
Formulation of Boron Encapsulated Smart Nanocapsules for Targeted Drug Delivery to the Brain




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
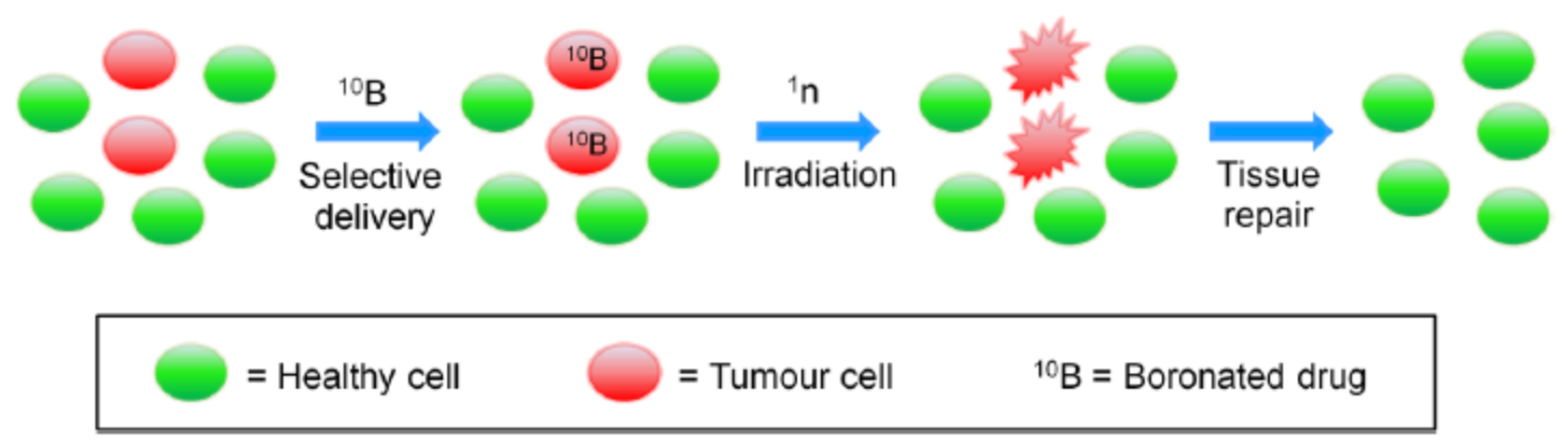
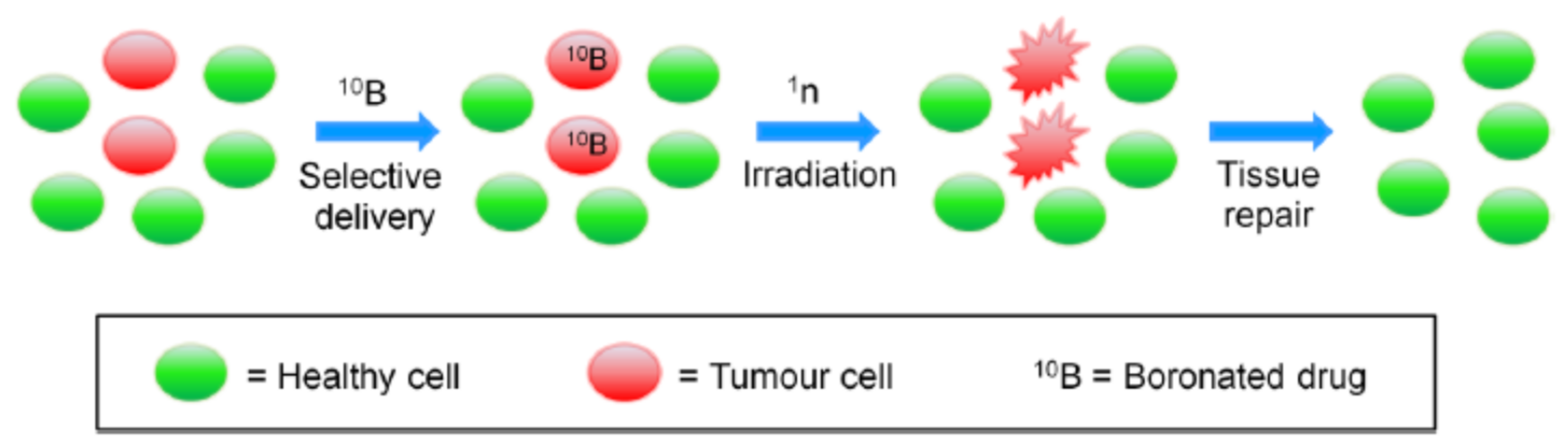
1.1. Boron Neutron Capture Therapy
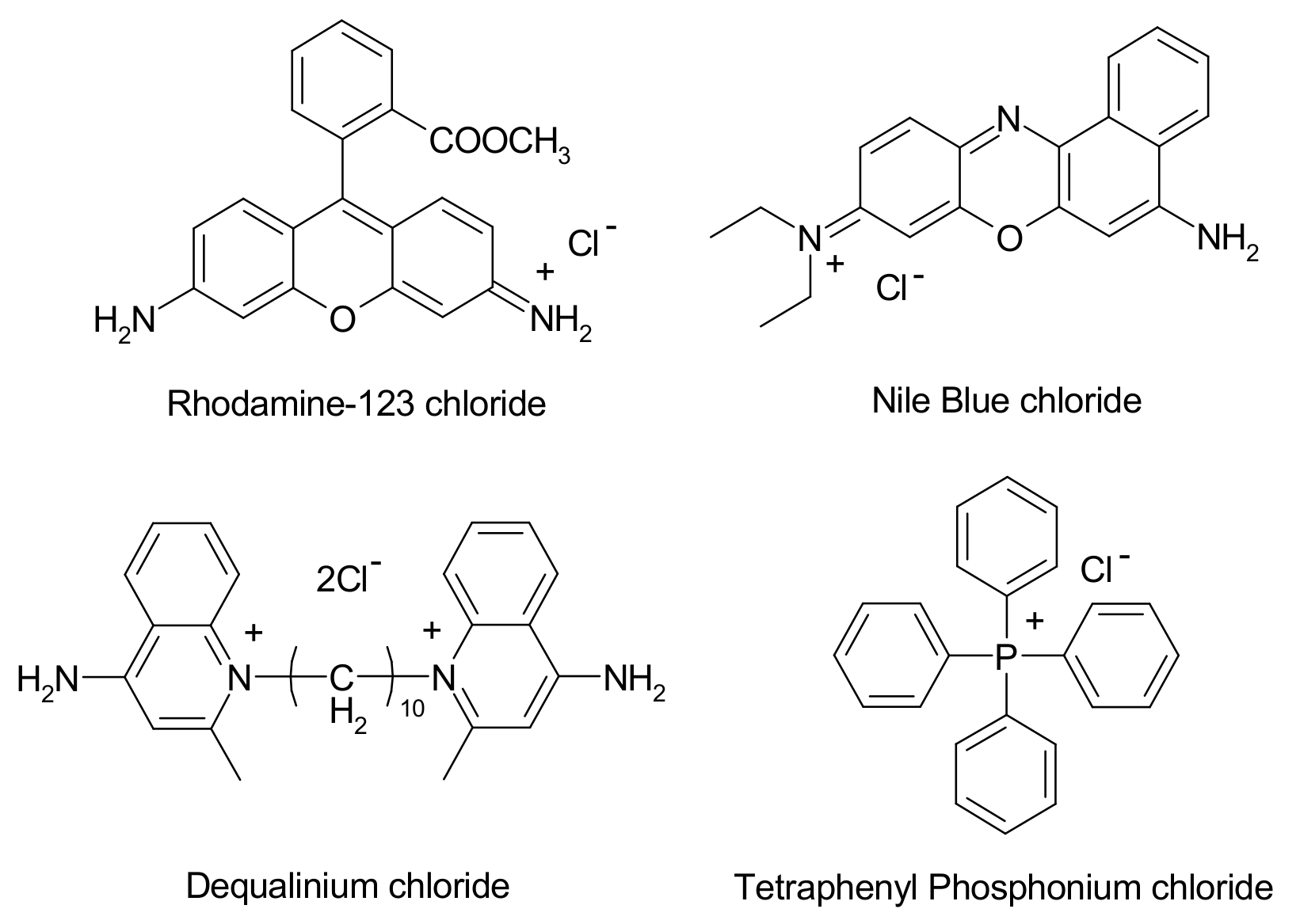
1.2. Mitochondrial Targeting
- Delivery across the BBB through the manipulation of the drug via a prodrug or nanodelivery system.
- Direct delivery to the brain: this can be performed through surgery or intra-cerebro-ventricular administration of a precalculated amount of drug to the brain. However, this is limited due to its invasive nature.
- Delivery of drug intranasally: the most promising delivery route due to its practical, noninvasive, rapid, and simple nature.
- Delivery of drug through the BBB via a two-component stimuli-responsive polymeric drug delivery system, which is enabled to release drug at a specified condition.
1.3. The Advantage of Using Nanotechnology
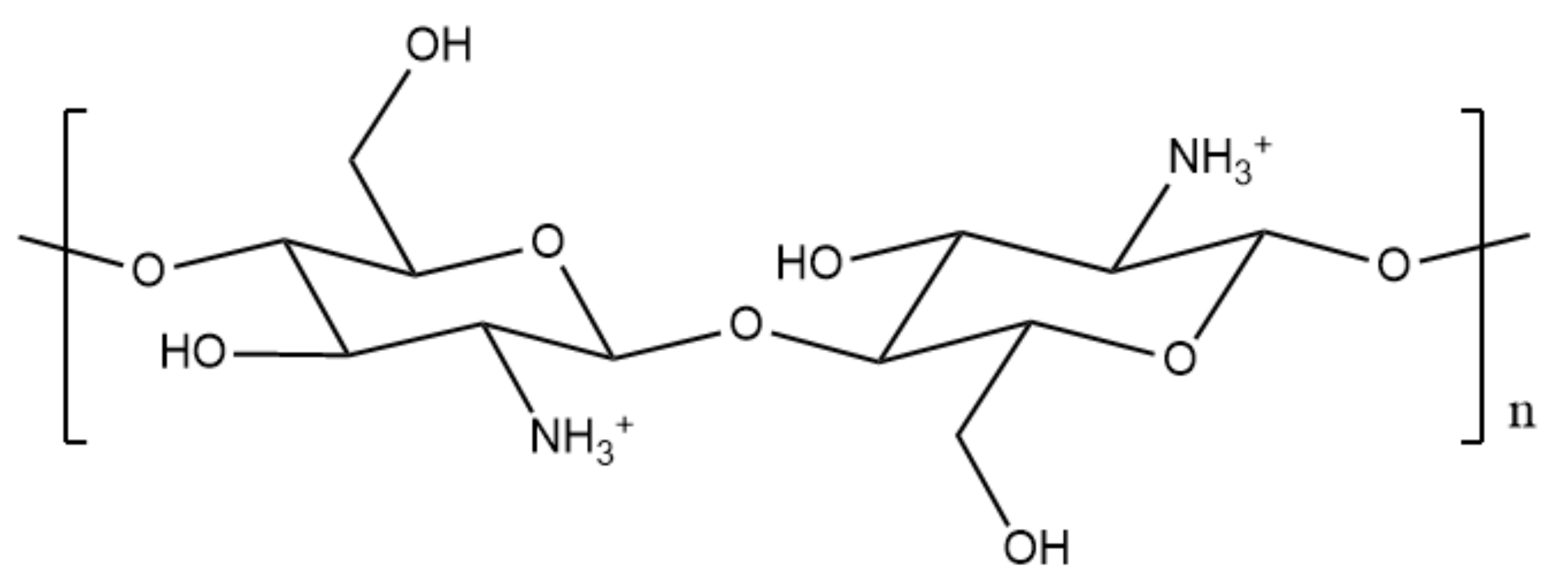
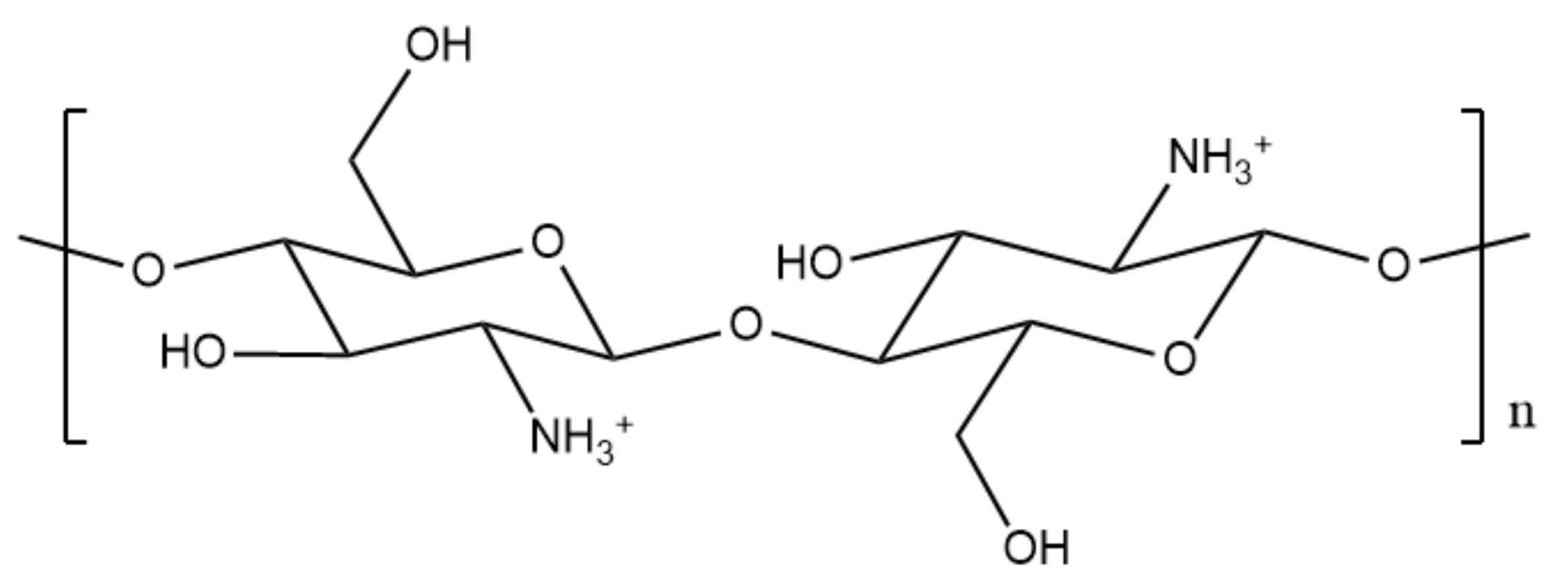
1.4. The Significance of Chitosan
1.5. Significance of Polypyrrole as a Stimuli-Responsive Polymer
2. Materials and Methods
2.1. Synthetic Methods
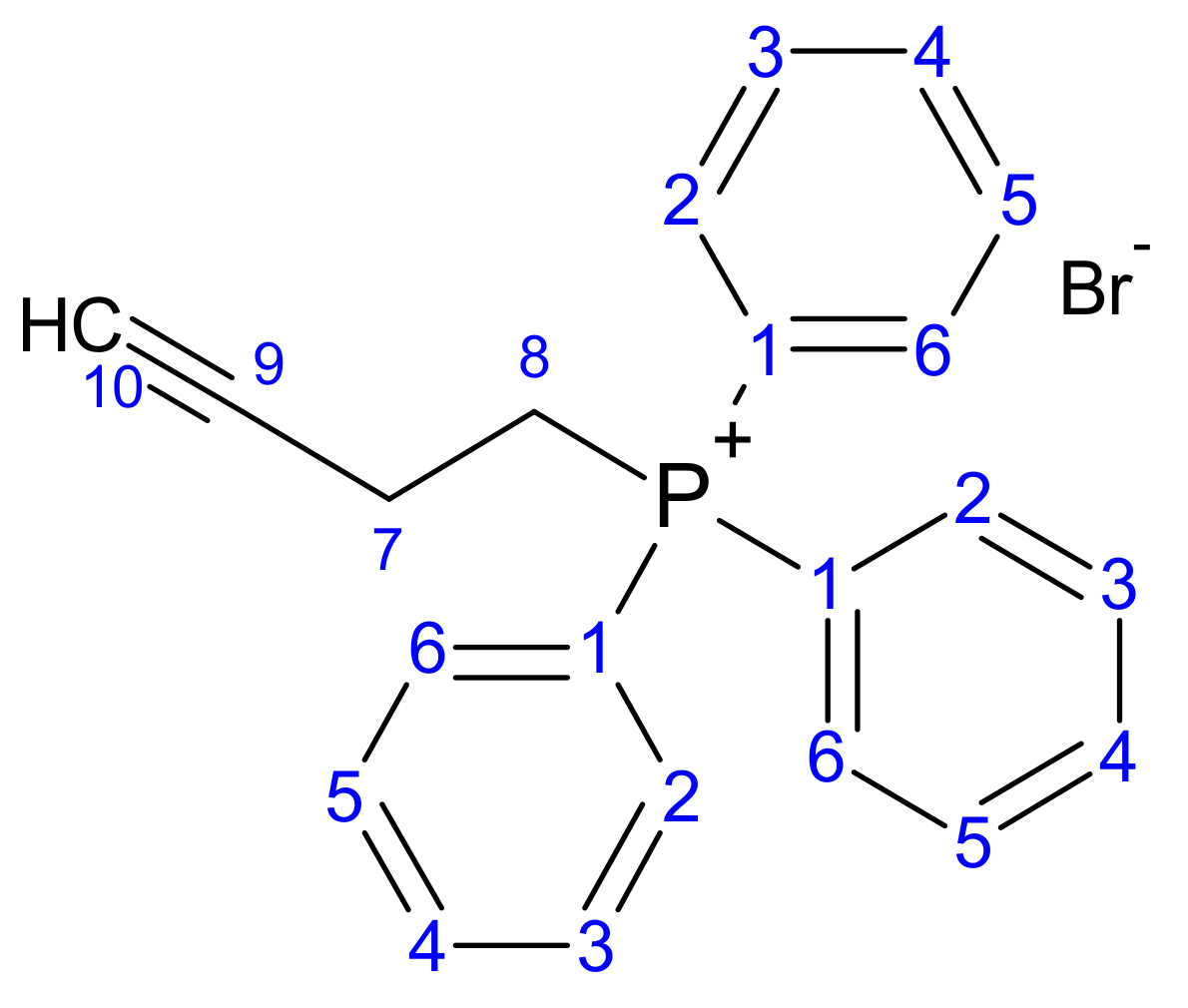
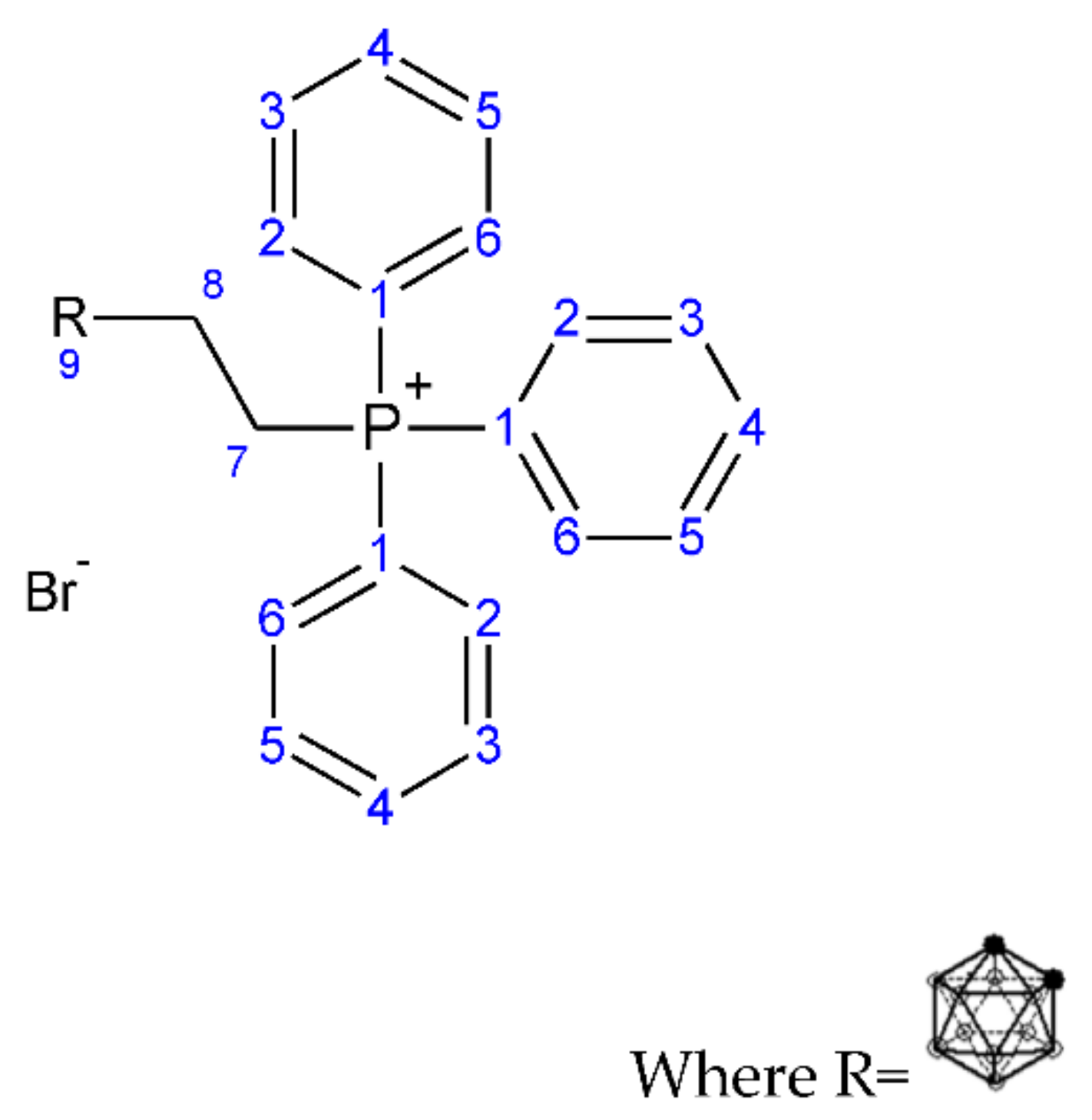
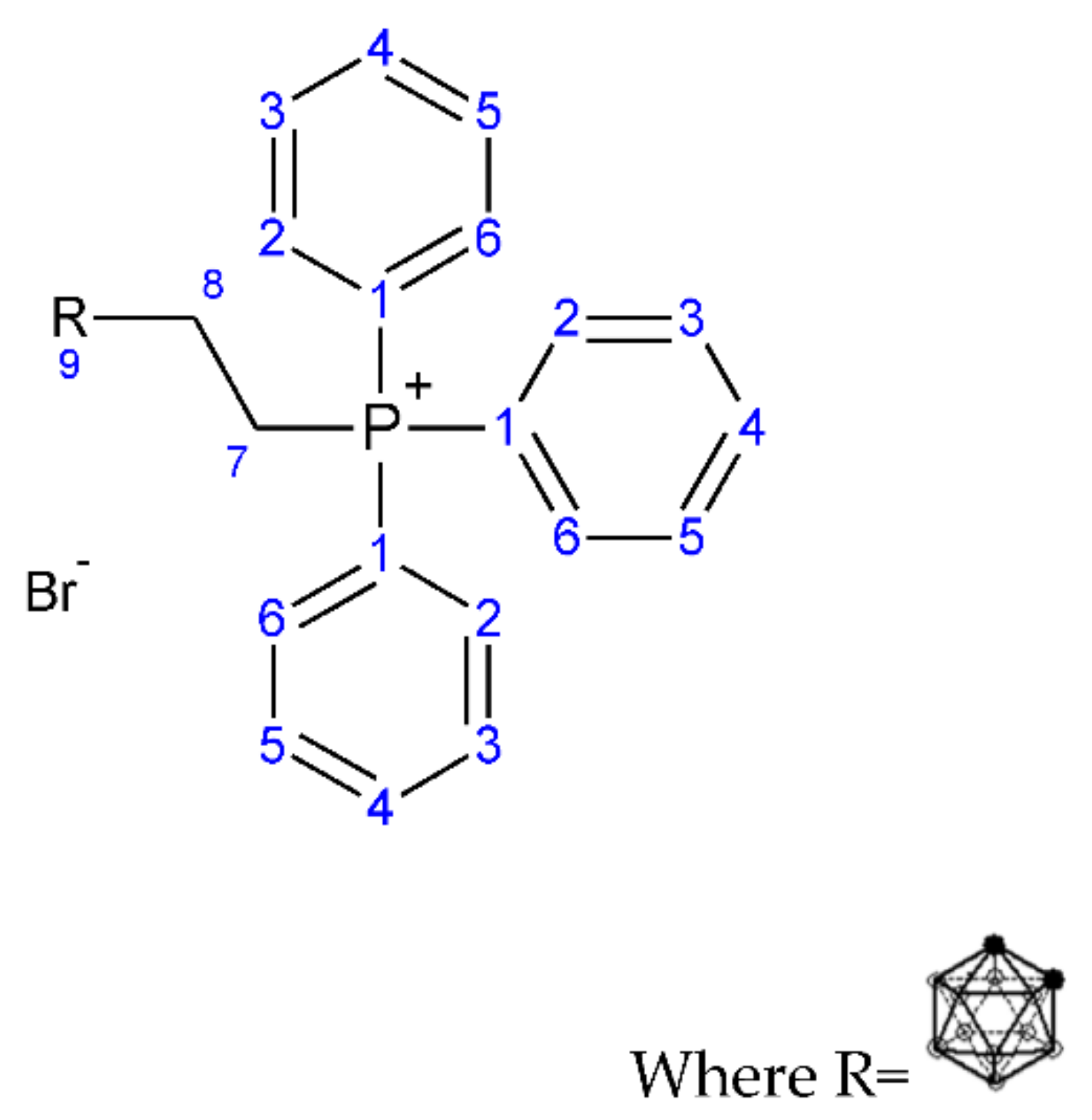
2.1.1. Synthesis of But-3-yn-1-yl (Triphenyl phosphonium) Bromide (1)
2.1.2. Synthesis of (3-Carboranyl-ethyl) (Triphenyl phosphonium) Bromide (2)
2.1.3. Synthesis of N9,N9-Diethyl-9H-benzo[a]phenoxazine-5,9-diamine
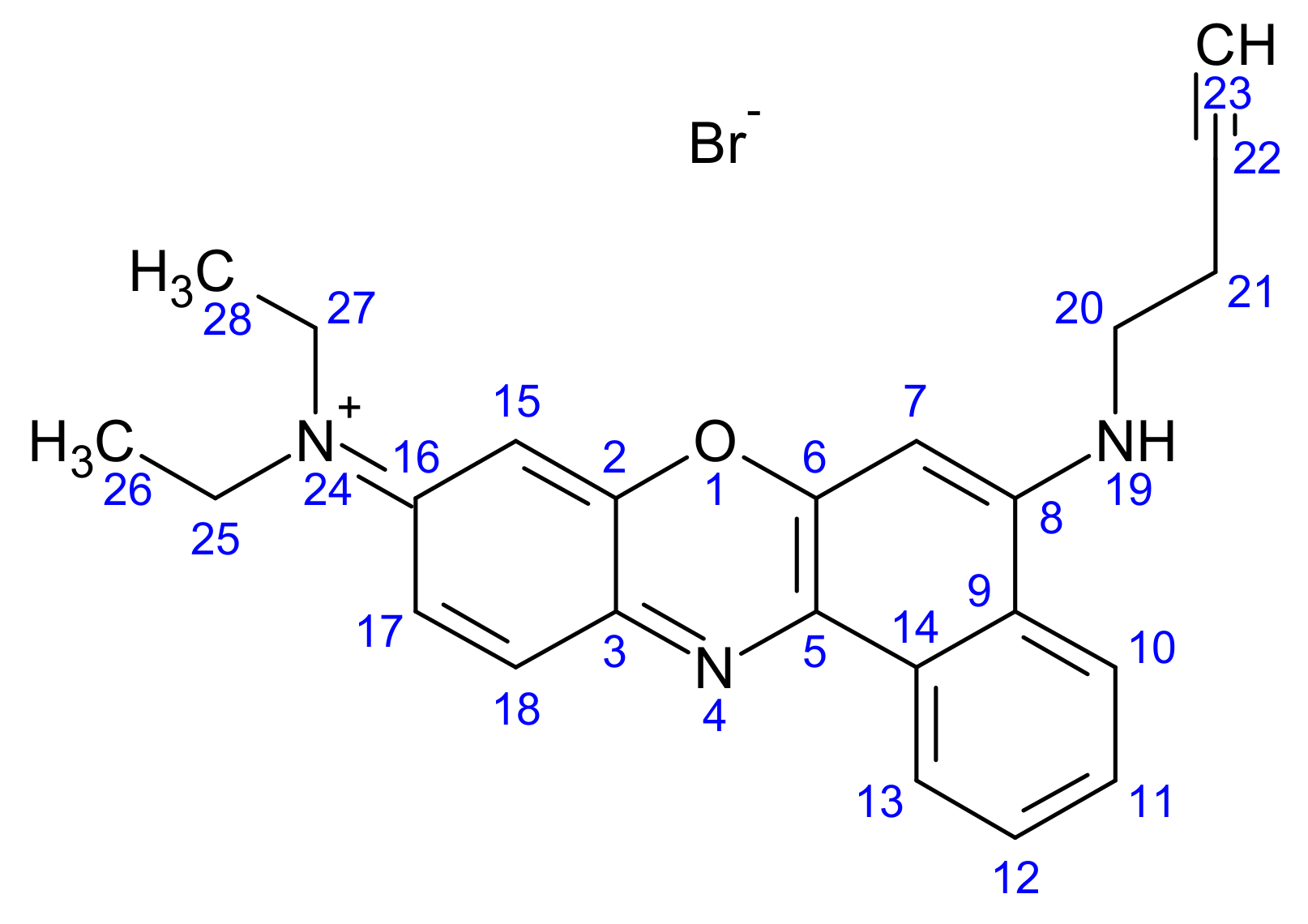
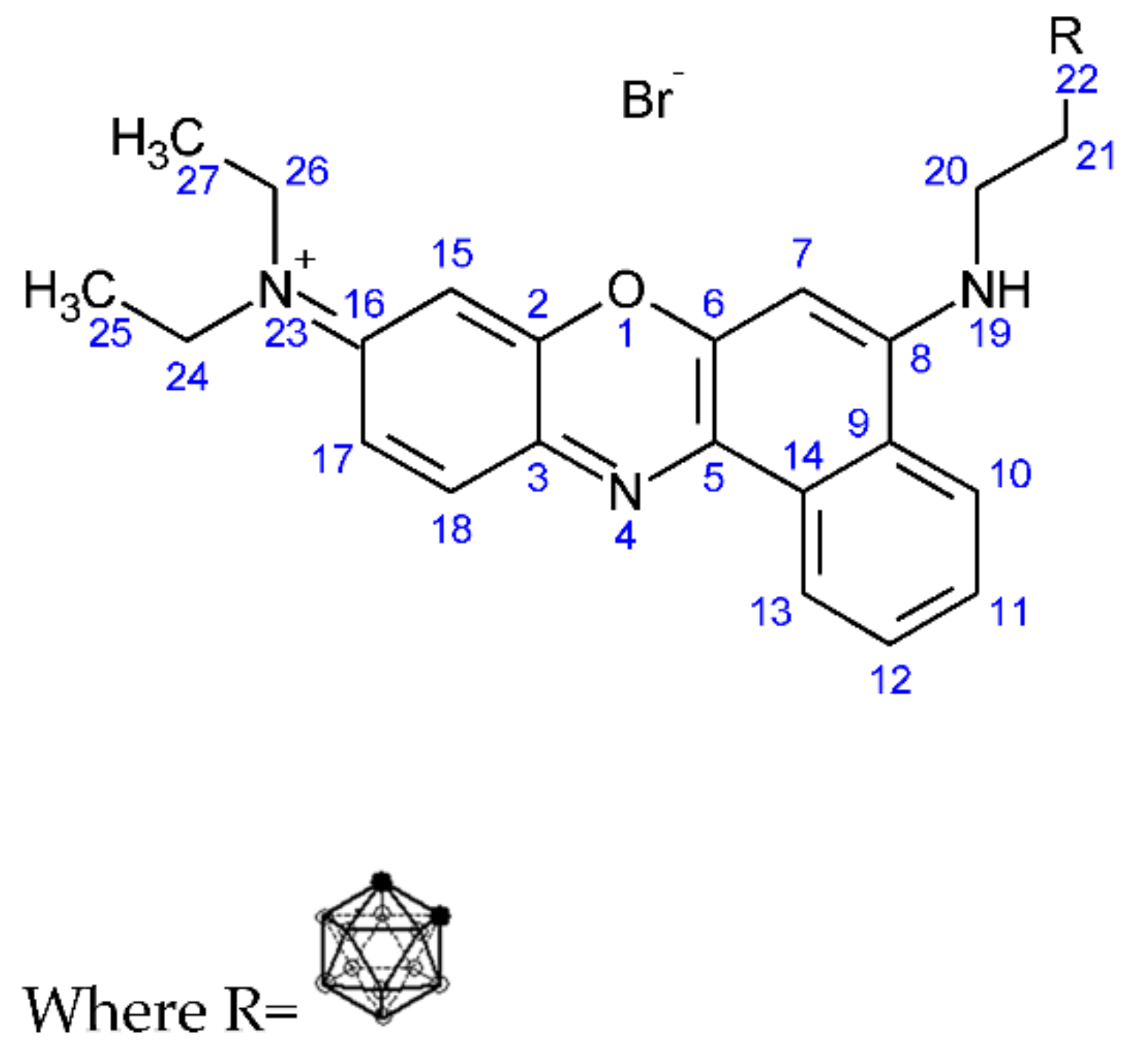
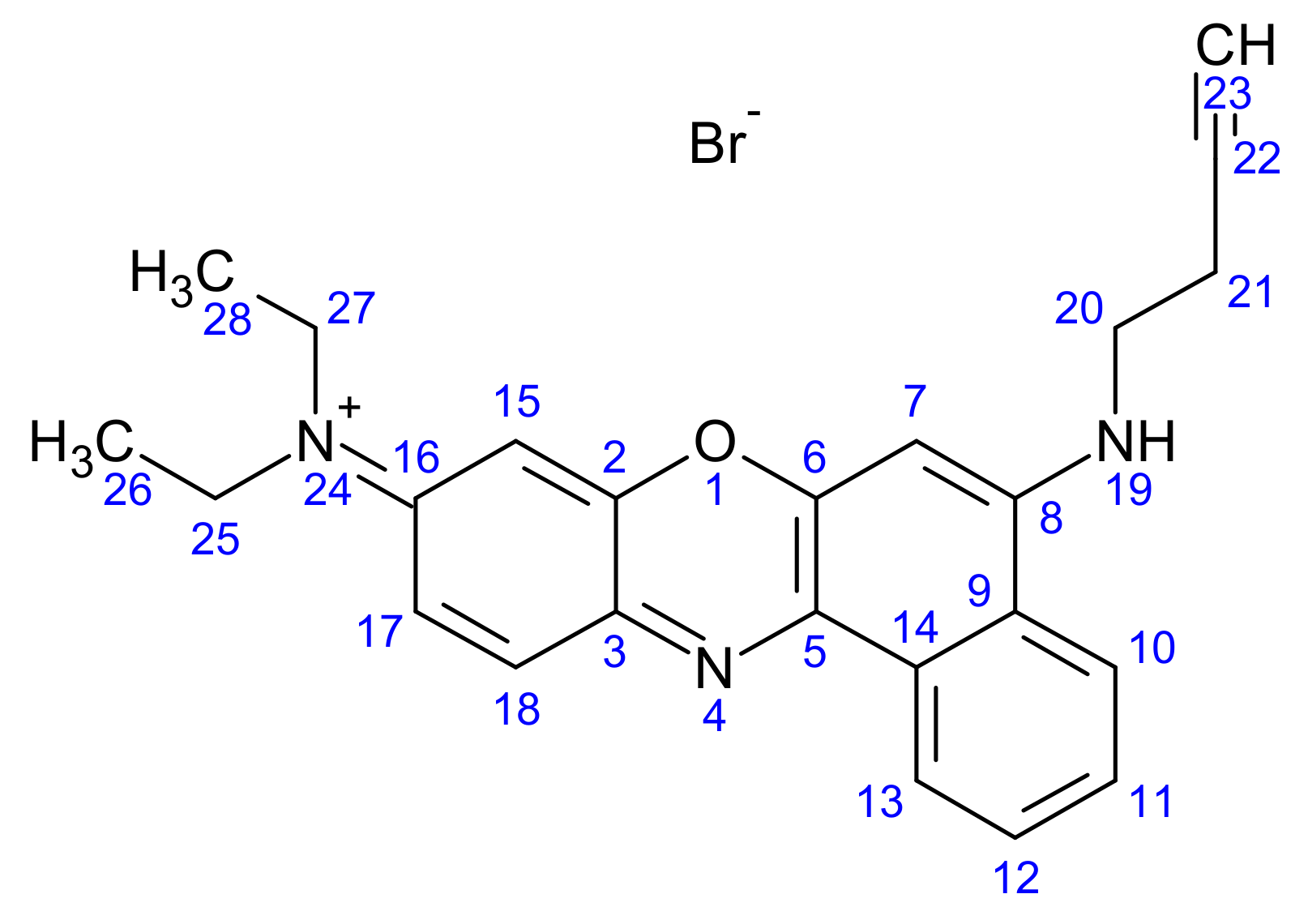
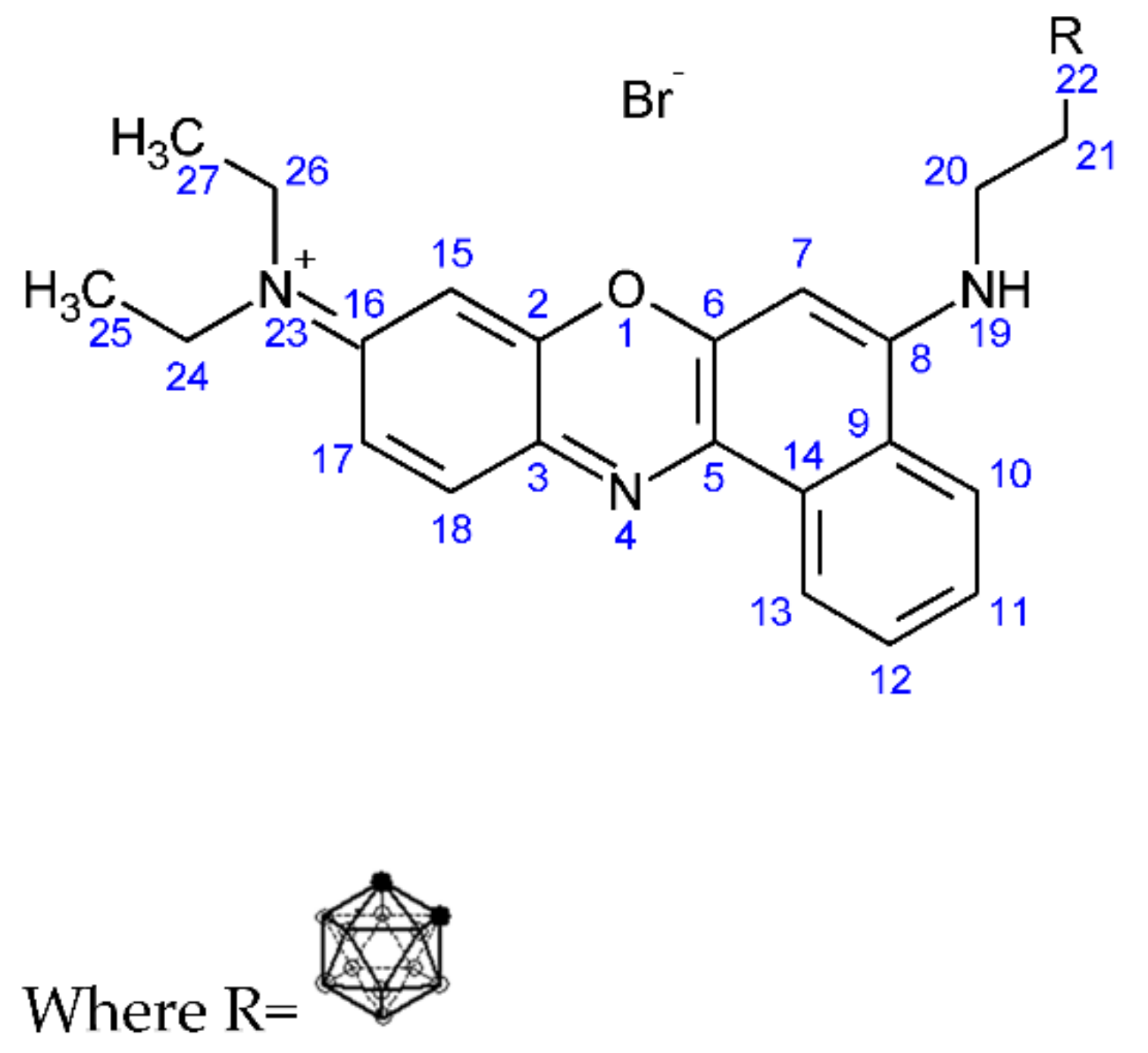
2.1.4. Synthesis of But-3-yn-1-yl (N9,N9-Diethyl-9H-benzo[a]phenoxazine-5,9-diamine) Bromide (3)
2.1.5. Synthesis of (3-Carboranylpropyl) (N9,N9-Diethyl-9H-benzo[a]phenoxazine-5,9-diamine) Bromide (4)
2.1.6. Synthesis of the 3,7-Diamino-5-phenylphenazin-5-ium
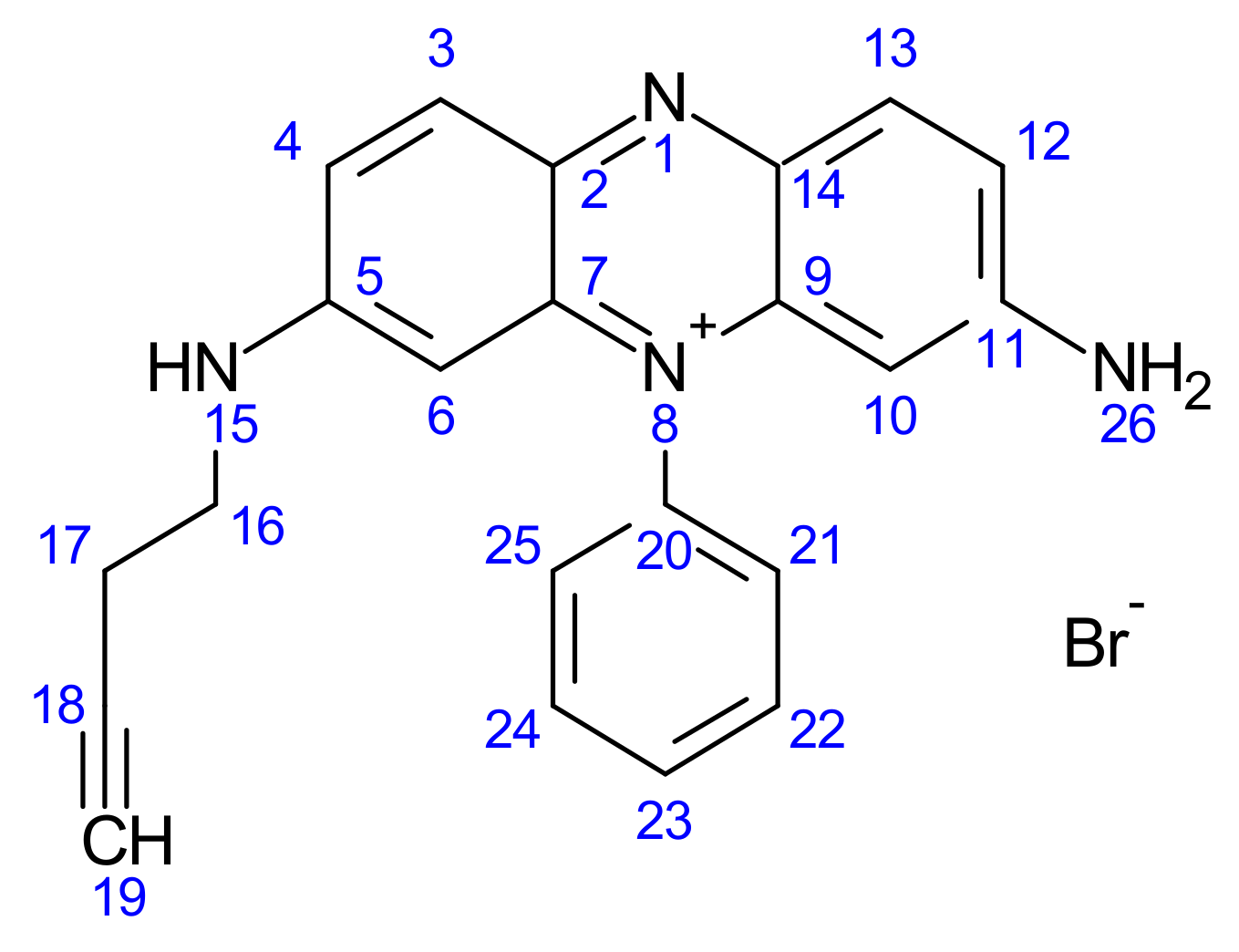
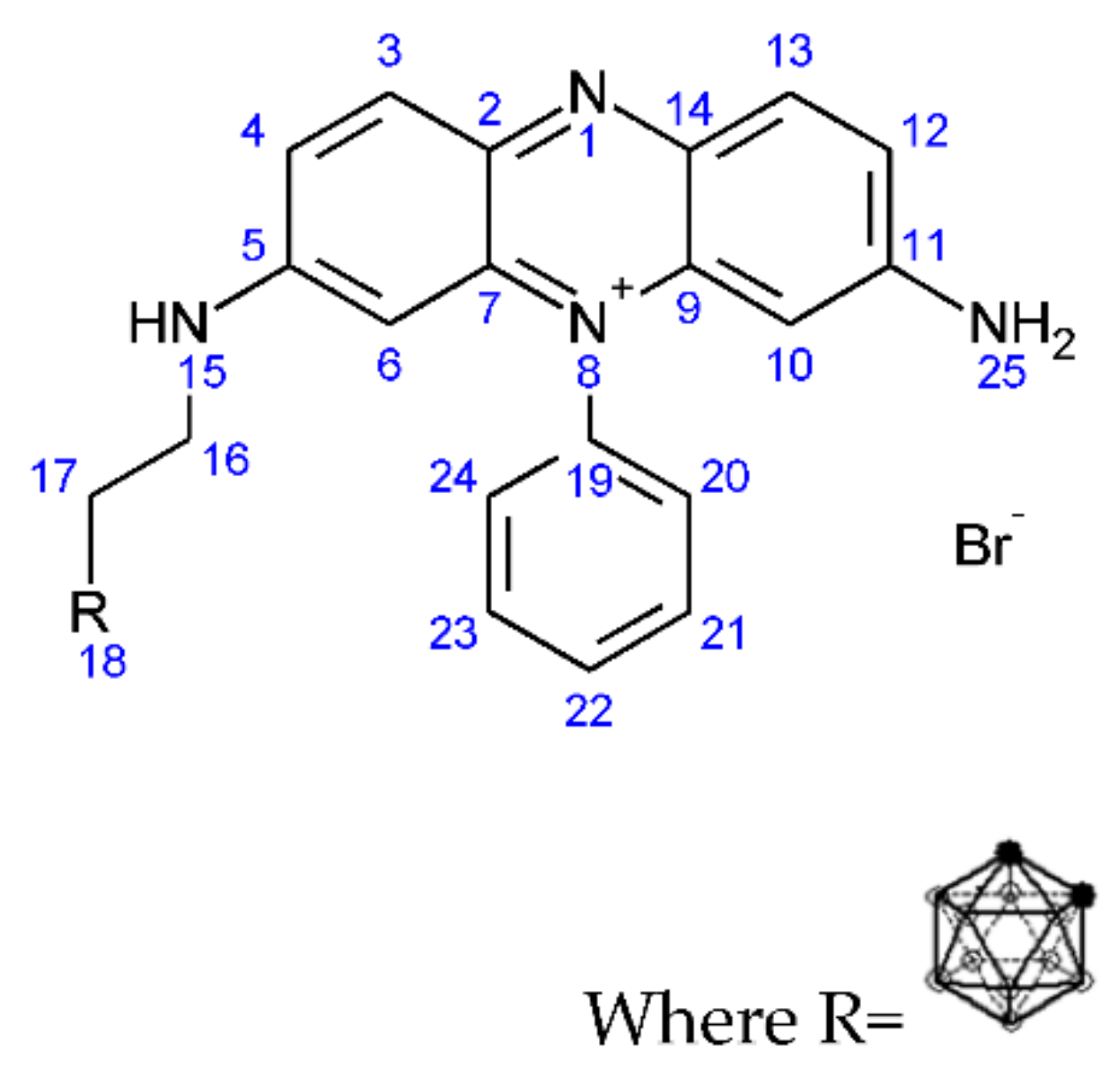
2.1.7. Synthesis of But-3-yn-1-yl 3,7-Diamino-5-phenylphenazin-5-ium Bromide (5)
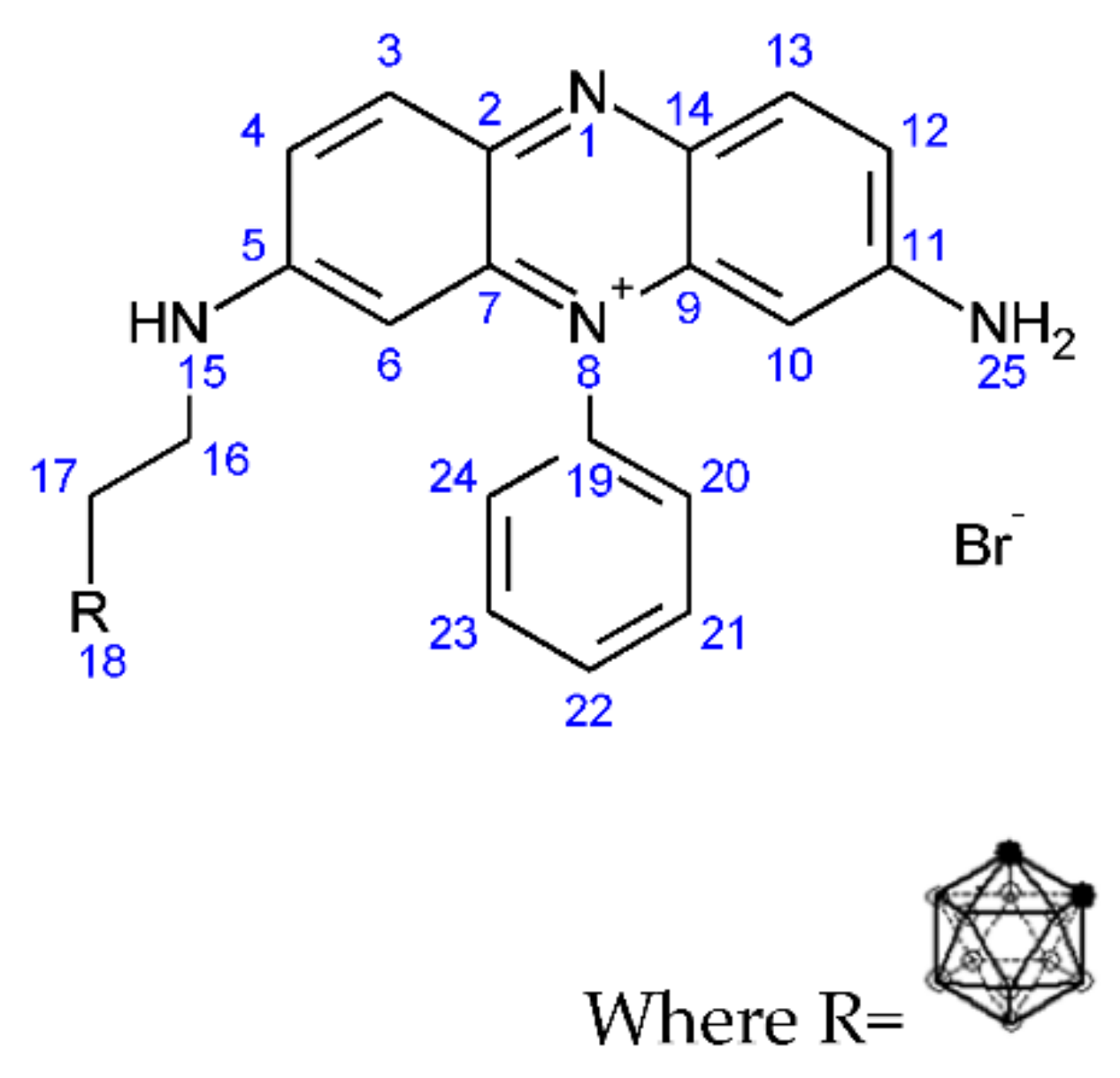
2.1.8. Synthesis of (3-Carboranylpropyl) (3,7-Diamino-5-phenylphenazin-5-ium) Bromide (6)
2.2. Formulation
2.2.1. Formulation of Nanoparticles via Ionotropic Gelation
2.2.2. Chitosan Nanoparticles Loaded with Carboranyl-DLCs
2.2.3. Synthesis of Drug Loaded AgCl@PPy-CS Nanoparticles
2.2.4. The Etching of the AgCl Core and Loading of Drugs in the Nano-Capsules
2.2.5. Characterisation of Formulated Nanoparticles
2.2.6. Boron Release Studies
2.3. Preliminary Cell Studies
2.3.1. In Vitro Cell Studies
2.3.2. Boron Uptake Assay
2.3.3. Neutral Red Assay
2.3.4. Passaging of Cells
2.3.5. Permeation and Uptake Studies
Permeation Studies
Uptake Studies
2.3.6. HEPES Transport Buffer
3. Results
3.1. Synthetic Methods
3.1.1. Synthesis of But-3-yn-1-yl (Triphenyl phosphonium) Bromide (1)
3.1.2. Synthesis of (3-Carboranylpropyl) (Triphenyl phosphonium) Bromide (2)
3.1.3. Synthesis of But-3-yn-1-yl N9,N9-Diethyl-9H-benzo[a]phenoxazine-5,9-diamine Bromide (3)
3.1.4. Synthesis of (3-Carboranylpropyl) (N9,N9-Diethyl-9H-benzo[a]phenoxazine-5,9-diamine) Bromide (4)
3.1.5. Synthesis of But-3-yn-1-yl 3,7-Diamino-5-phenylphenazin-5-ium Bromide (5)
3.1.6. Synthesis of (3-Carboranylpropyl) (3,7-Diamino-5-phenylphenazin-5-ium) Bromide (6)
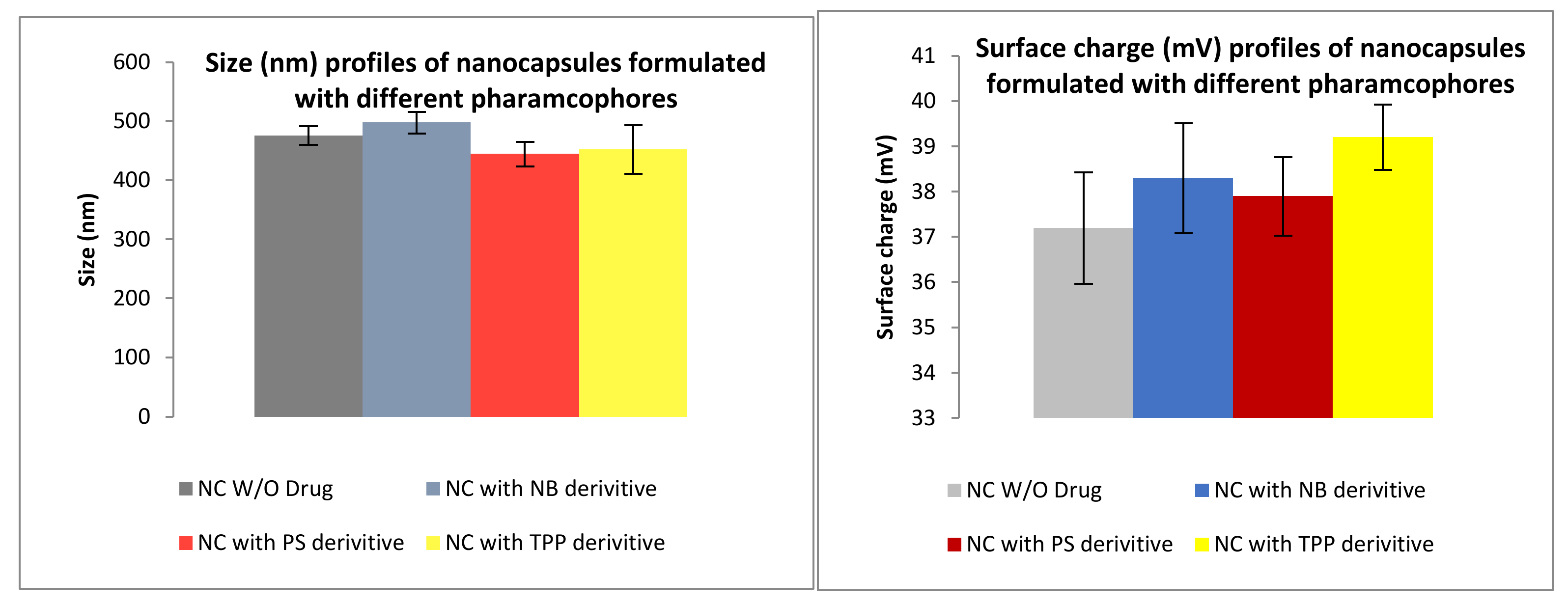
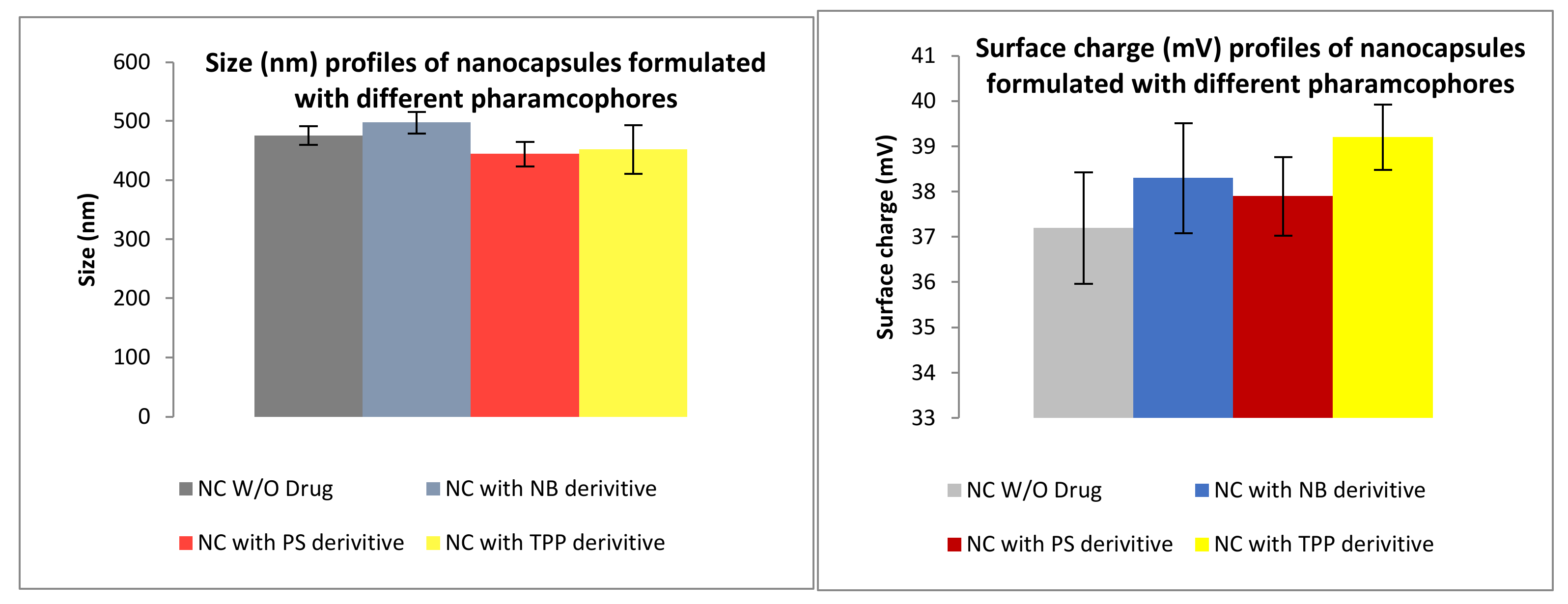
3.2. Formulation
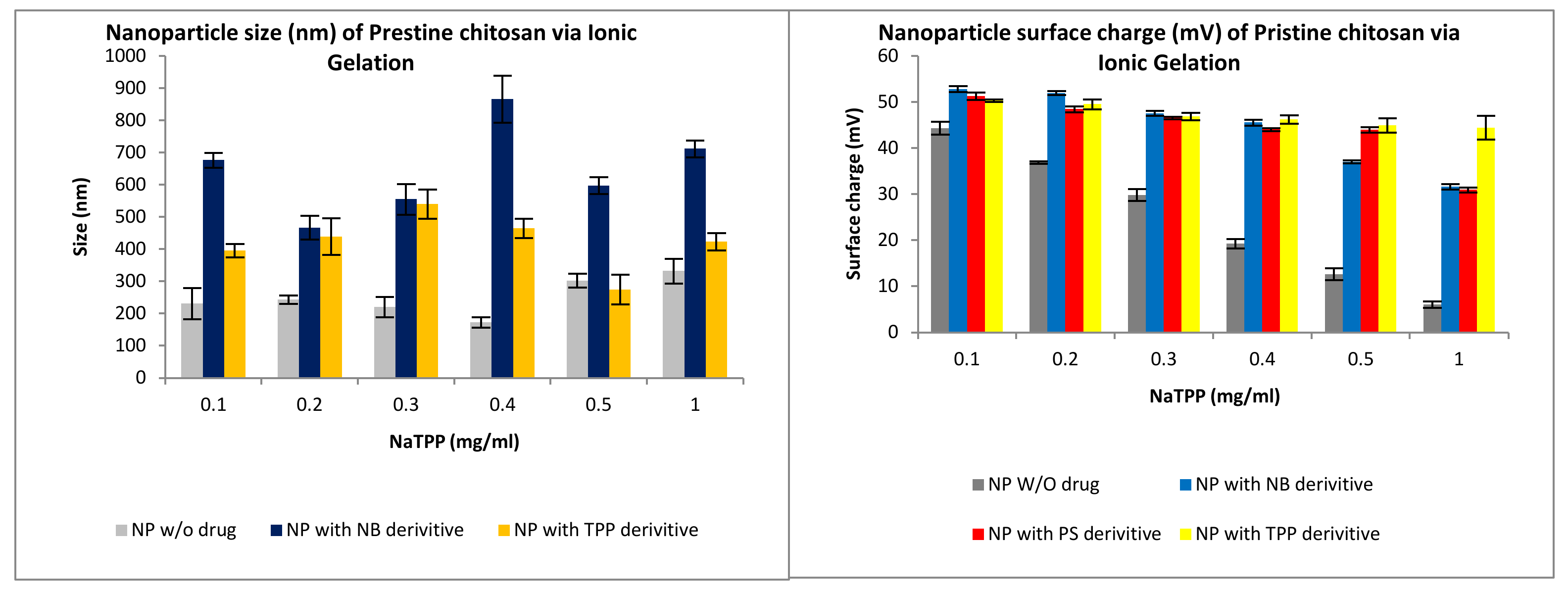
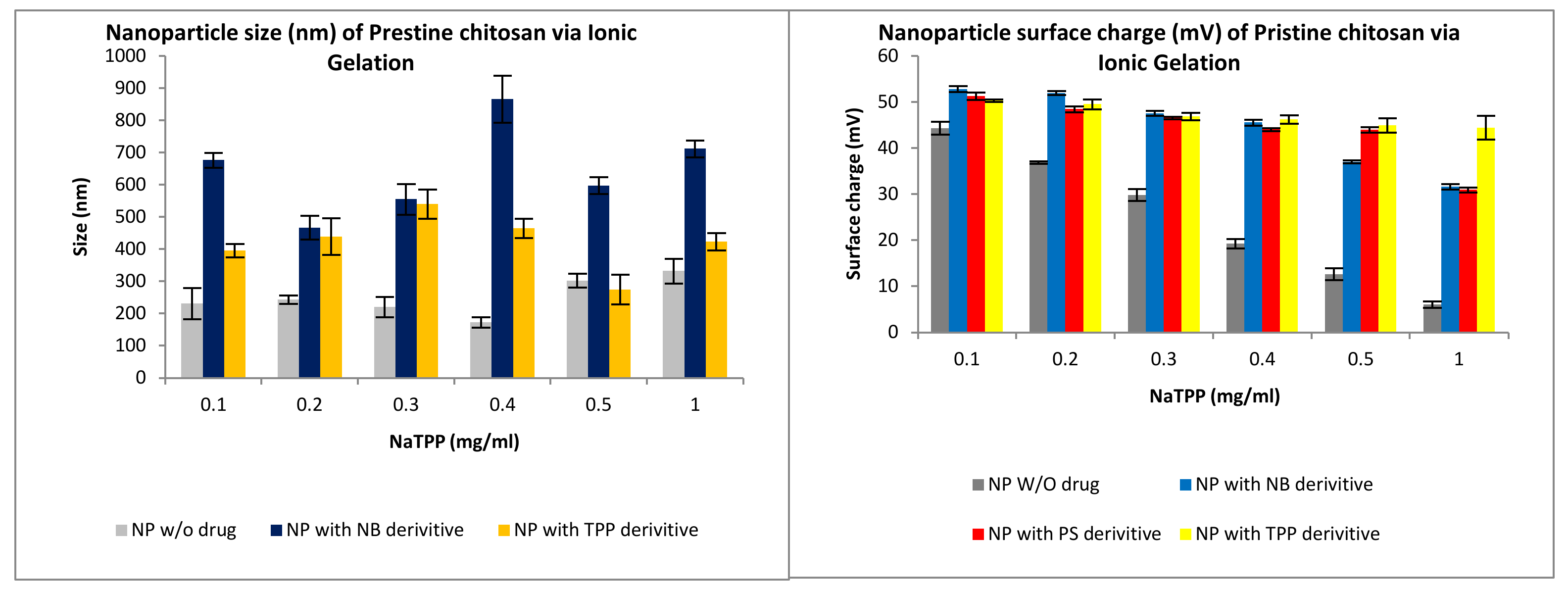
3.2.1. Size and Surface Charge Analysis via IG
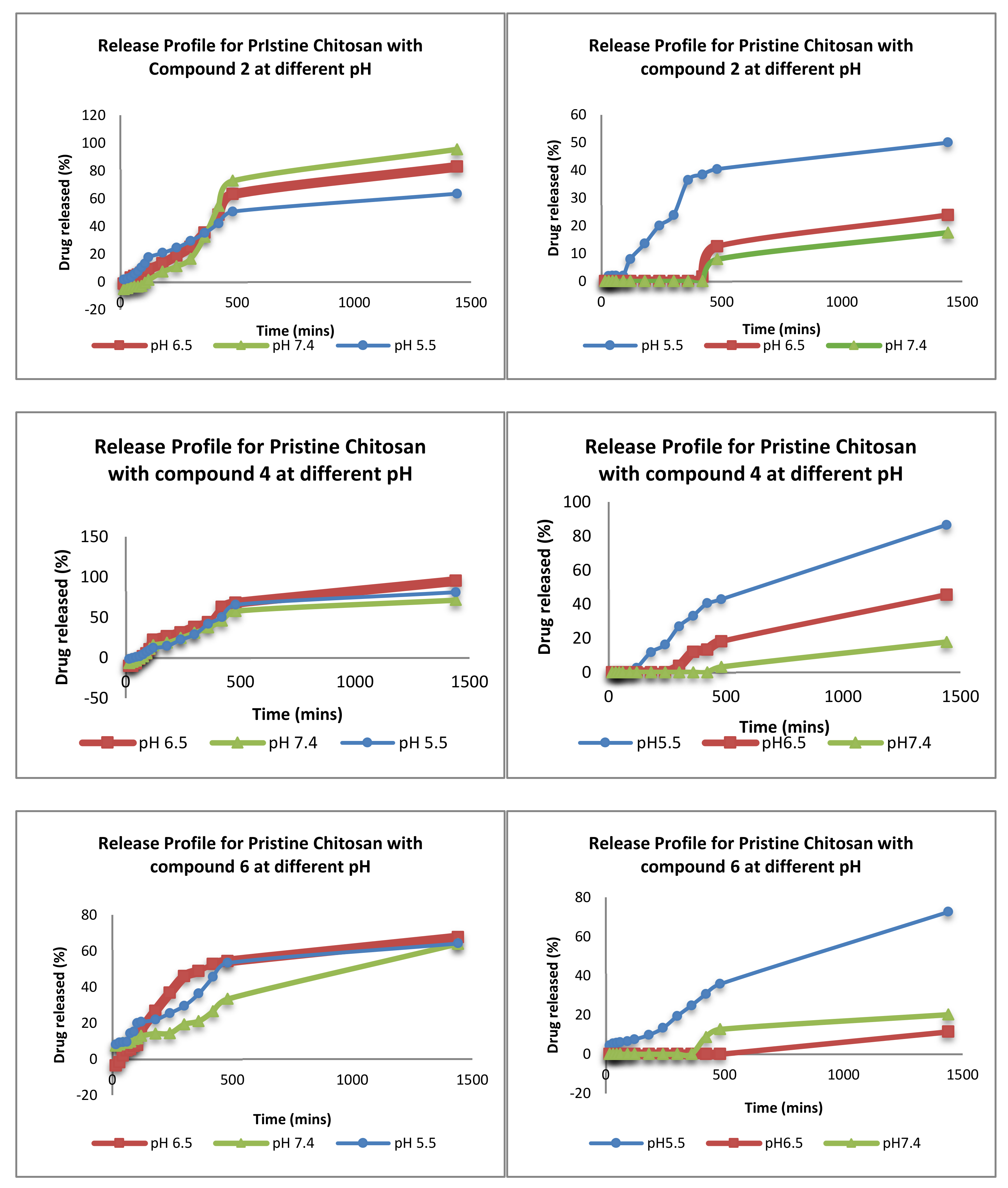
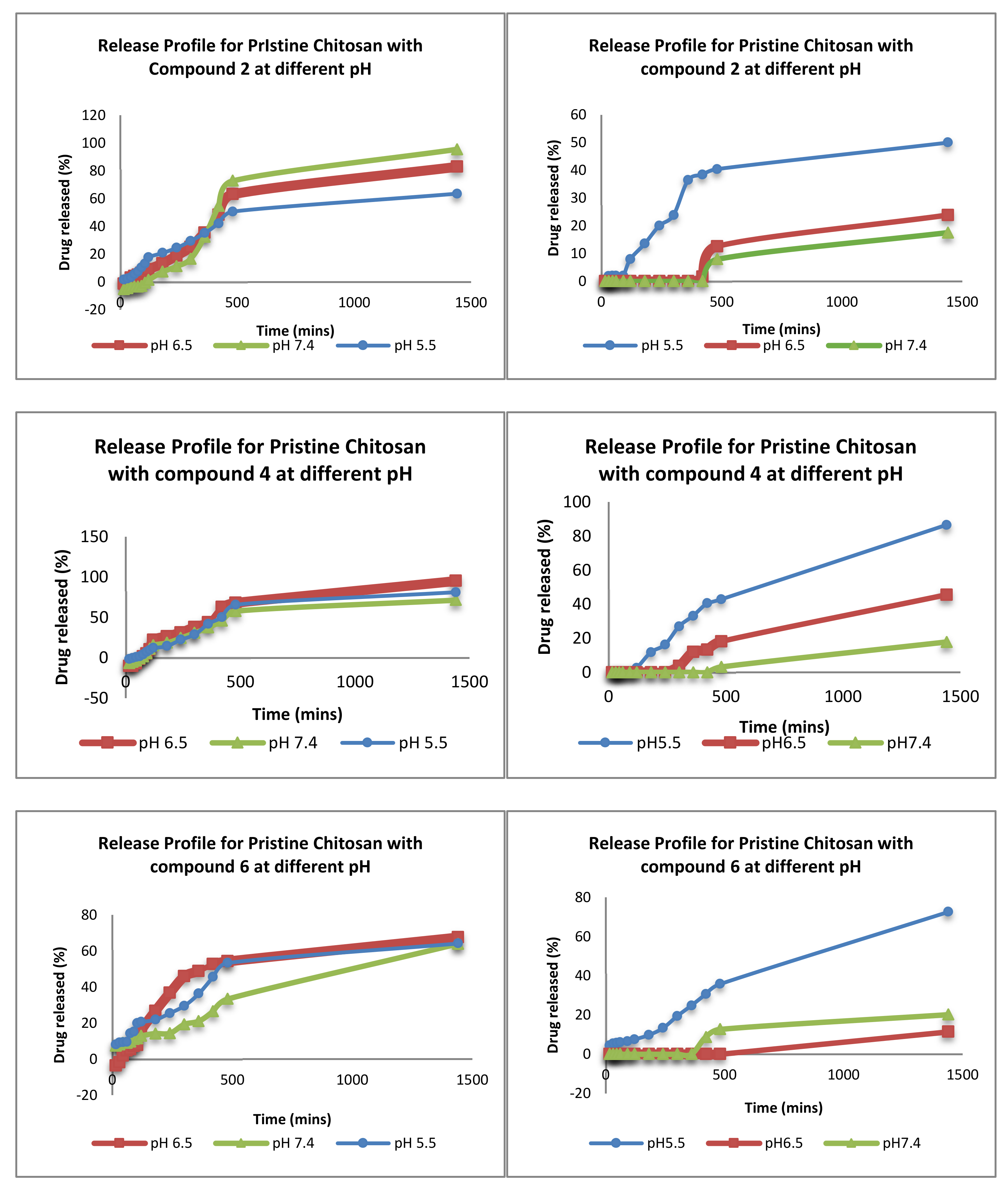
3.2.2. Ex-Vivo Release Studies
3.2.3. Scanning Electron Microscope for Loaded Nanoparticles
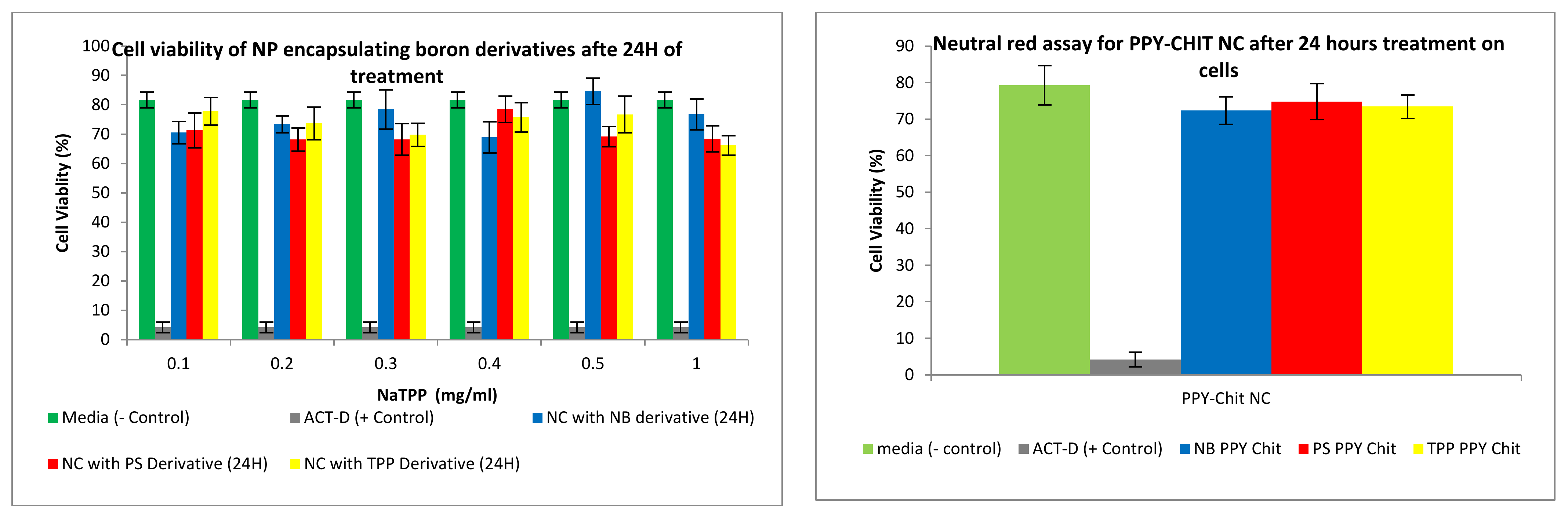
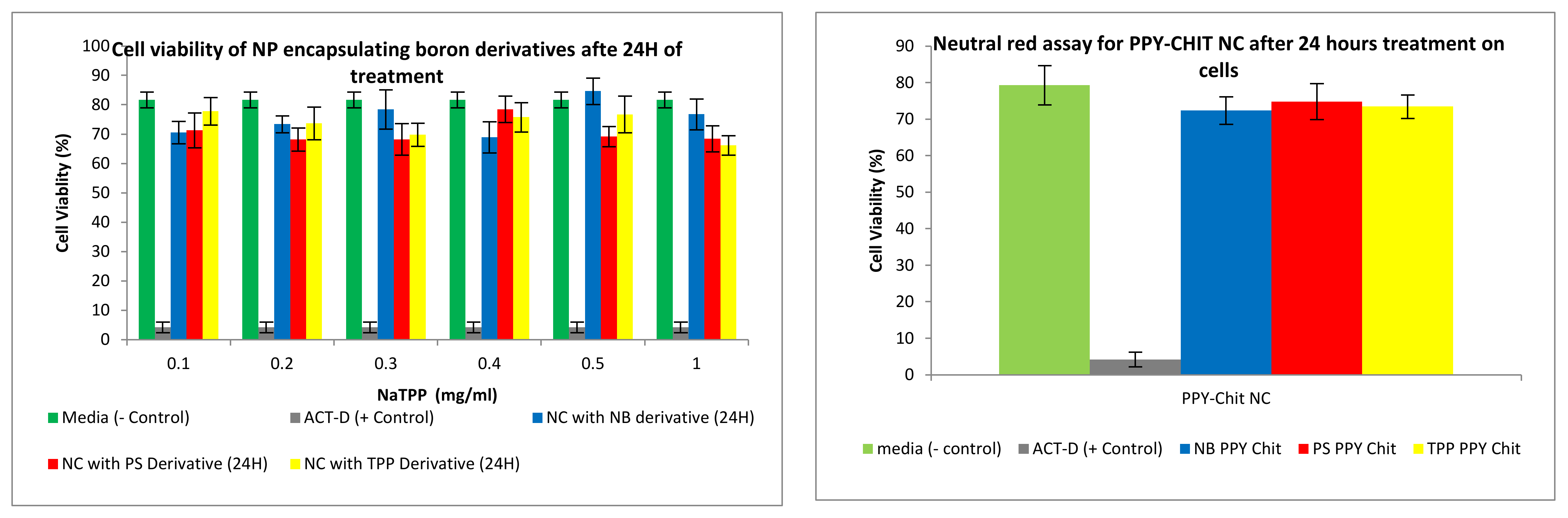
3.2.4. Neutral Red Toxicity Assays
3.3. Preliminary Cell Studies
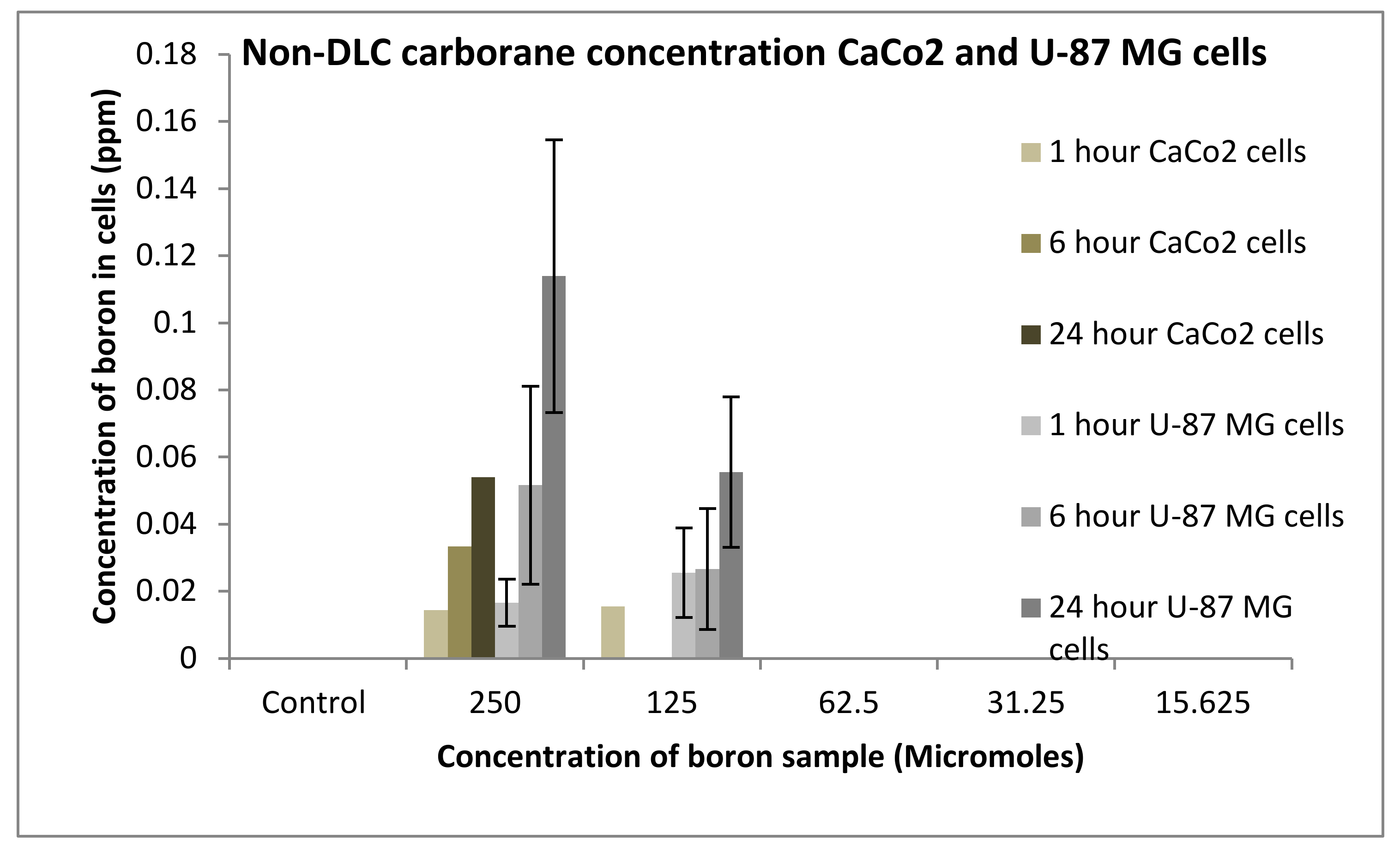
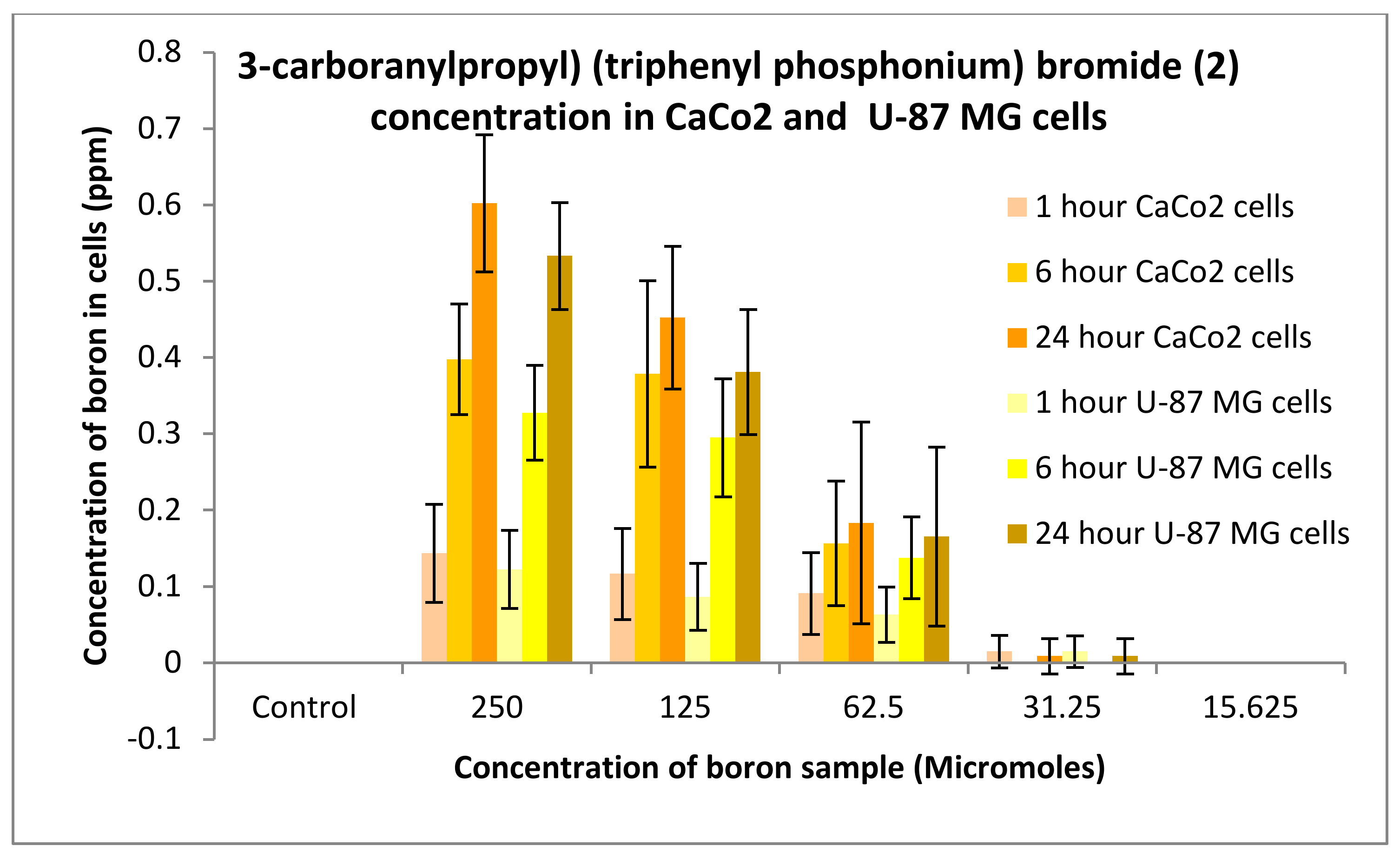
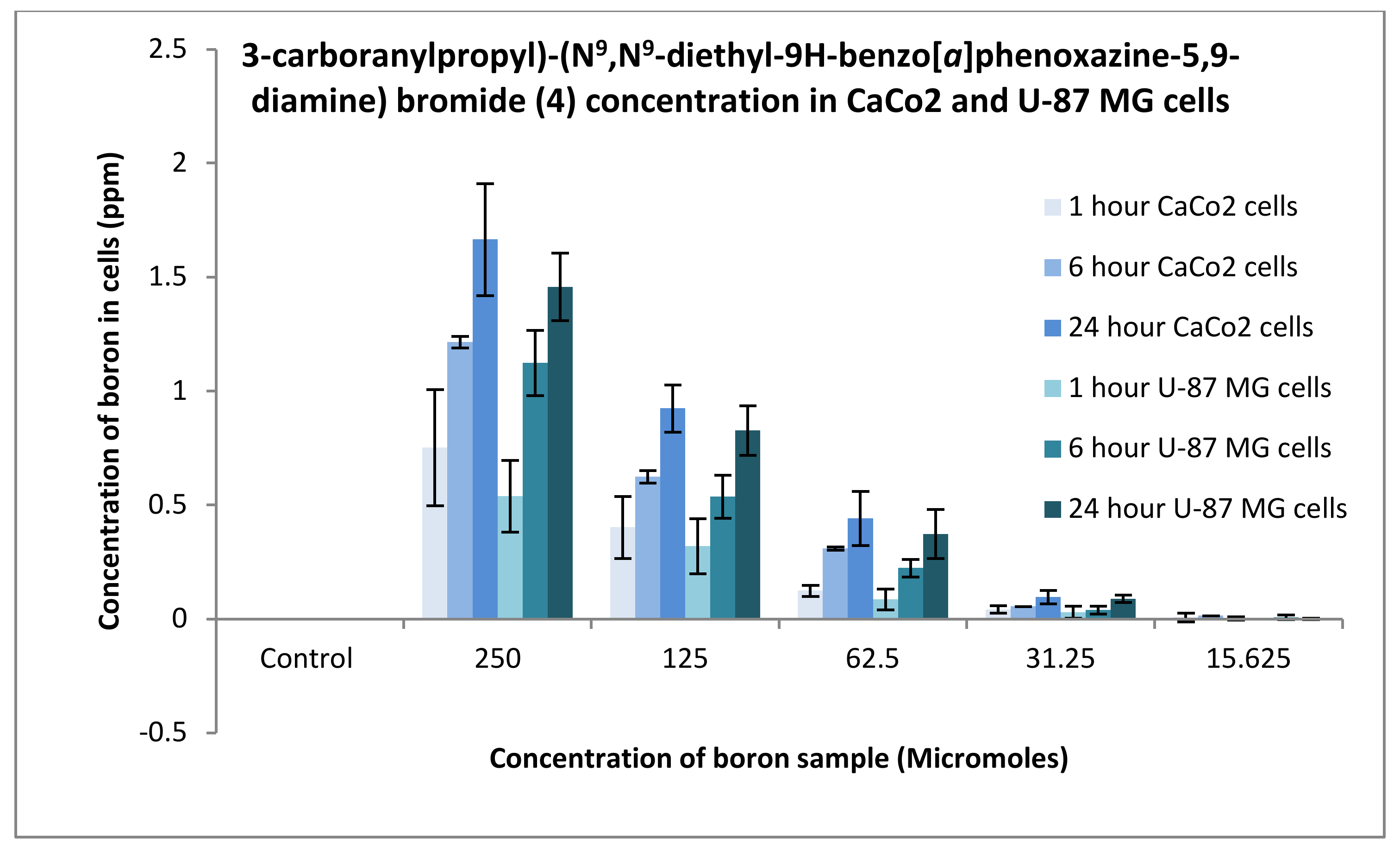
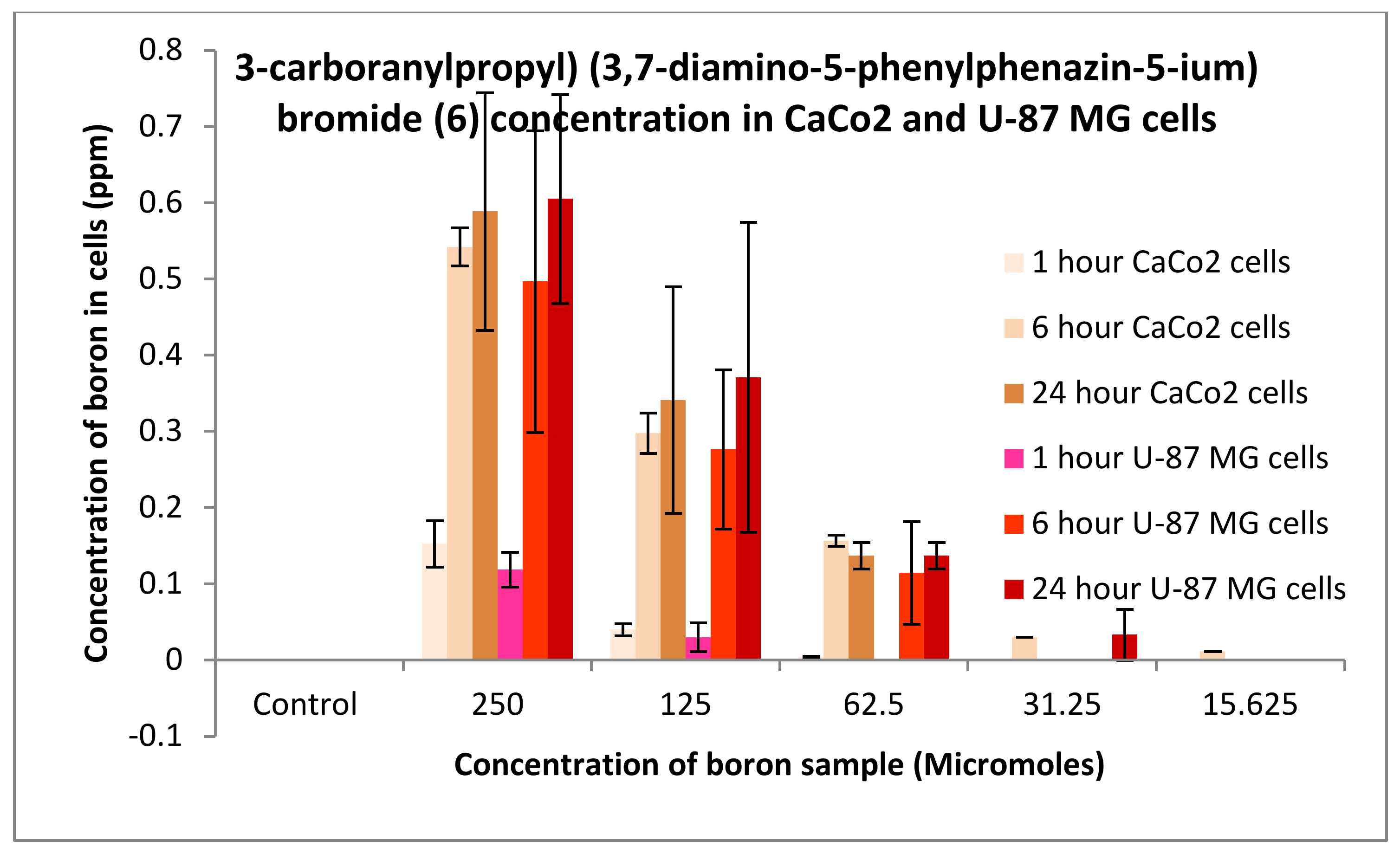
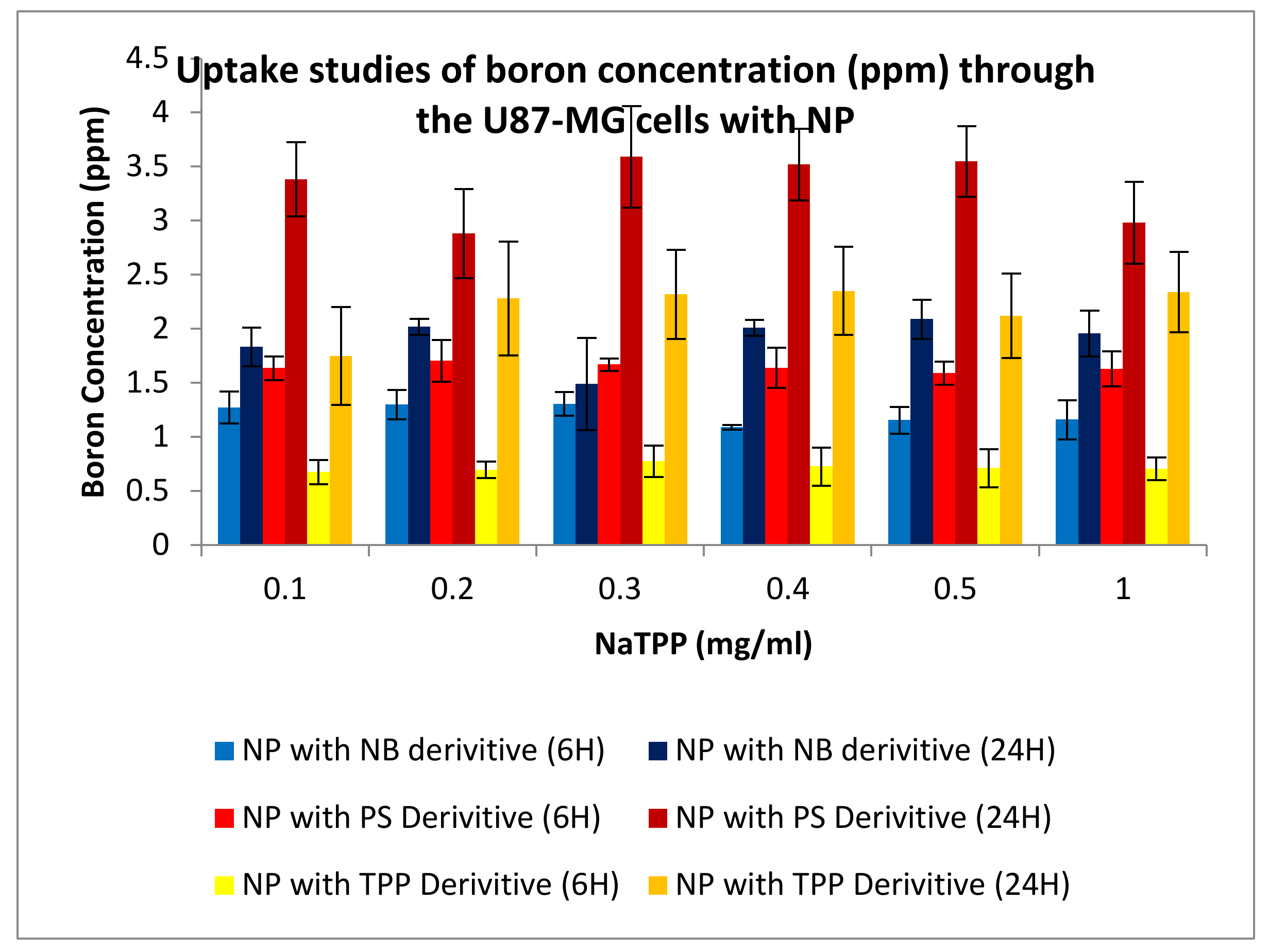
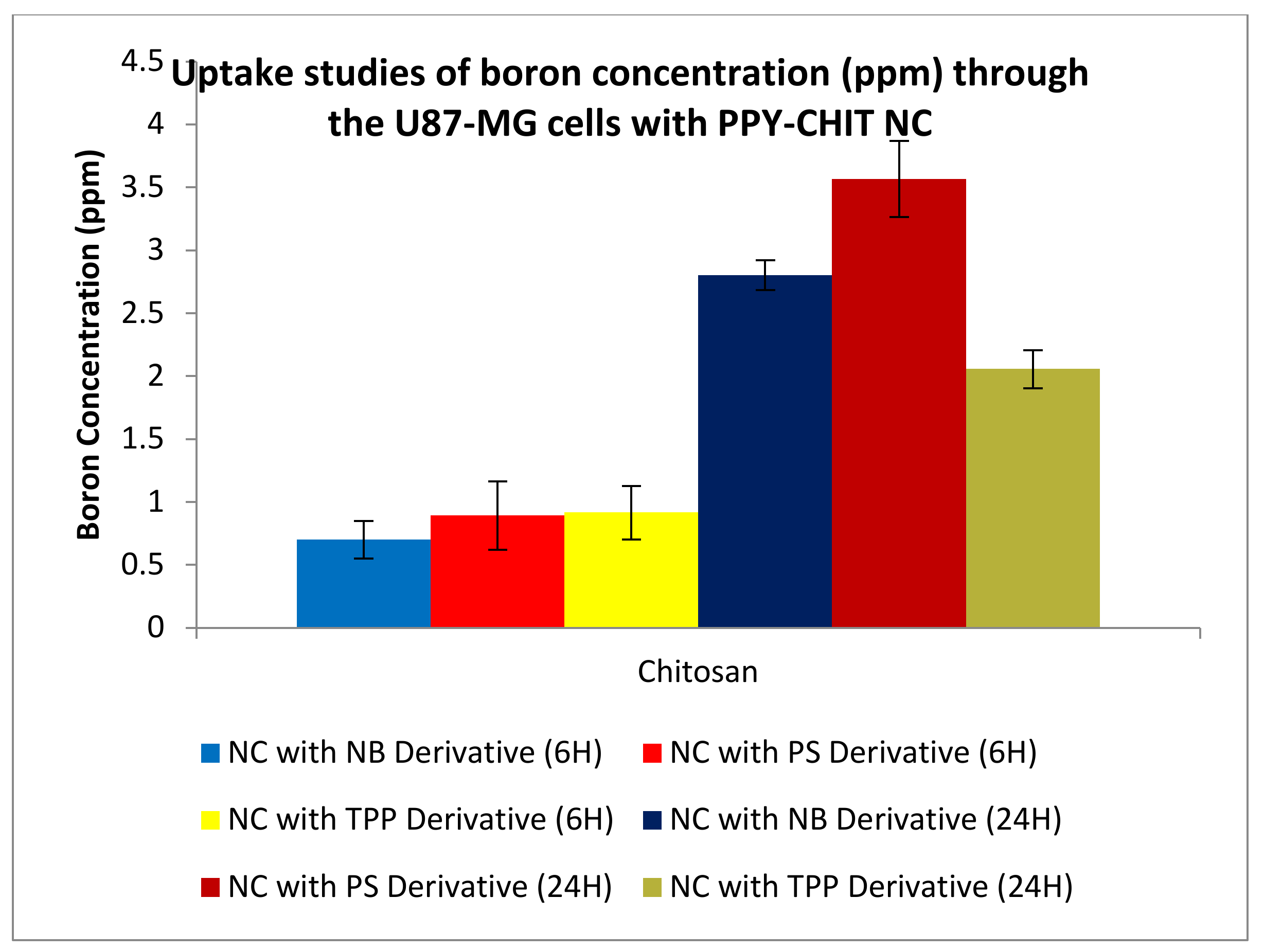
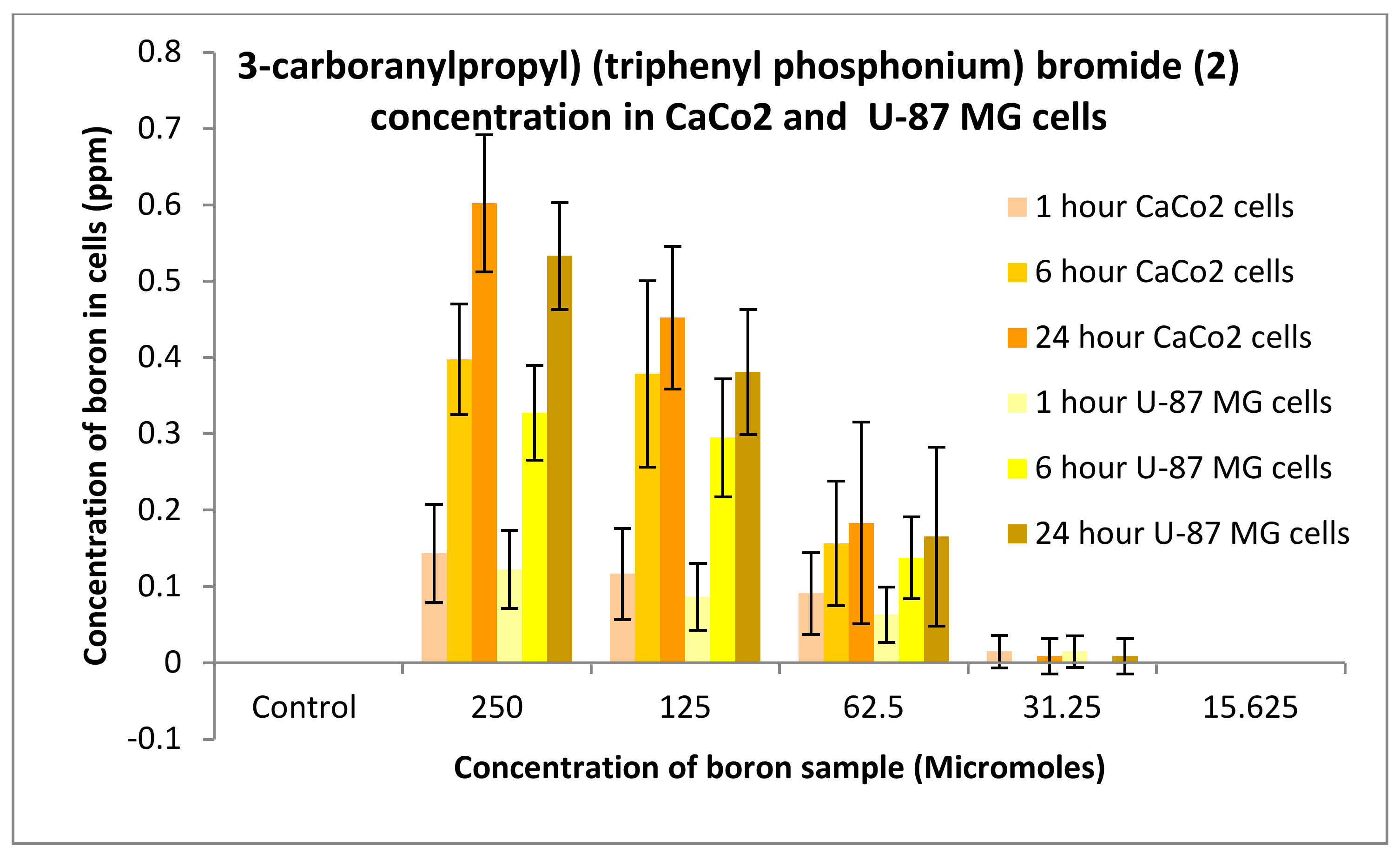
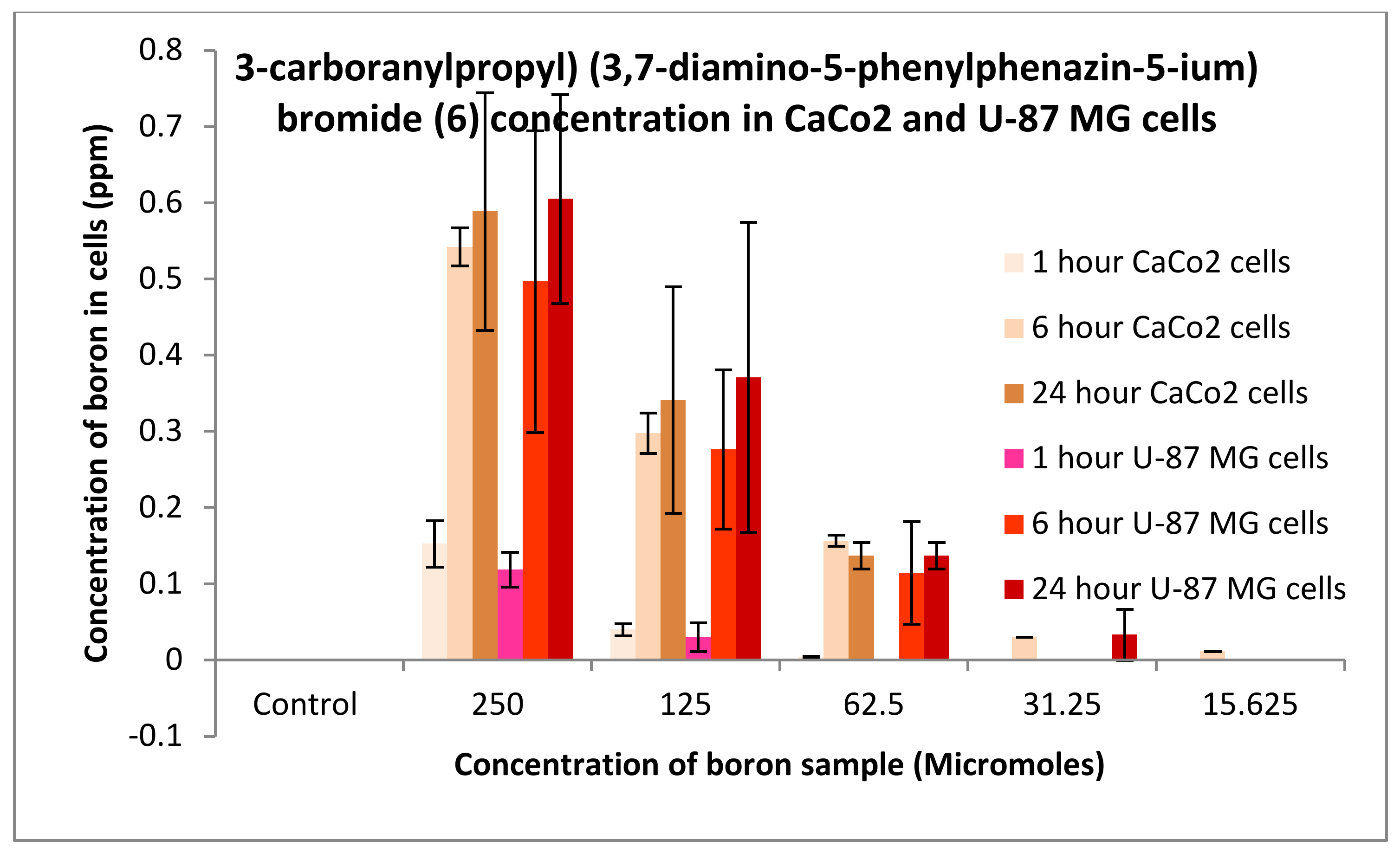
3.3.1. Boron Uptake Studies in CaCo2 Cells and U-87 MG Cells
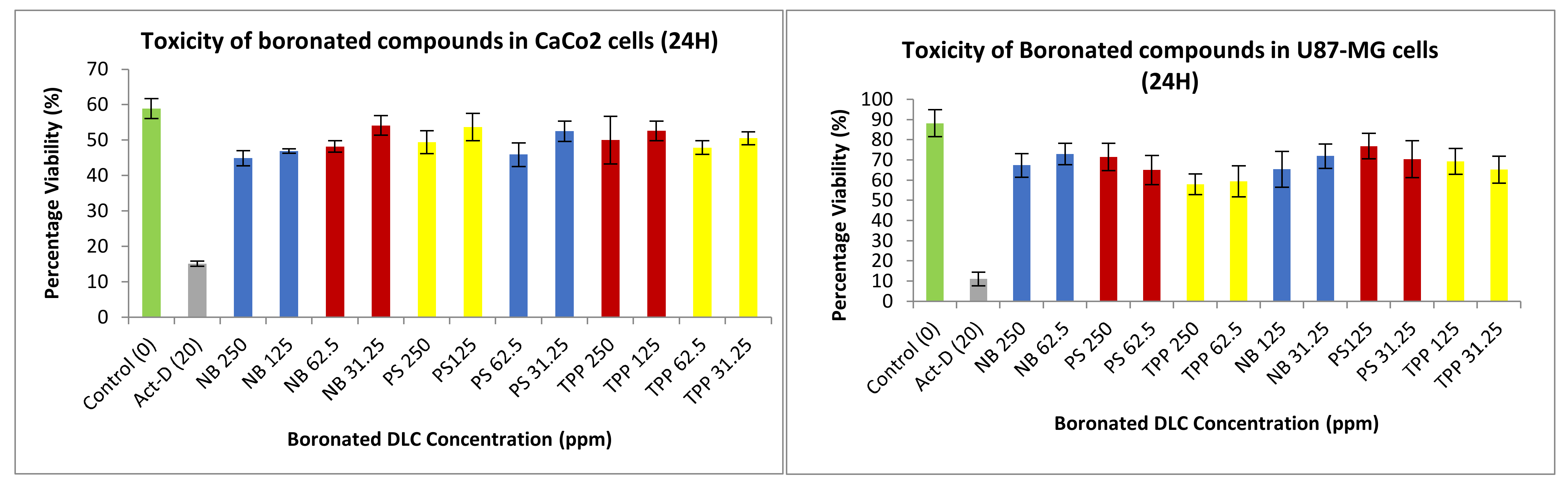
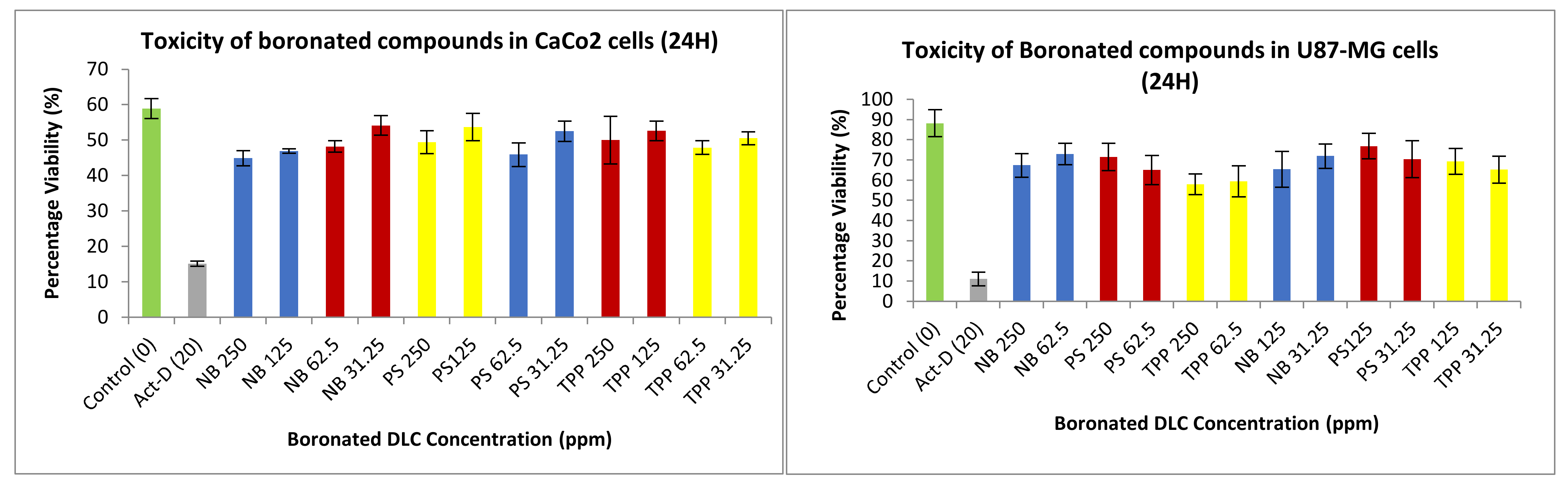
3.3.2. Neutral Red Toxicity Assays
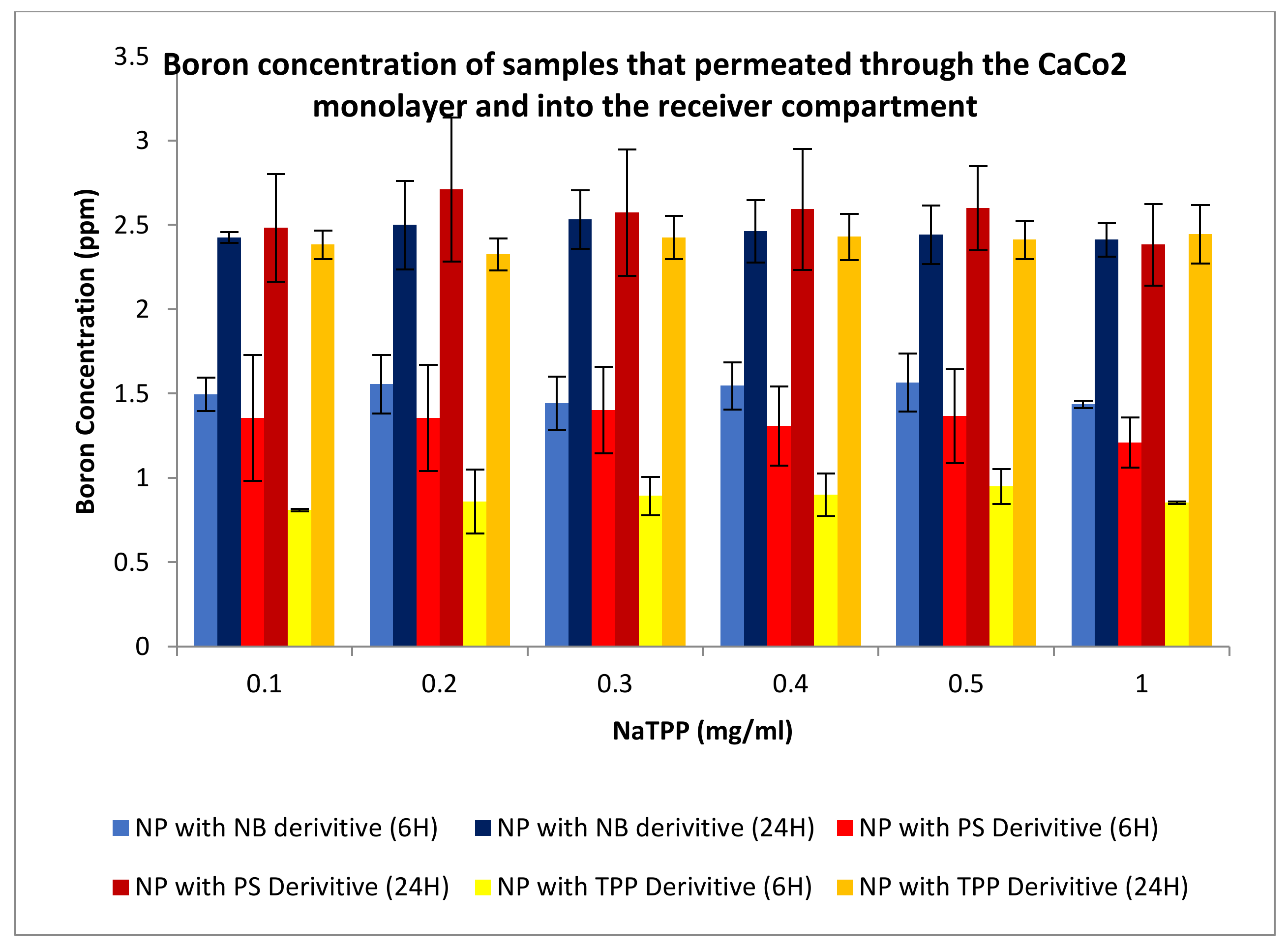
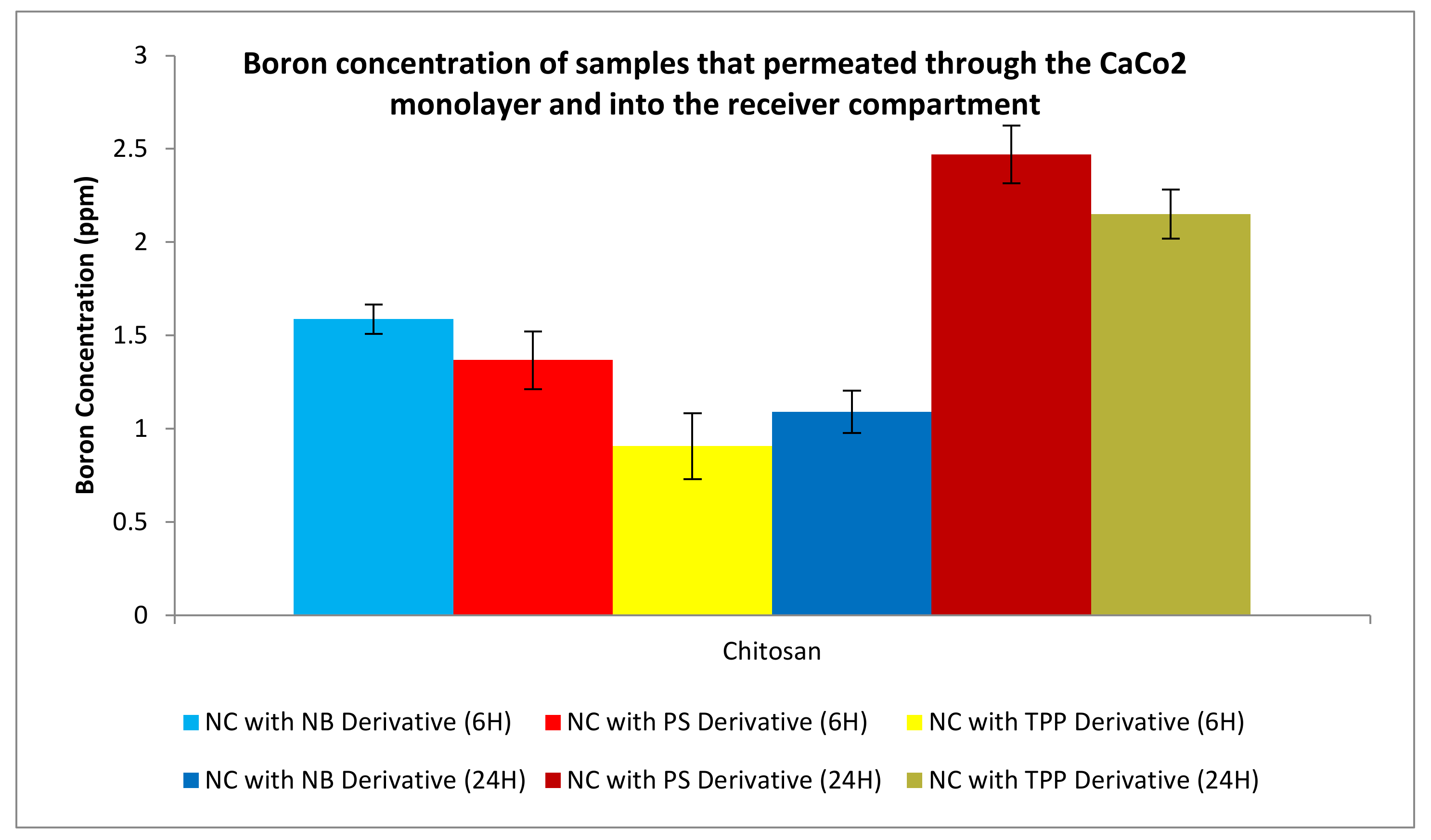
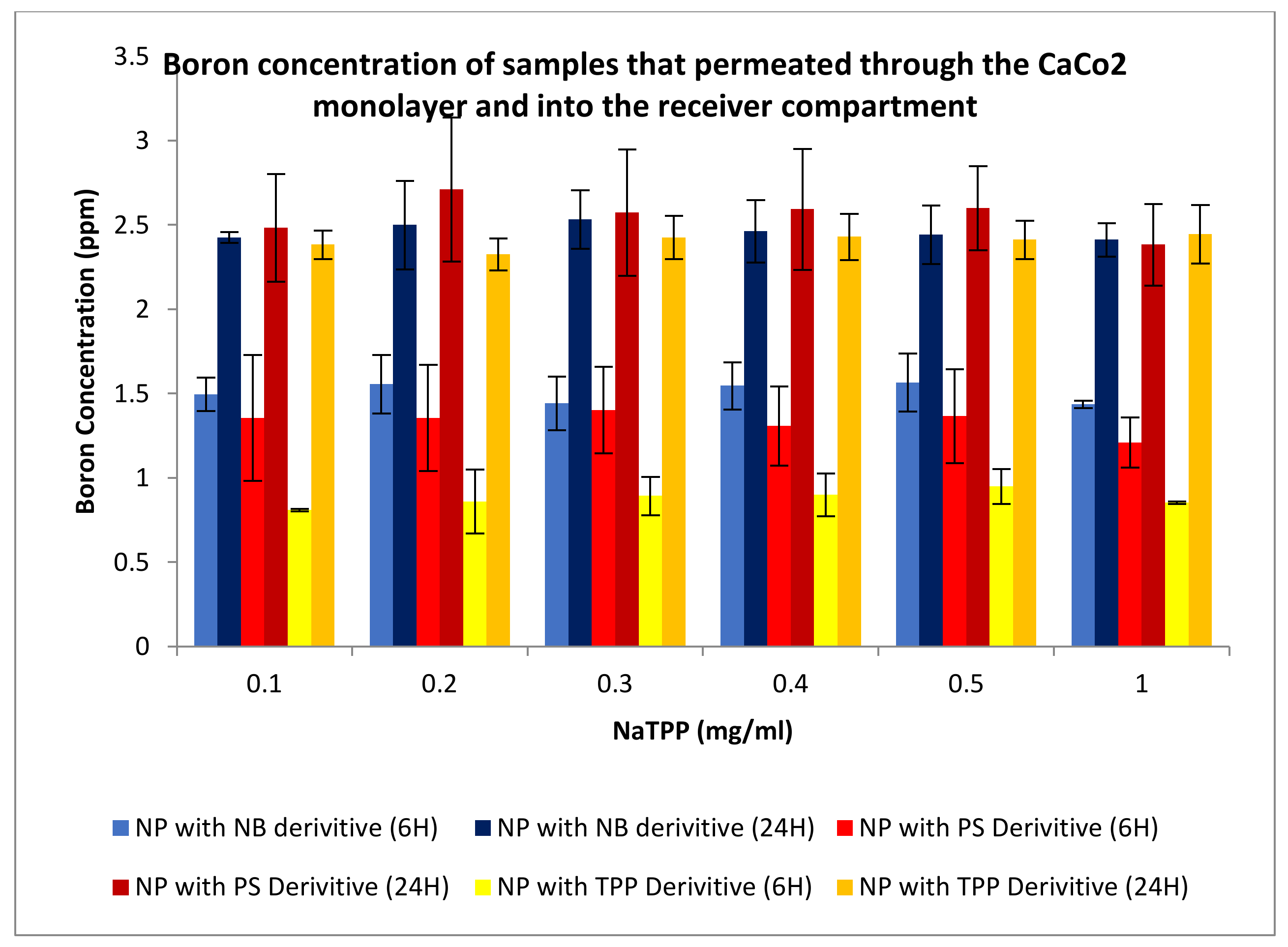
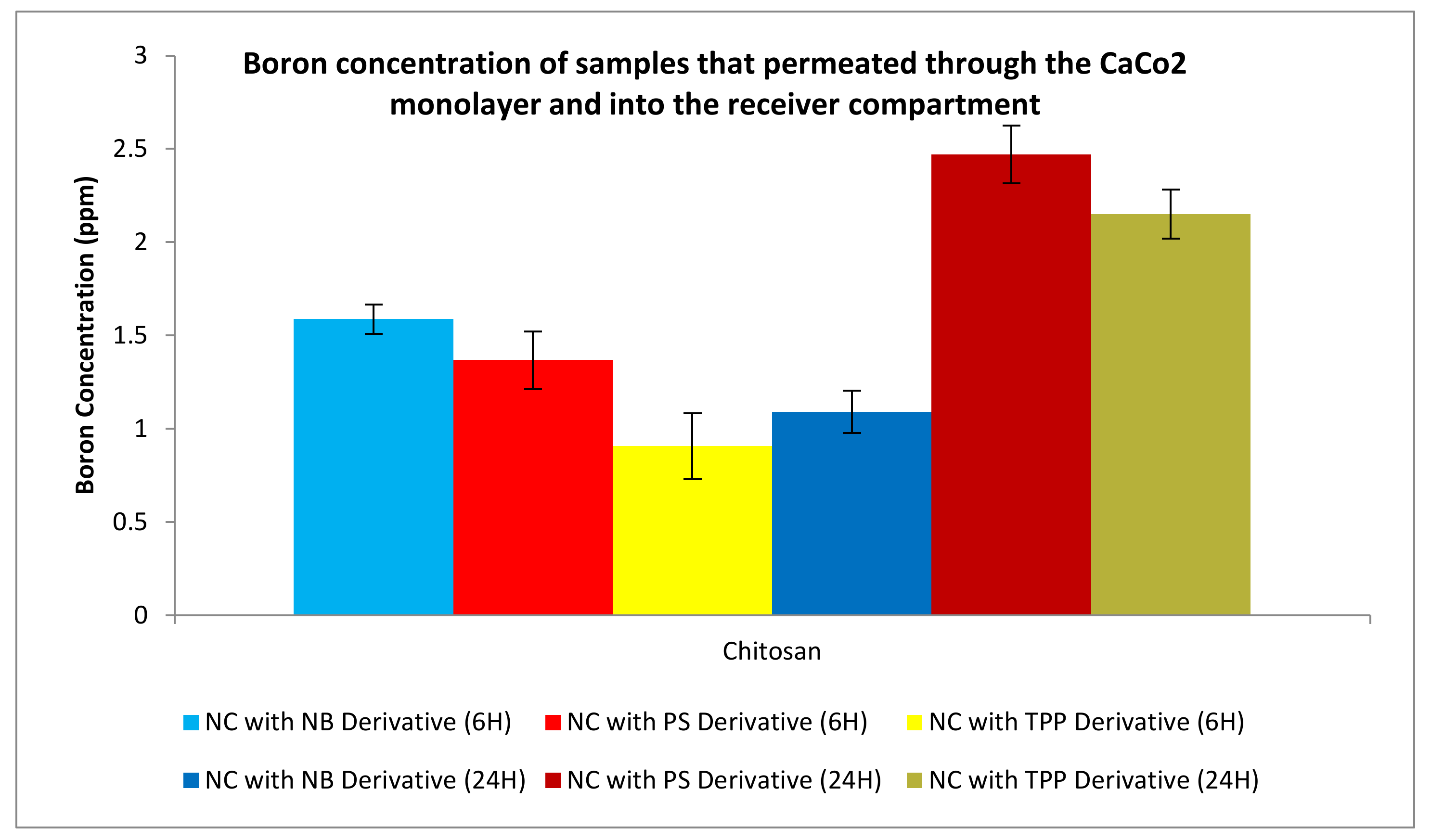
3.3.3. Permeation Studies
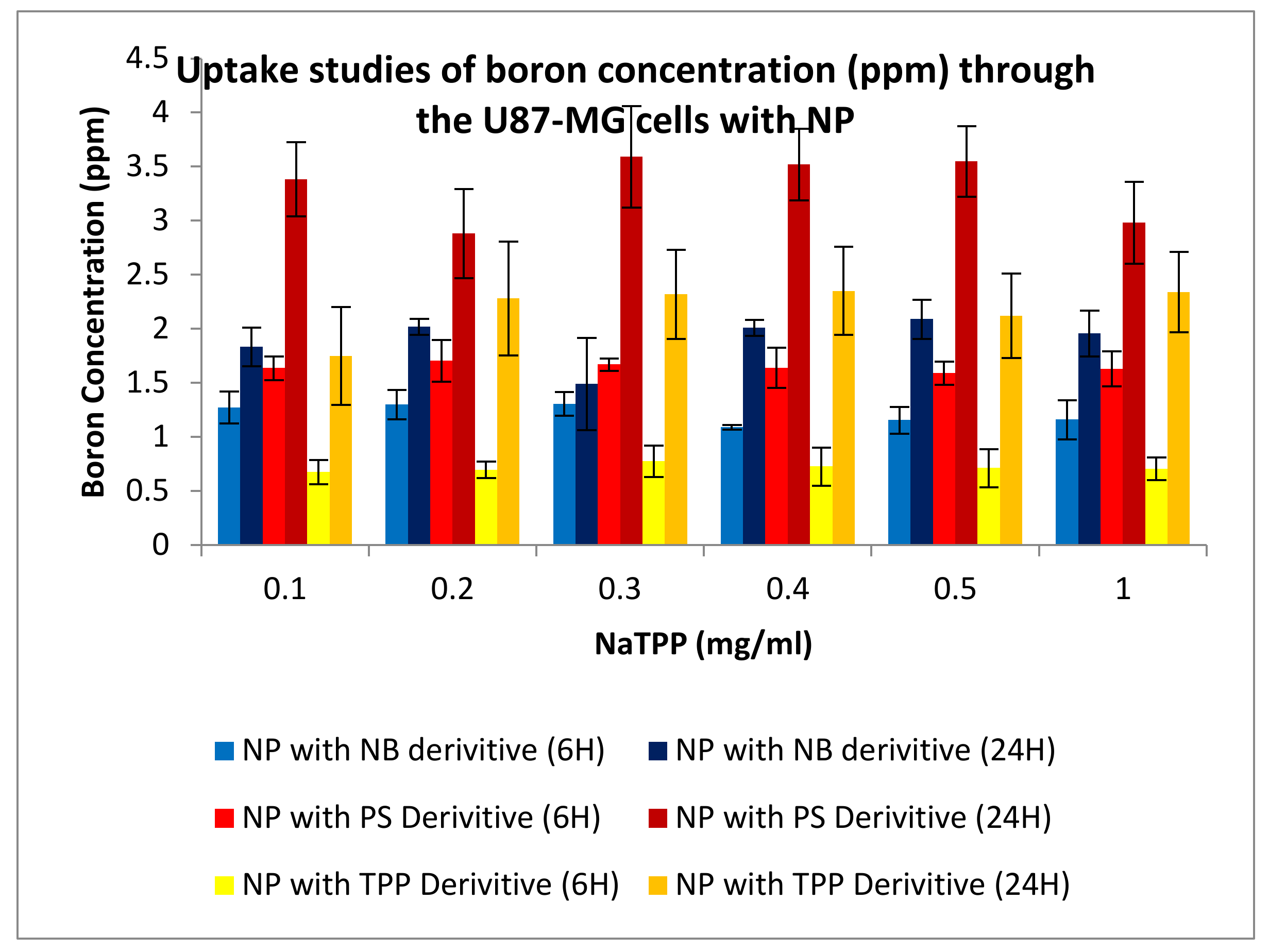
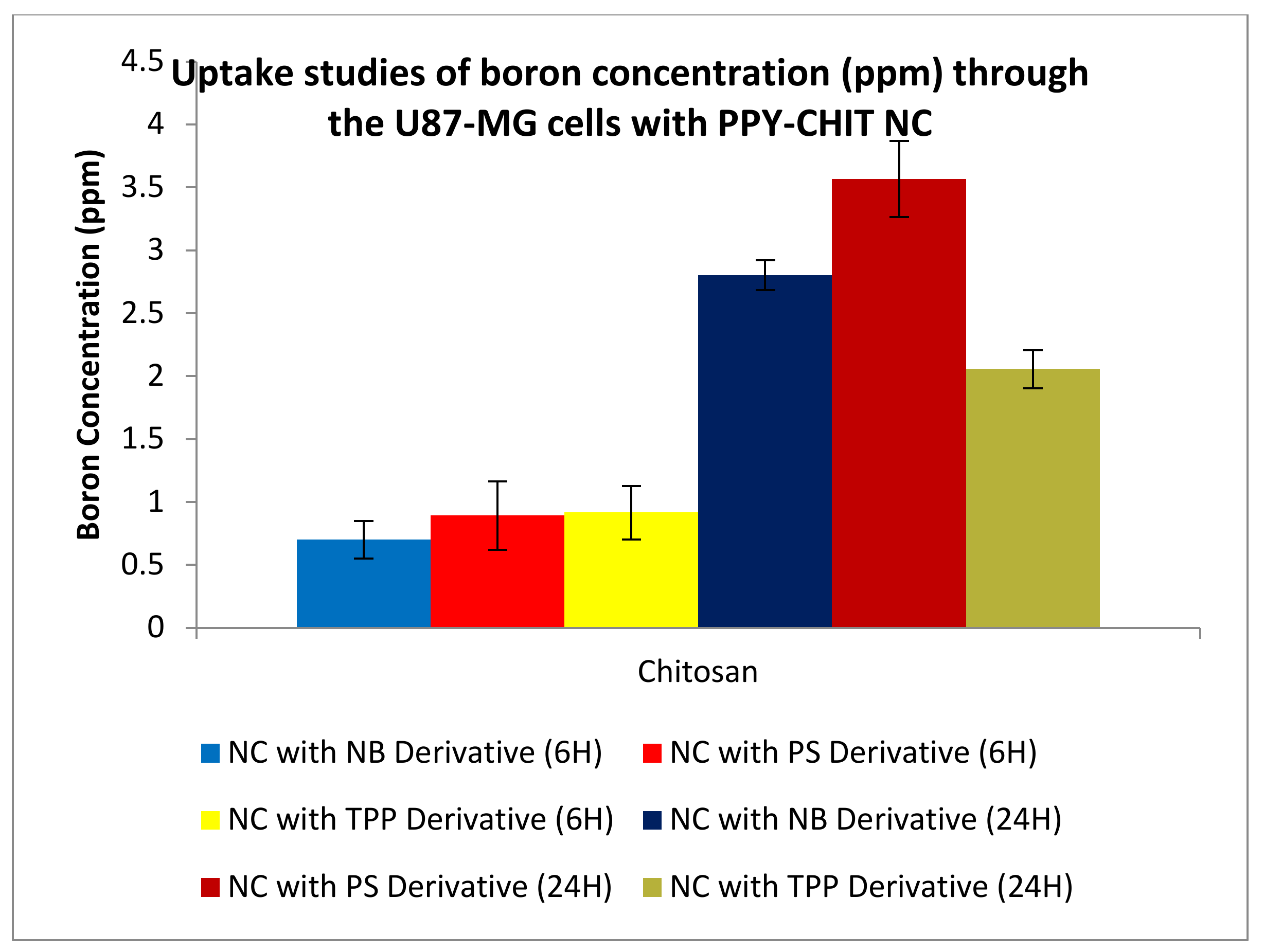
3.3.4. Uptake Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brain Basics: Know Your Brain, National Institute of Neurological Disorders and Stroke. 2018. Available online: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Know-Your-Brain (accessed on 14 October 2020).
- Aiello, L.C.; Wheeler, P. The Expensive-Tissue Hypothesis: The Brain and the Digestive System in Human and Primate Evolution. Curr. Anthropol. 1995, 36, 199–221. [Google Scholar] [CrossRef]
- Pakkenberg, B.; Gundersen, H.J. Neo-cortical neuron number in humans: Effect of sex and age. J. Comp. Neurol. 1997, 384, 312–320. [Google Scholar] [CrossRef]
- Mayer, F.; Mayer, N.; Chinn, L.; Pinsonneault, R.L.; Kroetz, D.; Bainton, R.J. Evolutionary conservation of vertebrate blood–brain barrier chemo-protective mechanisms in drosophila. J. Neurosci. 2009, 29, 3538–3550. [Google Scholar] [CrossRef]
- Harati, R.; Villégier, A.-S.; Banks, W.A.; Mabondzo, A. Susceptibility of juvenile and adult blood–brain barrier to endothelin-1: Regulation of P-glycoprotein and breast cancer resistance protein expression and transport activity. J. Neuroinflamm. 2012, 9, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease associated dysregulation, fluids barriers. Fluids Barriers CNS 2012, 9, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 2016, 4, 1154641–1154652. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Aschner, M.; Ghersi-Egea, J.F. Brain barrier systems: A new frontier in metal neuro-toxicological research. Toxicol. Appl. Pharmacol. 2003, 192, 1–11. [Google Scholar] [CrossRef] [Green Version]
- The Brain Tumour Society. Statistics for Brain Tumours in UK. 2018. Available online: https://www.thebraintumourcharity.org/get-involved/why-we-need-your-support/the-statistics-about-brain-tumours/ (accessed on 29 May 2020).
- Steliarova-Foucher, E.; O’Callaghan, M.; Ferlay, J.; Masuyer, E.; Rosso, S.; Forman, D.; Bray, F.; Comber, H. European Cancer Observatory: Cancer Incidence, Mortality, Prevalence and Survival in Europe. Eur. J. Cancer 2015, 51, 1131–1143. [Google Scholar] [CrossRef]
- Ostrom, Q.T. Brain Tumour Statistics. 2014. Available online: http://www.abta.org/about-us/news/brain-tumor-statistics/ (accessed on 29 May 2020).
- Visser, O.; Ardanaz, E.; Botta, L.; Sant, M.; Tavilla, A.; Minicozzi, P. Survival of adults with primary malignant brain tumours in Europe: Results of the EUROCARE-5 study. Eur. J. Cancer 2015, 51, 2231–2241. [Google Scholar] [CrossRef]
- The Brain Tumour Charity, Brain Tumour Prognosis. 2018. Available online: https://www.thebraintumourcharity.org/understanding-brain-tumours/getting-a-diagnosis/prognosis/prognosis-specific-brain-tumours/ (accessed on 14 October 2020).
- Treating a Malignant Brain Tumour. 2015. Available online: http://www.nhs.uk/Conditions/brain-tumour-malignant/Pages/Treatment.aspx (accessed on 29 May 2020).
- Sotiropoulou, P.A.; Christodoulou, M.S.; Silvani, A.; Herold-Mende, C.; Passarella, D. Chemical approaches to targeting drug resistance in cancer stem cells. Drug Discov. Today 2014, 19, 1547–1562. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, G.; Gomes, A.; Barbu, E.; Nevell, T.G.; Tsibouklis, J. Carborane-based derivatives of delocalised lipophilic cations for boron neutron capture therapy: Synthesis and preliminary in vitro evaluation. J. Mater. Chem. 2008, 18, 4864–4871. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer Stem Cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Gianpiero, C.; Anis, D.; Aikaterini, R.; Eirini, T.; Ioannis, V.S.; Dimitrios, F.G.; John, T. Boron-containing delocalised lipophilic cations for the selective targeting of cancer cells. Med. Chem. Commun. 2017, 8, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Sauerwein, W. Principles and history of neutron capture therapy. Strahlenther. Onkol. 1993, 169, 1–6. [Google Scholar]
- Raju, M.R. Particle radiotherapy: Historical developments and current status. Radiat. Res. 1996, 145, 391–407. [Google Scholar] [CrossRef]
- Davis, M.A.; Little, J.B. Relative biological effectiveness of the 10B (n, α) 7Li reaction in HeLa cells. Radiat. Res. 1970, 43, 534–553. [Google Scholar] [CrossRef]
- Schiffer, D.; Cavalla, P.; Pilkington, G.J. Brain Tumour Invasion: Biological, Clinical and Therapeutic Considerations; Mikkelsen, T., Bjerkvig, R., Laerum, O.D., Rosenbaum, M.L., Eds.; Wiley-Liss: New York, NY, USA, 1998; pp. 161–184. [Google Scholar]
- Yamamoto, T.; Nakai, K.; Kageji, T.; Kumada, H.; Endo, K.; Matsuda, M.; Shibata, Y.; Matsumura, A. Boron neutron capture therapy for newly diagnosed glioblastoma. Radiother. Oncol. 2009, 91, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Don, A.S.; Hogg, P.J. Mitochondria as cancer drug targets. Trends Mol. Med. 2004, 10, 372–378. [Google Scholar] [CrossRef]
- Weissig, V.; Cheng, S.M.; D’Souza, G.G. Mitochondrial pharmaceutics. Mitochondrion 2004, 3, 229–244. [Google Scholar] [CrossRef]
- Talegaonkar, S.; Mishra, P.R. Intranasal delivery: An approach to bypass the blood brain barrier. Indian J. Pharmacol. 2004, 36, 140–147. [Google Scholar]
- Illum, L. Nasal drug delivery—Possibilities, problems and solutions. J. Control. Release 2003, 87, 187–198. [Google Scholar] [CrossRef]
- Djupesland, P.G.; Messina, J.C.; Mahmoud, R.A. The nasal approach to delivering treatment for brain diseases: An anatomic, physiologic and delivery technology overview. Ther. Deliv. 2014, 5, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Welch, M.J.; Hawker, C.J.; Wooley, K.L. The advantages of nanoparticles for PET. J. Nucl. Med. 2009, 50, 1743–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akagi, T.; Matsusaki, M.; Akashi, M. Pharmaceutical and Medical Applications of Poly-Gamma-Glutamic Acid. Amino-Acid Homopolym. Occur. Nat. 2010, 15, 119–153. [Google Scholar]
- Wu, Y.; Yang, W.; Wang, C.; Hu, J.; Fu, S. Chitosan nanoparticles as a novel drug delivery system for ammonium glycyrrhizinate. Int. J. Pharm. 2005, 295, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Malmo, J.; Sandvig, A.; Vårum, K.M.; Strand, S.P. Nanoparticle mediated P-glycoprotein silencing for improved drug delivery across the blood-brain barrier: A siRNA-chitosan approach. PLoS ONE 2013, 81, e54182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Valdez, O.; Champagne-Hartley, R.; Saldivar-Guerra, E.; Champagne, P.; Cunningham, M.F. Modification of chitosan with polystyrene and poly (n-butyl acrylate) via nitroxide-mediated polymerization and grafting from approach in homogeneous media. Polym. Chem. 2015, 6, 2827–2836. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Elgadir, M.A.; Uddin, M.S.; Ferdosh, S.; Adam, A.; Chowdhury, A.J.; Sarker, M.Z. Impact of chitosan composites and chitosan nanoparticle composites on various drug delivery Systems: A review. J. Food Drug Anal. 2015, 23, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Dey, B. Chitosan microspheres in novel drug delivery systems. Indian J. Pharm. Sci. 2011, 73, 355. [Google Scholar] [PubMed]
- Ramya, R.; Jayachandran, V.; Kwon, K.S.; Sudha, P.N. Biomedical Applications of Chitosan: An Overview. J. Biomater. Tissue Eng. 2012, 2, 100–111. [Google Scholar] [CrossRef]
- Pillay, V.; Tsai, T.S.; Choonara, Y.E.; du Toit, L.C.; Kumar, P.; Modi, G.; Naidoo, D.; Tomar, L.K.; Tyagi, C.; Ndesendo, V.M. A review of integrating electroactive polymers as responsive systems for specialized drug delivery applications. J. Biomed. Mater. Res. Part A 2014, 102, 2039–2054. [Google Scholar] [CrossRef] [PubMed]
- Uppalapati, D.; Boyd, B.J.; Garg, S.; Travas-Sejdic, J.; Svirskis, D. Conducting polymers with defined micro- or nanostructures for drug delivery. Biomaterials 2016, 111, 149–162. [Google Scholar] [CrossRef]
- Lu, X.; Chao, D.; Chen, J.; Zhang, W.; Wei, Y. Preparation and characterization of inorganic/organic hybrid nanocomposites based on Au nanoparticles and polypyrrole. Mater. Lett. 2006, 60, 2851–2854. [Google Scholar] [CrossRef]
- Cheng, D.; Xia, H.; Chan, H.S.O. Facile Fabrication of AgCl@Polypyrrole-Chitosan Core-Shell Nanoparticles and Polymeric Hollow Nanospheres. Langmuir 2004, 20, 9909–9912. [Google Scholar] [CrossRef]
- Lockman, P.R.; Koziara, J.M.; Mumper, R.J.; Allen, D.D. Nanoparticle surface surface charges alter blood–brain barrier integrity and permeability. J. Drug Target. 2004, 12, 635–641. [Google Scholar] [CrossRef]
- Gao, X.; Qian, J.; Zheng, S.; Changyi, Y.; Zhang, J.; Ju, S. Overcoming the blood–brain barrier for delivering drugs into the brain by using adenosine receptor nano-agonist. ACS Nano 2014, 8, 3678–3689. [Google Scholar] [CrossRef]
- Jallouli, Y.; Paillard, A.; Chang, J.; Sevin, E.; Betbeder, D. Influence of surface surface charge and inner composition of porous nanoparticles to cross blood–brain barrier in vitro. Int. J. Pharm. 2007, 344, 103–109. [Google Scholar] [CrossRef]
- Goldsmith, M.; Abramovitz, L.; Peer, D. Precision nanomedicine in neurodegenerative diseases. ACS Nano 2014, 8, 1958–1965. [Google Scholar] [CrossRef]
- Barbu, E.; Molnar, E.; Tsibouklis, J.; Gorecki, D.C. The potential for nanoparticle-based drug delivery to the brain: Overcoming the blood–brain barrier. Expert Opin. Drug Deliv. 2009, 6, 553–565. [Google Scholar] [CrossRef]
- Lockman, P.R.; Mumper, R.J.; Khan, M.A.; Allen, D.D. Nanoparticle technology for drug delivery across the blood–brain barrier. Drug Dev. Ind. Pharm. 2002, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Etame, A.B.; Smith, C.A.; Chan, W.C.; Rutka, J.T. Design and potential application of PEGylated gold nanoparticles with size-dependent permeation through brain microvasculature. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Hanada, S.; Fujioka, K.; Inoue, Y.; Kanaya, F.; Manome, Y.; Yamamoto, K. Cell-based in vitro blood–brain barrier model can rapidly evaluate nanoparticles’ brain permeability in association with particle size and surface modification. Int. J. Mol. Sci. 2014, 15, 1812–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonavane, G.; Tomoda, K.; Makino, K. Biodistribution of colloidal gold nanoparticles after intravenous administration: Effect of particle size. Colloids Surf. B Biointerfaces 2008, 66, 274–280. [Google Scholar] [CrossRef]
- Decuzzi, P.; Godin, B.; Tanaka, T.; Lee, S.Y.; Chiappini, C.; Liu, X. Size and shape effects in the bio-distribution of intravascularly injected particles. J. Control. Release 2010, 141, 320–327. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daou, A.; Alany, R.G.; Calabrese, G. Formulation of Boron Encapsulated Smart Nanocapsules for Targeted Drug Delivery to the Brain. Appl. Sci. 2021, 11, 10738. https://doi.org/10.3390/app112210738
Daou A, Alany RG, Calabrese G. Formulation of Boron Encapsulated Smart Nanocapsules for Targeted Drug Delivery to the Brain. Applied Sciences. 2021; 11(22):10738. https://doi.org/10.3390/app112210738
Chicago/Turabian StyleDaou, Anis, Raid G. Alany, and Gianpiero Calabrese. 2021. "Formulation of Boron Encapsulated Smart Nanocapsules for Targeted Drug Delivery to the Brain" Applied Sciences 11, no. 22: 10738. https://doi.org/10.3390/app112210738
APA StyleDaou, A., Alany, R. G., & Calabrese, G. (2021). Formulation of Boron Encapsulated Smart Nanocapsules for Targeted Drug Delivery to the Brain. Applied Sciences, 11(22), 10738. https://doi.org/10.3390/app112210738