Role of the Binding Motifs in the Energy Level Alignment and Conductance of Amine-Gold Linked Molecular Junctions within DFT and DFT + Σ



Abstract
1. Introduction
2. Methods
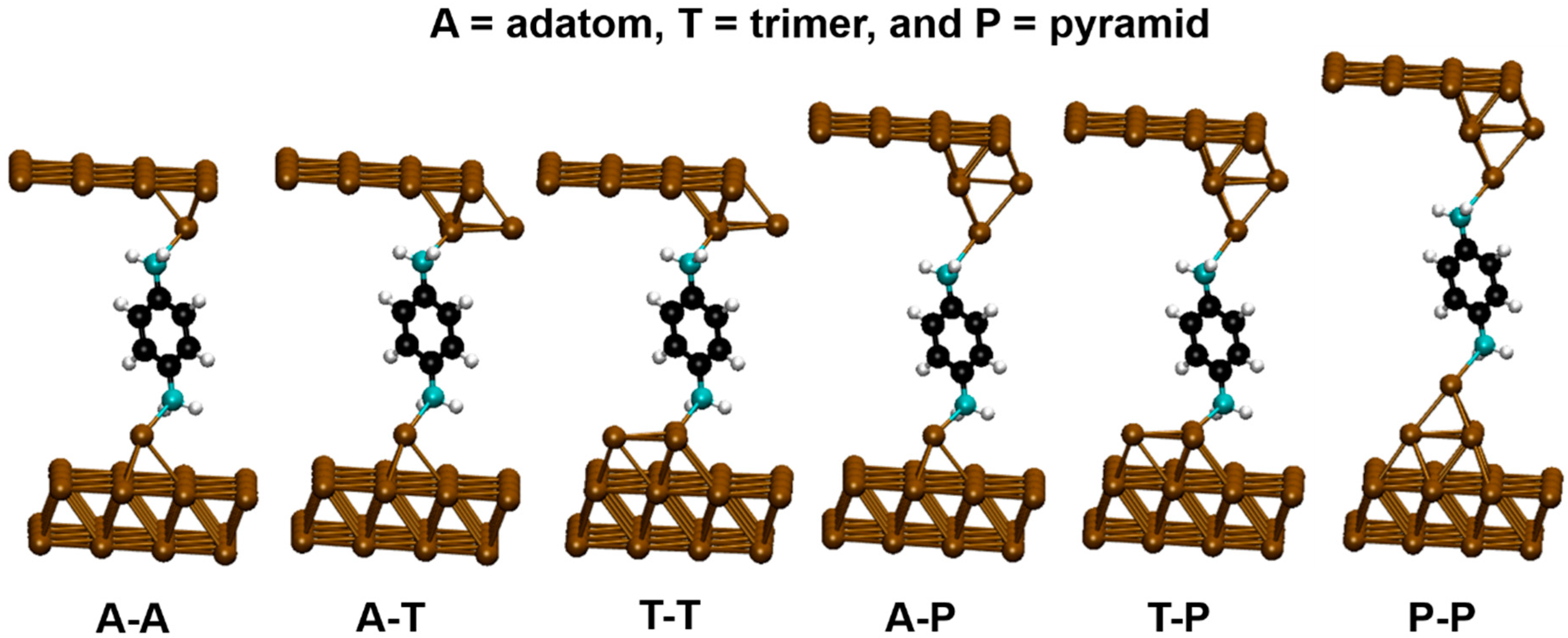
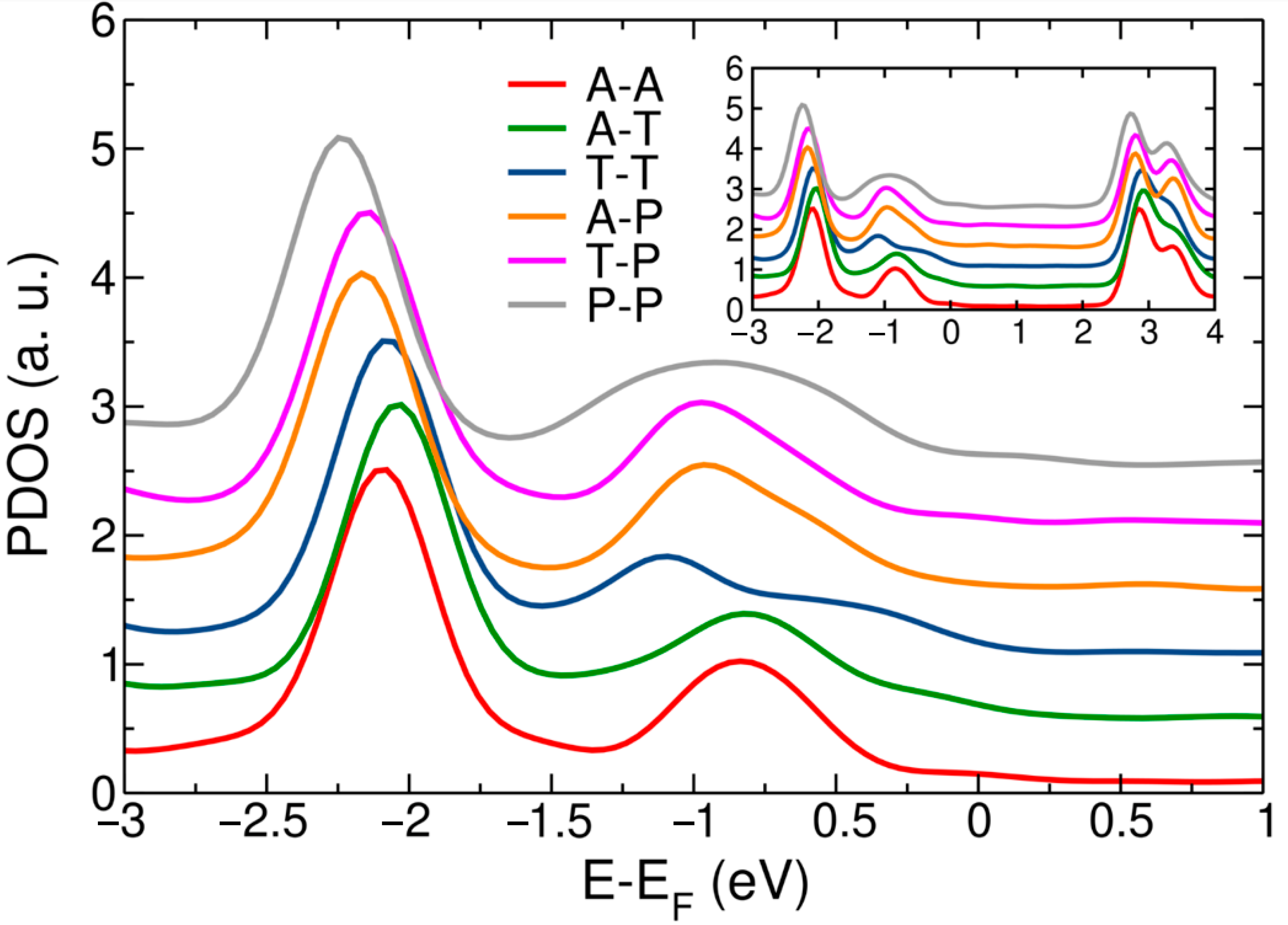
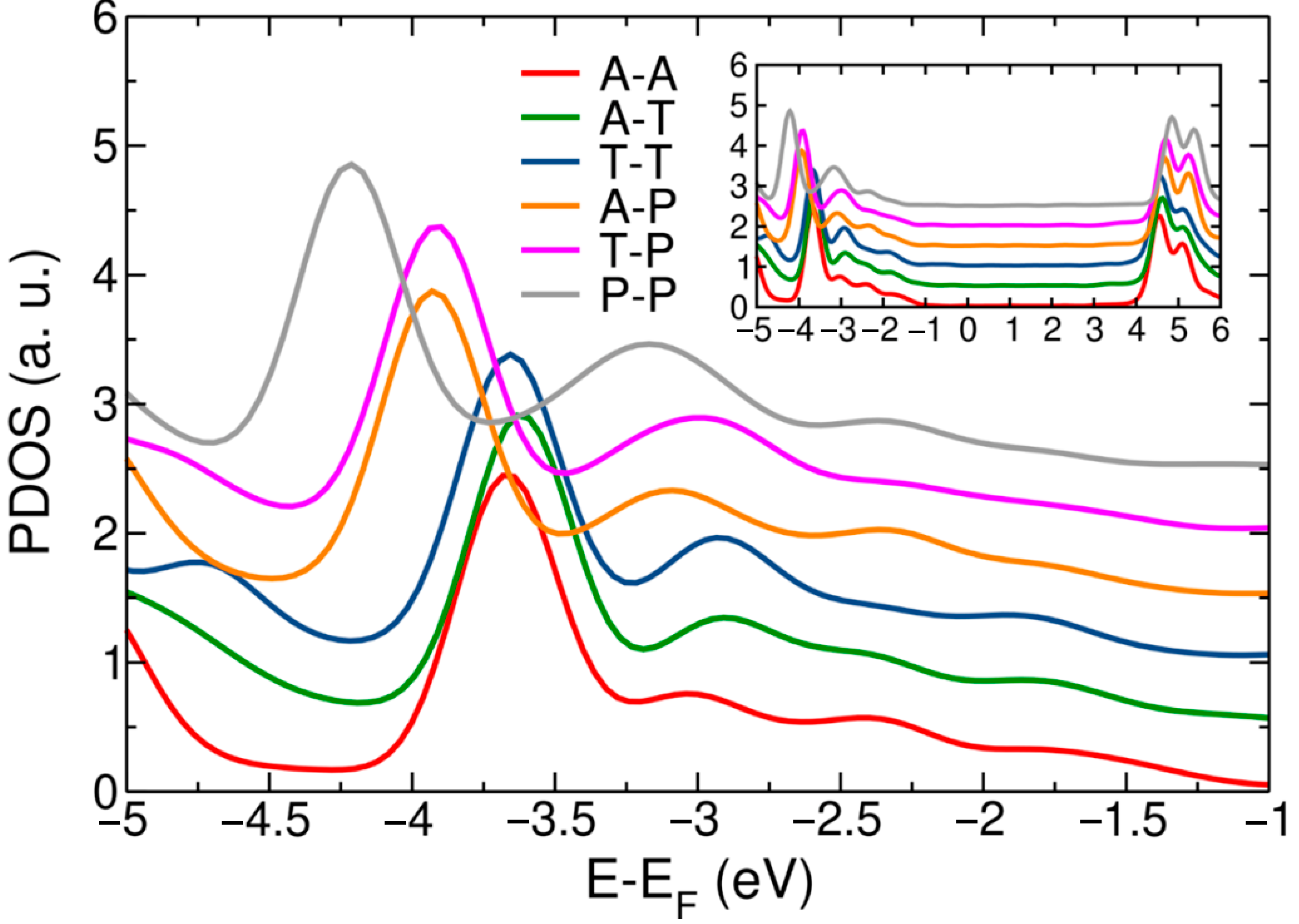
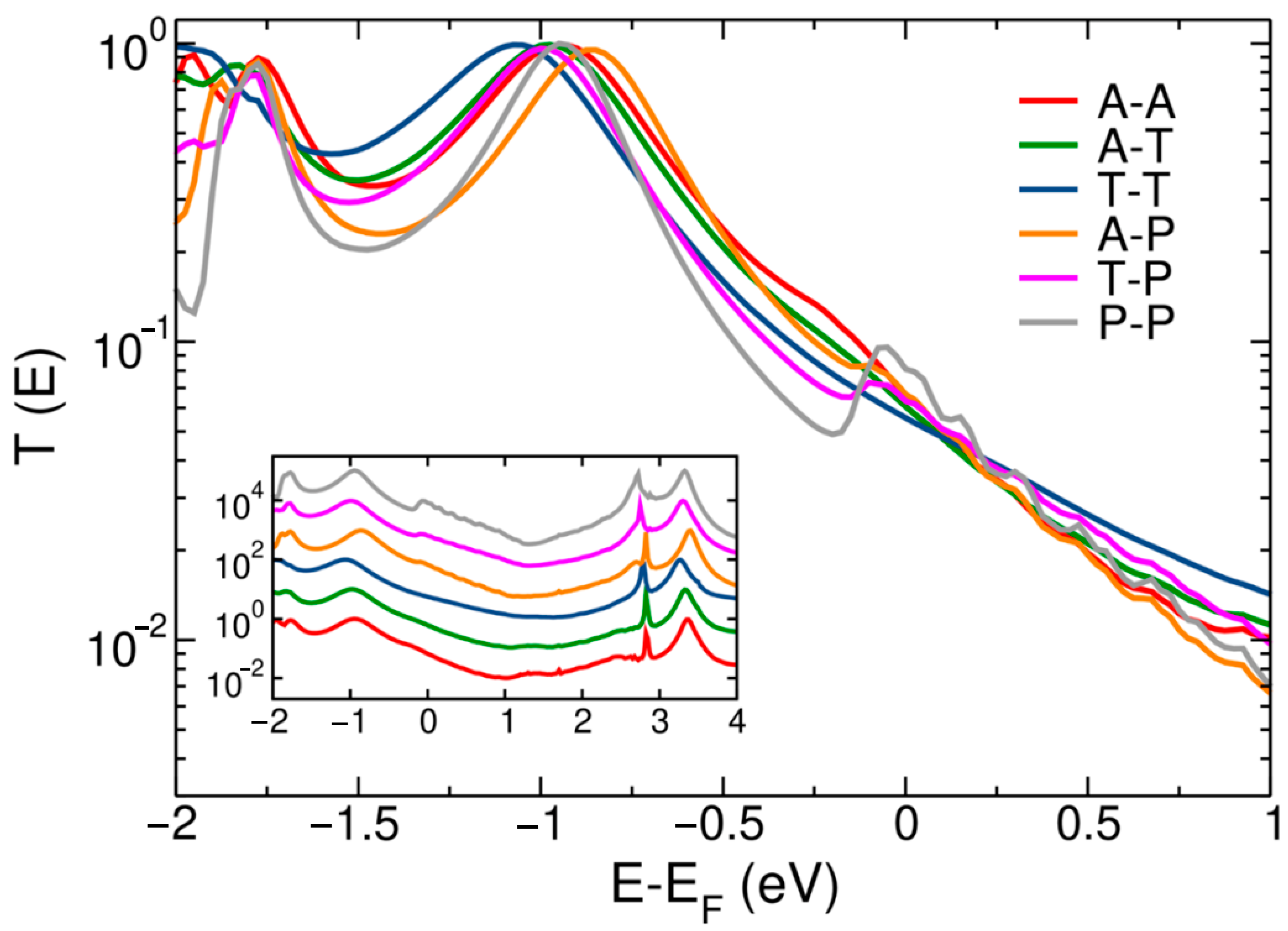
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aviram, A.; Ratner, M.A. Molecular rectifiers. Chem. Phys. Lett. 1974, 29, 277–283. [Google Scholar] [CrossRef]
- Nitzan, A.; Ratner, M.A. Electron transport in molecular wire junctions. Science 2003, 300, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.C.; Scheer, E. Molecular Electronics: An Introduction to Theory and Experimen; World Scientific: Singapore, 2010; Volume 1, ISBN 978-981-4282-58-1. [Google Scholar]
- Xu, B.; Tao, N.J. Measurement of Single-Molecule Resistance by Repeated Formation of Molecular Junctions. Science 2003, 301, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, L.; Klare, J.E.; Tam, I.W.; Nuckolls, C.; Hybertsen, M.S.; Steigerwald, M.L. Single-molecule circuits with well-defined molecular conductance. Nano Lett. 2006, 6, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Teramae, Y.; Kurokawa, S.; Sakai, A. High-conductance states of single benzenedithiol molecules. Appl. Phys. Lett. 2006, 89, 163111. [Google Scholar] [CrossRef]
- Martin, C.A.; Ding, D.; Van Der Zant, H.S.J.; Van Ruitenbeek, J.M. Lithographic mechanical break junctions for single-molecule measurements in vacuum: Possibilities and limitations. New J. Phys. 2008, 10, 65008. [Google Scholar] [CrossRef]
- Haiss, W.; Wang, C.; Grace, I.; Batsanov, A.S.; Schiffrin, D.J.; Higgins, S.J.; Bryce, M.R.; Lambert, C.J.; Nichols, R.J. Precision control of single-molecule electrical junctions. Nat. Mater. 2006, 5, 995. [Google Scholar] [CrossRef]
- Kim, Y.; Pietsch, T.; Erbe, A.; Belzig, W.; Scheer, E. Benzenedithiol: A Broad-Range Single-Channel Molecular Conductor. Nano Lett. 2011, 11, 3734–3738. [Google Scholar] [CrossRef]
- González, M.T.; Wu, S.; Huber, R.; van der Molen, S.J.; Schönenberger, C.; Calame, M. Electrical Conductance of Molecular Junctions by a Robust Statistical Analysis. Nano Lett. 2006, 6, 2238–2242. [Google Scholar] [CrossRef]
- Kiguchi, M. Single-Molecule Electronics: An Introduction to Synthesis, Measurement and Theory; Kiguchi, M., Ed.; Springer: Singapore, 2016; ISBN 9789811007248. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Brandbyge, M.; Mozos, J.L.; Ordejón, P.; Taylor, J.; Stokbro, K. Density-functional method for nonequilibrium electron transport. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 65, 1654011–16540117. [Google Scholar] [CrossRef]
- Zang, Y.; Pinkard, A.; Liu, Z.-F.; Neaton, J.B.; Steigerwald, M.L.; Roy, X.; Venkataraman, L. Electronically Transparent Au–N Bonds for Molecular Junctions. J. Am. Chem. Soc. 2017, 139, 14845–14848. [Google Scholar] [CrossRef] [PubMed]
- Leary, E.; Zotti, L.A.; Miguel, D.; Márquez, I.R.; Palomino-Ruiz, L.; Cuerva, J.M.; Rubio-Bollinger, G.; González, M.T.; Agrait, N. The Role of Oligomeric Gold–Thiolate Units in Single-Molecule Junctions of Thiol-Anchored Molecules. J. Phys. Chem. C 2018, 122, 3211–3218. [Google Scholar] [CrossRef]
- Li, H.; Su, T.A.; Camarasa-Gómez, M.; Hernangómez-Pérez, D.; Henn, S.E.; Pokorný, V.; Caniglia, C.D.; Inkpen, M.S.; Korytár, R.; Steigerwald, M.L.; et al. Silver Makes Better Electrical Contacts to Thiol-Terminated Silanes than Gold. Angew. Chem. Int. Ed. 2017, 56, 14145–14148. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.; Chen, S.Z.; Yan, L.; Chen, K.Q.; Shang, X.; Liu, P.N.; Lin, N. Negative Differential Conductance in Polyporphyrin Oligomers with Nonlinear Backbones. J. Am. Chem. Soc. 2018, 140, 570–573. [Google Scholar] [CrossRef]
- Zang, Y.; Ray, S.; Fung, E.-D.; Borges, A.; Garner, M.H.; Steigerwald, M.L.; Solomon, G.C.; Patil, S.; Venkataraman, L. Resonant Transport in Single Diketopyrrolopyrrole Junctions. J. Am. Chem. Soc. 2018, 140, 13167–13170. [Google Scholar] [CrossRef]
- Stefani, D.; Weiland, K.J.; Skripnik, M.; Hsu, C.; Perrin, M.L.; Mayor, M.; Pauly, F.; van der Zant, H.S.J. Large Conductance Variations in a Mechanosensitive Single-Molecule Junction. Nano Lett. 2018, 18, 5981–5988. [Google Scholar] [CrossRef]
- Schneebeli, S.; Kamenetska, M.; Foss, F.; Vazquez, H.; Skouta, R.; Hybertsen, M.; Venkataraman, L.; Breslow, R. The electrical properties of biphenylenes. Org. Lett. 2010, 12, 4114–4117. [Google Scholar] [CrossRef]
- Perdew, J.P.; Levy, M. Physical Content of the Exact Kohn-Sham Orbital Energies: Band Gaps and Derivative Discontinuities. Phys. Rev. Lett. 1983, 51, 1884–1887. [Google Scholar] [CrossRef]
- Kümmel, S.; Kronik, L. Orbital-dependent density functionals: Theory and applications. Rev. Mod. Phys. 2008, 80, 3–60. [Google Scholar] [CrossRef]
- Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation gaps of finite-sized systems from optimally tuned range-separated hybrid functionals. J. Chem. Theory Comput. 2012, 8, 1515–1531. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.J.; Schllter, M. Density-functional theory of the energy gap. Phys. Rev. Lett. 1983, 51, 1888–1891. [Google Scholar] [CrossRef]
- Quek, S.Y.; Venkataraman, L.; Choi, H.J.; Louie, S.G.; Hybertsen, M.S.; Neaton, J.B. Amine—Gold linked single-molecule circuits: Experiment and theory. Nano Lett. 2007, 7, 3477–3482. [Google Scholar] [CrossRef] [PubMed]
- Quek, S.Y.; Choi, H.J.; Louie, S.G.; Neaton, J.B. Length dependence of conductance in aromatic single-molecule junctions. Nano Lett. 2009, 9, 3949–3953. [Google Scholar] [CrossRef]
- Li, G.; Rangel, T.; Liu, Z.F.; Cooper, V.R.; Neaton, J.B. Energy level alignment of self-assembled linear chains of benzenediamine on Au(111) from first principles. Phys. Rev. B 2016, 93, 125429. [Google Scholar] [CrossRef]
- Markussen, T.; Jin, C.; Thygesen, K.S. Quantitatively accurate calculations of conductance and thermopower of molecular junctions. Phys. Status Solidi Basic Res. 2013, 250, 2394–2402. [Google Scholar] [CrossRef]
- Zotti, L.A.; Bürkle, M.; Pauly, F.; Lee, W.; Kim, K.; Jeong, W.; Asai, Y.; Reddy, P.; Cuevas, J.C. Heat dissipation and its relation to thermopower in single-molecule junctions. New J. Phys. 2014, 16, 15004. [Google Scholar] [CrossRef]
- Jin, C.; Strange, M.; Markussen, T.; Solomon, G.C.; Thygesen, K.S. Energy level alignment and quantum conductance of functionalized metal-molecule junctions: Density functional theory versus GW calculations. J. Chem. Phys. 2013, 139, 184307. [Google Scholar] [CrossRef]
- Jin, C.; Thygesen, K.S. Dynamical image-charge effect in molecular tunnel junctions: Beyond energy level alignment. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 89, 41102. [Google Scholar] [CrossRef]
- Neaton, J.B.; Hybertsen, M.S.; Louie, S.G. Renormalization of molecular electronic levels at metal-molecule interfaces. Phys. Rev. Lett. 2006, 97, 216405. [Google Scholar] [CrossRef]
- Dell’Angela, M.; Kladnik, G.; Cossaro, A.; Verdini, A.; Kamenetska, M.; Tamblyn, I.; Quek, S.Y.; Neaton, J.B.; Cvetko, D.; Morgante, A.; et al. Relating energy level alignment and amine-linked single molecule junction conductance. Nano Lett. 2010, 10, 2470–2474. [Google Scholar] [CrossRef] [PubMed]
- Su, T.A.; Neupane, M.; Steigerwald, M.L.; Venkataraman, L.; Nuckolls, C. Chemical principles of single-molecule electronics. Nat. Rev. Mater. 2016, 1, 16002. [Google Scholar] [CrossRef]
- Arasu, N.P.; Vázquez, H. Direct Au-C contacts based on biphenylene for single molecule circuits. Phys. Chem. Chem. Phys. 2018, 20, 10378–10383. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Whalley, A.C.; Kamenetska, M.; Steigerwald, M.L.; Hybertsen, M.S.; Nuckolls, C.; Venkataraman, L. Contact chemistry and single-molecule conductance: A comparison of phosphines, methyl sulfides, and amines. J. Am. Chem. Soc. 2007, 129, 15768–15769. [Google Scholar] [CrossRef]
- Kamenetska, M.; Quek, S.Y.; Whalley, A.C.; Steigerwald, M.L.; Choi, H.J.; Louie, S.G.; Nuckolls, C.; Hybertsen, M.S.; Neaton, J.B.; Venkataraman, L. Conductance and Geometry of Pyridine-Linked Single-Molecule Junctions. J. Am. Chem. Soc. 2010, 132, 6817–6821. [Google Scholar] [CrossRef]
- Mishchenko, A.; Zotti, L.A.; Vonlanthen, D.; Bürkle, M.; Pauly, F.; Cuevas, J.C.; Mayor, M.; Wandlowski, T. Single-Molecule Junctions Based on Nitrile-Terminated Biphenyls: A Promising New Anchoring Group. J. Am. Chem. Soc. 2011, 133, 184–187. [Google Scholar] [CrossRef]
- Tour, J.M.; Jones, L.; Pearson, D.L.; Lamba, J.J.S.; Burgin, T.P.; Whitesides, G.M.; Allara, D.L.; Parikh, A.N.; Atre, S. Self-Assembled Monolayers and Multilayers of Conjugated Thiols, alpha, omega.-Dithiols, and Thioacetyl-Containing Adsorbates. Understanding Attachments between Potential Molecular Wires and Gold Surfaces. J. Am. Chem. Soc. 1995, 117, 9529–9534. [Google Scholar] [CrossRef]
- González, M.T.; Leary, E.; García, R.; Verma, P.; Herranz, M.Á.; Rubio-Bollinger, G.; Martín, N.; Agraït, N. Break-Junction Experiments on Acetyl-Protected Conjugated Dithiols under Different Environmental Conditions. J. Phys. Chem. C 2011, 115, 17973–17978. [Google Scholar] [CrossRef]
- Inkpen, M.S.; Liu, Z.-F.; Li, H.; Campos, L.M.; Neaton, J.B.; Venkataraman, L. Non-chemisorbed gold–sulfur binding prevails in self-assembled monolayers. Nat. Chem. 2019, 11, 351–358. [Google Scholar] [CrossRef]
- Paik, W.; Han, S.; Shin, W.; Kim, Y. Adsorption of Carboxylic Acids on Gold by Anodic Reaction. Langmuir 2003, 19, 4211–4216. [Google Scholar] [CrossRef]
- Chen, F.; Li, X.; Hihath, J.; Huang, Z.; Tao, N. Effect of Anchoring Groups on Single-Molecule Conductance: Comparative Study of Thiol-, Amine-, and Carboxylic-Acid-Terminated Molecules. J. Am. Chem. Soc. 2006, 128, 15874–15881. [Google Scholar] [CrossRef] [PubMed]
- Quek, S.Y.; Kamenetska, M.; Steigerwald, M.L.; Choi, H.J.; Louie, S.G.; Hybertsen, M.S.; Neaton, J.B.; Venkataraman, L. Mechanically controlled binary conductance switching of a single-molecule junction. Nat. Nanotechnol. 2009, 4, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Su, T.A.; Widawsky, J.R.; Li, H.; Klausen, R.S.; Leighton, J.L.; Steigerwald, M.L.; Venkataraman, L.; Nuckolls, C. Silicon ring strain creates high-conductance pathways in single-molecule circuits. J. Am. Chem. Soc. 2013, 135, 18331–18334. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, M.; Ohto, T.; Fujii, S.; Sugiyasu, K.; Nakajima, S.; Takeuchi, M.; Nakamura, H. Single molecular resistive switch obtained via sliding multiple anchoring points and varying effective wire length. J. Am. Chem. Soc. 2014, 136, 7327–7332. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Montes, E.; Suzuki, S.; Fujii, S.; Nishino, T.; Tsukagoshi, K.; Ikeda, K.; Kano, H.; Nakamura, H.; Vázquez, H.; et al. Identifying the molecular adsorption site of a single molecule junction through combined Raman and conductance studies. Chem. Sci. 2019, 10, 6261–6269. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, M.; Krag, C.; Frederiksen, T.; Brandbyge, M. Conductance of Alkanedithiol Single-Molecule Junctions: A Molecular Dynamics Study. Nano Lett. 2009, 9, 117–121. [Google Scholar] [CrossRef]
- Isshiki, Y.; Fujii, S.; Nishino, T.; Kiguchi, M. Fluctuation in Interface and Electronic Structure of Single-Molecule Junctions Investigated by Current versus Bias Voltage Characteristics. J. Am. Chem. Soc. 2018, 140, 3760–3767. [Google Scholar] [CrossRef]
- Zotti, L.A.; Kirchner, T.; Cuevas, J.-C.; Pauly, F.; Huhn, T.; Scheer, E.; Erbe, A. Revealing the Role of Anchoring Groups in the Electrical Conduction Through Single-Molecule Junctions. Small 2010, 6, 1529–1535. [Google Scholar] [CrossRef]
- Venkataraman, L.; Klare, J.E.; Nuckolls, C.; Hybertsen, M.S.; Steigerwald, M.L. Dependence of single-molecule junction conductance on molecular conformation. Nature 2006, 442, 904–907. [Google Scholar] [CrossRef]
- Kim, T.; Vázquez, H.; Hybertsen, M.S.; Venkataraman, L. Conductance of molecular junctions formed with silver electrodes. Nano Lett. 2013, 13, 3358–3364. [Google Scholar] [CrossRef]
- Chen, Y.; Tamblyn, I.; Quek, S.Y. Energy Level Alignment at Hybridized Organic-Metal Interfaces: The Role of Many-Electron Effects. J. Phys. Chem. C 2017, 121, 13125–13134. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order- N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- García, A.; Papior, N.; Akhtar, A.; Artacho, E.; Blum, V.; Bosoni, E.; Brandimarte, P.; Brandbyge, M.; Cerdá, J.I.; Corsetti, F.; et al. Siesta: Recent developments and applications. J. Chem. Phys. 2020, 152, 204108. [Google Scholar] [CrossRef] [PubMed]
- Papior, N.; Lorente, N.; Frederiksen, T.; García, A.; Brandbyge, M. Improvements on non-equilibrium and transport Green function techniques: The next-generation TRANSIESTA. Comput. Phys. Commun. 2017, 212, 8–24. [Google Scholar] [CrossRef]
- Román-Pérez, G.; Soler, J.M. Efficient implementation of a van der waals density functional: Application to double-wall carbon nanotubes. Phys. Rev. Lett. 2009, 103, 96102. [Google Scholar] [CrossRef]
- Datta, S. Quantum Transport: Atom to Transistor; Cambridge University Press: Cambridge, UK, 2005; ISBN 9781139164313. [Google Scholar]
- Capozzi, B.; Chen, Q.; Darancet, P.; Kotiuga, M.; Buzzeo, M.; Neaton, J.B.; Nuckolls, C.; Venkataraman, L. Tunable Charge Transport in Single-Molecule Junctions via Electrolytic Gating. Nano Lett. 2014, 14, 1400–1404. [Google Scholar] [CrossRef]
- Liu, Z.F.; Wei, S.; Yoon, H.; Adak, O.; Ponce, I.; Jiang, Y.; Jang, W.D.; Campos, L.M.; Venkataraman, L.; Neaton, J.B. Control of single-molecule junction conductance of porphyrins via a transition-metal center. Nano Lett. 2014, 14, 5365–5370. [Google Scholar] [CrossRef]
- Flores, F.; Ortega, J.; Vázquez, H. Modelling energy level alignment at organic interfaces and density functional theory. Phys. Chem. Chem. Phys. 2009, 11, 8658–8675. [Google Scholar] [CrossRef]
- Smith, N.V.; Chen, C.T.; Weinert, M. Distance of the image plane from metal surfaces. Phys. Rev. B 1989, 40, 7565–7573. [Google Scholar] [CrossRef]
- Schneider, N.L.; Néel, N.; Andersen, N.P.; Lü, J.T.; Brandbyge, M.; Kröger, J.; Berndt, R. Spectroscopy of transmission resonances through a C60junction. J. Phys. Condens. Matter 2014, 27, 15001. [Google Scholar] [CrossRef]
- Solomon, G.C.; Andrews, D.Q.; Hansen, T.; Goldsmith, R.H.; Wasielewski, M.R.; Van Duyne, R.P.; Ratner, M.A. Understanding quantum interference in coherent molecular conduction. J. Chem. Phys. 2008, 129, 54701. [Google Scholar] [CrossRef]
- Fujii, S.; Marqués-González, S.; Shin, J.Y.; Shinokubo, H.; Masuda, T.; Nishino, T.; Arasu, N.P.; Vázquez, H.; Kiguchi, M. Highly-conducting molecular circuits based on antiaromaticity. Nat. Commun. 2017, 8, 15984. [Google Scholar] [CrossRef] [PubMed]
- Refaely-Abramson, S.; Liu, Z.F.; Bruneval, F.; Neaton, J.B. First-Principles Approach to the Conductance of Covalently Bound Molecular Junctions. J. Phys. Chem. C 2019, 123, 6379–6387. [Google Scholar] [CrossRef]
- Delmas, V.; Diez-Cabanes, V.; van Dyck, C.; Scheer, E.; Costuas, K.; Cornil, J. On the reliability of acquiring molecular junction parameters by Lorentzian fitting of I/V curves. Phys. Chem. Chem. Phys. 2020, 22, 26702–26706. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (eV) | |
|---|---|
| Adatom–Adatom | 1.22 |
| Adatom–Trimer | 1.22 |
| Trimer–Trimer | 1.22 |
| Adatom–Pyramid | 1.03 |
| Trimer–Pyramid | 1.03 |
| Pyramid–Pyramid | 0.81 |
| ΣHOMO (eV) | |
|---|---|
| Adatom–Adatom | −1.77 |
| Adatom–Trimer | −1.77 |
| Trimer–Trimer | −1.77 |
| Adatom–Pyramid | −1.96 |
| Trimer–Pyramid | −1.96 |
| Pyramid–Pyramid | −2.18 |
| HOMO Peak Position (eV) | ||
|---|---|---|
| DFT | DFT + Σ | |
| Adatom–Adatom | −0.84 | −3.04 |
| Adatom–Trimer | −0.83 | −2.91 |
| Trimer–Trimer | −1.09 | −2.93 |
| Adatom–Pyramid | −0.96 | −3.01 |
| Trimer–Pyramid | −0.97 | −2.98 |
| Pyramid–Pyramid | −0.93 | −3.17 |
| Conductance (10−2 G0) | |||
|---|---|---|---|
| DFT-Landauer | Lorentzian (DFT) | Lorentzian (DFT + Σ) | |
| Adatom–Adatom | 6.61 (10.33×) | 6.66 (10.40×) | 1.00 (1.56×) |
| Adatom–Trimer | 6.07 (9.48×) | 6.64 (10.38×) | 1.03 (1.61×) |
| Trimer–Trimer | 5.55 (8.68×) | 6.25 (9.77×) | 1.08 (1.69×) |
| Adatom–Pyramid | 6.66 (10.41×) | 5.66 (8.85×) | 0.64 (1.00×) |
| Trimer–Pyramid | 6.33 (9.88×) | 4.62 (7.21×) | 0.63 (1.00×) |
| Pyramid–Pyramid | 8.10 (12.65×) | 3.23 (5.05×) | 0.35 (1.82×) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montes, E.; Vázquez, H. Role of the Binding Motifs in the Energy Level Alignment and Conductance of Amine-Gold Linked Molecular Junctions within DFT and DFT + Σ. Appl. Sci. 2021, 11, 802. https://doi.org/10.3390/app11020802
Montes E, Vázquez H. Role of the Binding Motifs in the Energy Level Alignment and Conductance of Amine-Gold Linked Molecular Junctions within DFT and DFT + Σ. Applied Sciences. 2021; 11(2):802. https://doi.org/10.3390/app11020802
Chicago/Turabian StyleMontes, Enrique, and Héctor Vázquez. 2021. "Role of the Binding Motifs in the Energy Level Alignment and Conductance of Amine-Gold Linked Molecular Junctions within DFT and DFT + Σ" Applied Sciences 11, no. 2: 802. https://doi.org/10.3390/app11020802
APA StyleMontes, E., & Vázquez, H. (2021). Role of the Binding Motifs in the Energy Level Alignment and Conductance of Amine-Gold Linked Molecular Junctions within DFT and DFT + Σ. Applied Sciences, 11(2), 802. https://doi.org/10.3390/app11020802