 7.421 | 7.381 | 7.345 | - | |||
| Interlayer distance (Å) | BL-BL | - | - | - | - | - | - | 2.549 | 2.439 | 1.977 | ||
| BL-QL | - | 2.466 | 2.450 | 2.476 | 2.434 | 2.425 | 2.419 | 2.338 | - | |||
| QL-QL | 2.598 | 2.534 | 2.510 | 2.540 | 2.471 | - | - | - | - | |||
| Exc | Phase | SOI | EVBM | kVBM | ECBM | kCBM | Type | |||
|---|---|---|---|---|---|---|---|---|---|---|
| PBE | Bi2Te3 | X | −0.136 | 0.000 0.000 0.000 | 0.136 | 0.000 0.000 0.000 | 0.272 | 0.272 | 0.000 0.000 0.000 | SC |
| O | −0.061 | 0.140 0.000 0.500 | 0.068 | 0.090 0.000 0.000 | 0.129 | 0.135 | 0.130 0.000 0.500 | SC | ||
| Bi4Te5 | X | 0.006 | 0.033 0.033 0.500 | −0.059 | 0.000 0.000 0.500 | −0.065 | 0.005 | 0.010 0.000 0.500 | SM | |
| O | 0.012 | 0.170 0.000 0.000 | −0.035 | 0.090 0.000 0.000 | −0.047 | 0.004 | 0.010 0.000 0.500 | SM | ||
| Bi6Te7 | X | 0.002 | 0.033 0.033 0.000 | −0.043 | 0.007 0.007 0.000 | −0.045 | 0.029 | 0.020 0.000 0.000 | SM | |
| O | 0.006 | 0.180 0.000 0.500 | −0.018 | 0.000 0.000 0.500 | −0.024 | 0.008 | 0.020 0.000 0.000 | SM | ||
| Bi8Te9 | X | −0.004 | 0.033 0.033 0.500 | −0.030 | 0.007 0.007 0.500 | −0.026 | 0.027 | 0.020 0.000 0.500 | SM | |
| O | 0.001 | 0.170 0.000 0.500 | −0.019 | 0.000 0.000 0.500 | −0.020 | 0.001 | 0.000 0.000 0.500 | SM | ||
| BiTe | X | 0.017 | 0.060 0.000 0.000 | −0.023 | 0.013 0.013 0.500 | −0.040 | 0.035 | 0.060 0.000 0.000 | SM | |
| O | −0.064 | 0.180 0.000 0.500 | 0.053 | 0.027 0.027 0.500 | 0.117 | 0.152 | 0.040 0.000 0.500 | SC | ||
| Bi4Te3 | X | 0.030 | 0.110 0.000 0.000 | −0.036 | 0.027 0.027 0.500 | −0.066 | 0.021 | 0.027 0.027 0.500 | SM | |
| O | 0.108 | 0.000 0.000 0.000 | −0.084 | 0.500 0.000 0.000 | −0.192 | 0.001 | 0.020 0.000 0.000 | SM | ||
| Bi2Te | X | 0.025 | 0.067 0.067 0.000 | −0.051 | 0.033 0.033 0.000 | −0.076 | 0.038 | 0.033 0.033 0.000 | SM | |
| O | 0.138 | 0.000 0.000 0.000 | −0.041 | 0.500 0.000 0.500 | −0.179 | 0.030 | 0.030 0.000 0.000 | SM | ||
| Bi7Te3 | X | 0.077 | 0.150 0.000 0.500 | −0.068 | 0.027 0.027 0.500 | −0.145 | 0.021 | 0.027 0.027 0.500 | SM | |
| O | 0.041 | 0.000 0.000 0.500 | −0.093 | 0.500 0.000 0.500 | −0.134 | 0.025 | 0.020 0.020 0.500 | SM | ||
| Bi2 | X | 0.066 | 0.047 0.047 0.500 | −0.197 | 0.480 0.000 0.500 | −0.263 | 0.020 | 0.047 0.047 0.500 | SM | |
| O | 0.113 | 0.000 0.000 0.500 | −0.048 | 0.500 0.000 0.500 | −0.161 | 0.079 | 0.500 0.000 0.500 | SM |
| Exc | Phase | SOI | EVBM | kVBM | ECBM | kCBM | Type | |||
|---|---|---|---|---|---|---|---|---|---|---|
| LDA | Bi2Te3 | X | −0.098 | 0.000 0.000 0.000 | 0.098 | 0.000 0.000 0.000 | 0.196 | 0.196 | 0.000 0.000 0.000 | SC |
| O | −0.055 | 0.140 0.000 0.500 | 0.056 | 0.130 0.000 0.500 | 0.111 | 0.115 | 0.130 0.000 0.500 | SC | ||
| Bi4Te5 | X | 0.027 | 0.040 0.040 0.500 | −0.078 | 0.007 0.007 0.500 | −0.105 | 0.017 | 0.007 0.007 0.500 | SM | |
| O | −0.004 | 0.180 0.000 0.000 | −0.028 | 0.090 0.000 0.000 | −0.024 | 0.005 | 0.000 0.000 0.500 | SM | ||
| Bi6Te7 | X | 0.021 | 0.040 0.040 0.500 | −0.057 | 0.020 0.000 0.000 | −0.078 | 0.007 | 0.013 0.013 0.000 | SM | |
| O | −0.009 | 0.000 0.000 0.000 | −0.013 | 0.100 0.000 0.000 | −0.004 | 0.006 | 0.010 0.000 0.000 | SM | ||
| Bi8Te9 | X | −0.004 | 0.070 0.000 0.500 | −0.019 | 0.013 0.013 0.500 | −0.015 | 0.007 | 0.013 0.013 0.500 | SM | |
| O | −0.005 | 0.000 0.000 0.000 | −0.002 | 0.000 0.000 0.000 | 0.003 | 0.003 | 0.000 0.000 0.000 | SC | ||
| BiTe | X | 0.024 | 0.060 0.000 0.000 | −0.028 | 0.020 0.020 0.500 | −0.052 | 0.013 | 0.060 0.000 0.000 | SM | |
| O | −0.062 | 0.060 0.000 0.000 | 0.048 | 0.027 0.027 0.500 | 0.110 | 0.134 | 0.040 0.000 0.500 | SC | ||
| Bi4Te3 | X | 0.025 | 0.110 0.000 0.000 | −0.039 | 0.027 0.027 0.500 | −0.064 | 0.036 | 0.027 0.027 0.500 | SM | |
| O | 0.081 | 0.000 0.000 0.000 | −0.077 | 0.500 0.000 0.000 | −0.158 | 0.001 | 0.020 0.000 0.500 | SM | ||
| Bi2Te | X | 0.044 | 0.067 0.067 0.000 | −0.083 | 0.033 0.033 0.000 | −0.127 | 0.005 | 0.033 0.033 0.000 | SM | |
| O | 0.120 | 0.000 0.000 0.000 | −0.045 | 0.500 0.000 0.500 | −0.165 | 0.021 | 0.020 0.000 0.000 | SM | ||
| Bi7Te3 | X | 0.085 | 0.170 0.000 0.000 | −0.068 | 0.033 0.033 0.500 | −0.153 | 0.010 | 0.033 0.033 0.500 | SM | |
| O | 0.041 | 0.000 0.000 0.000 | −0.095 | 0.500 0.000 0.500 | −0.136 | 0.009 | 0.000 0.000 0.000 | SM | ||
| Bi2 | X | 0.073 | 0.053 0.053 0.500 | −0.207 | 0.500 0.000 0.500 | −0.280 | 0.011 | 0.500 0.000 0.500 | SM | |
| O | 0.076 | 0.000 0.000 0.500 | −0.038 | 0.500 0.000 0.500 | −0.114 | 0.089 | 0.500 0.000 0.500 | SM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Ryu, B.; Park, S. Structural Analysis, Phase Stability, Electronic Band Structures, and Electric Transport Types of (Bi2)m(Bi2Te3)n by Density Functional Theory Calculations. Appl. Sci. 2021, 11, 11341. https://doi.org/10.3390/app112311341
Park S, Ryu B, Park S. Structural Analysis, Phase Stability, Electronic Band Structures, and Electric Transport Types of (Bi2)m(Bi2Te3)n by Density Functional Theory Calculations. Applied Sciences. 2021; 11(23):11341. https://doi.org/10.3390/app112311341
Chicago/Turabian StylePark, Sungjin, Byungki Ryu, and SuDong Park. 2021. "Structural Analysis, Phase Stability, Electronic Band Structures, and Electric Transport Types of (Bi2)m(Bi2Te3)n by Density Functional Theory Calculations" Applied Sciences 11, no. 23: 11341. https://doi.org/10.3390/app112311341
APA StylePark, S., Ryu, B., & Park, S. (2021). Structural Analysis, Phase Stability, Electronic Band Structures, and Electric Transport Types of (Bi2)m(Bi2Te3)n by Density Functional Theory Calculations. Applied Sciences, 11(23), 11341. https://doi.org/10.3390/app112311341