Attenuation Factors in Molecular Electronics: Some Theoretical Concepts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
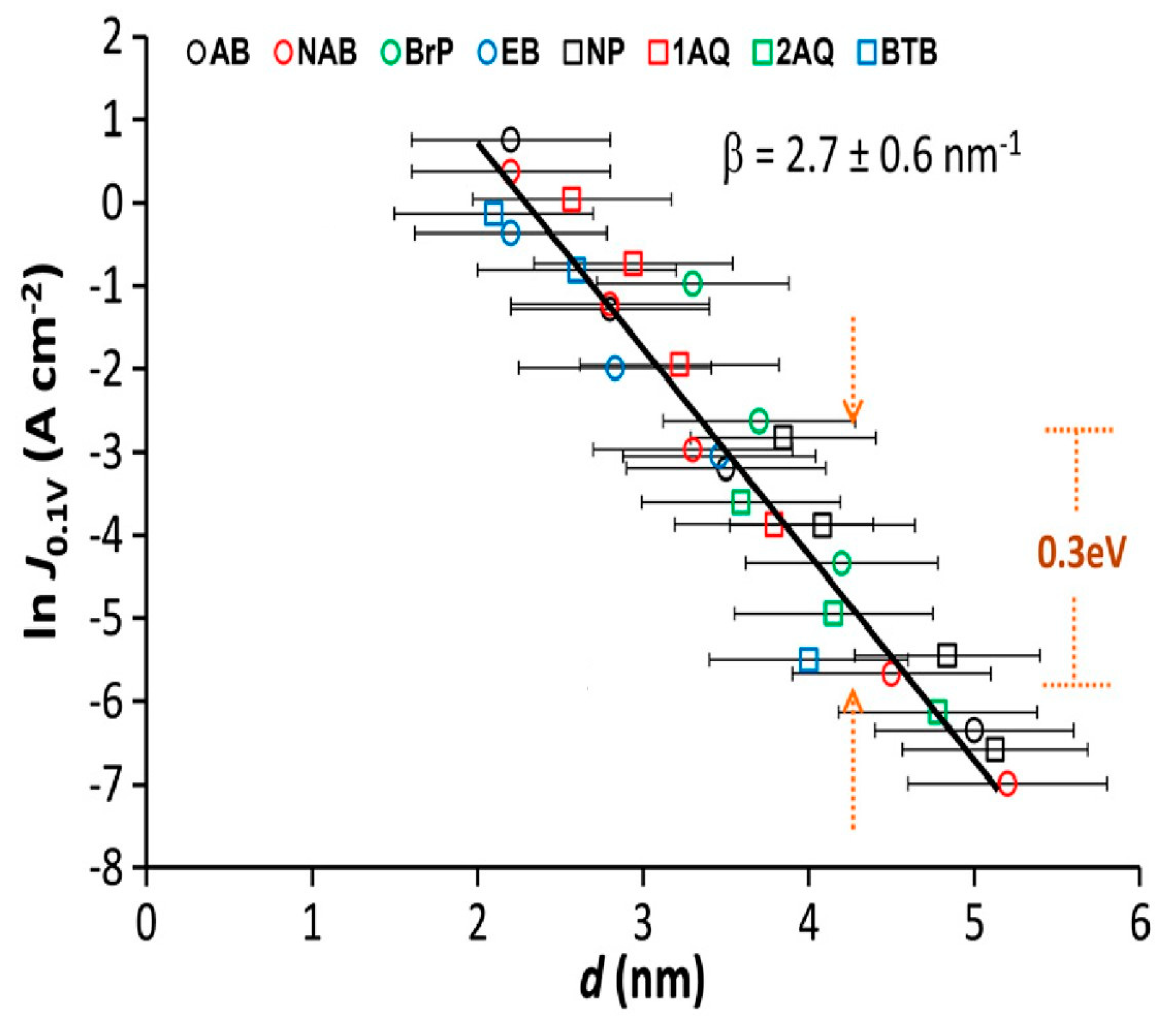
2. Some Attenuation Factors of Standard Molecular Junctions
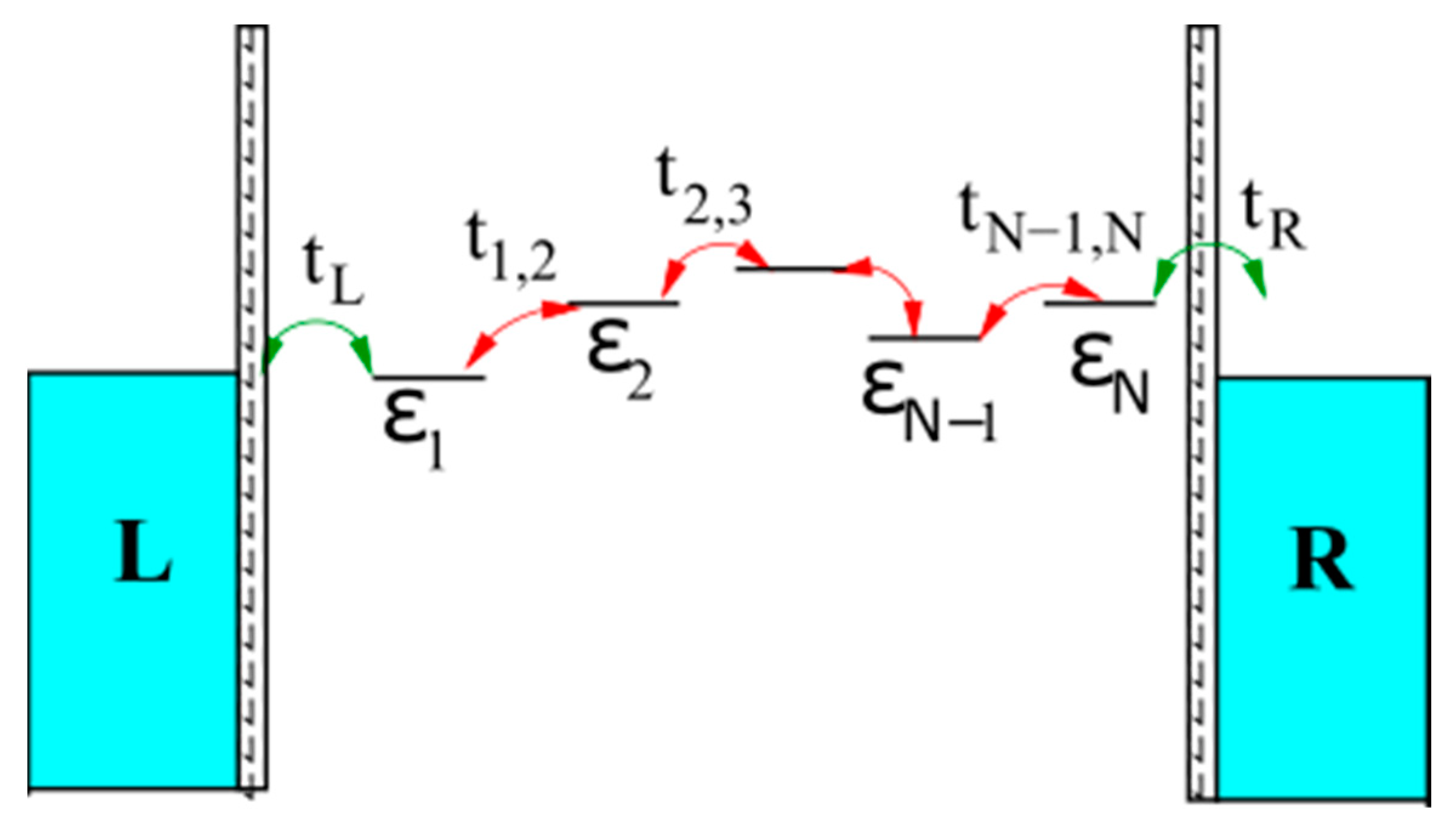
3. Theoretical Approaches
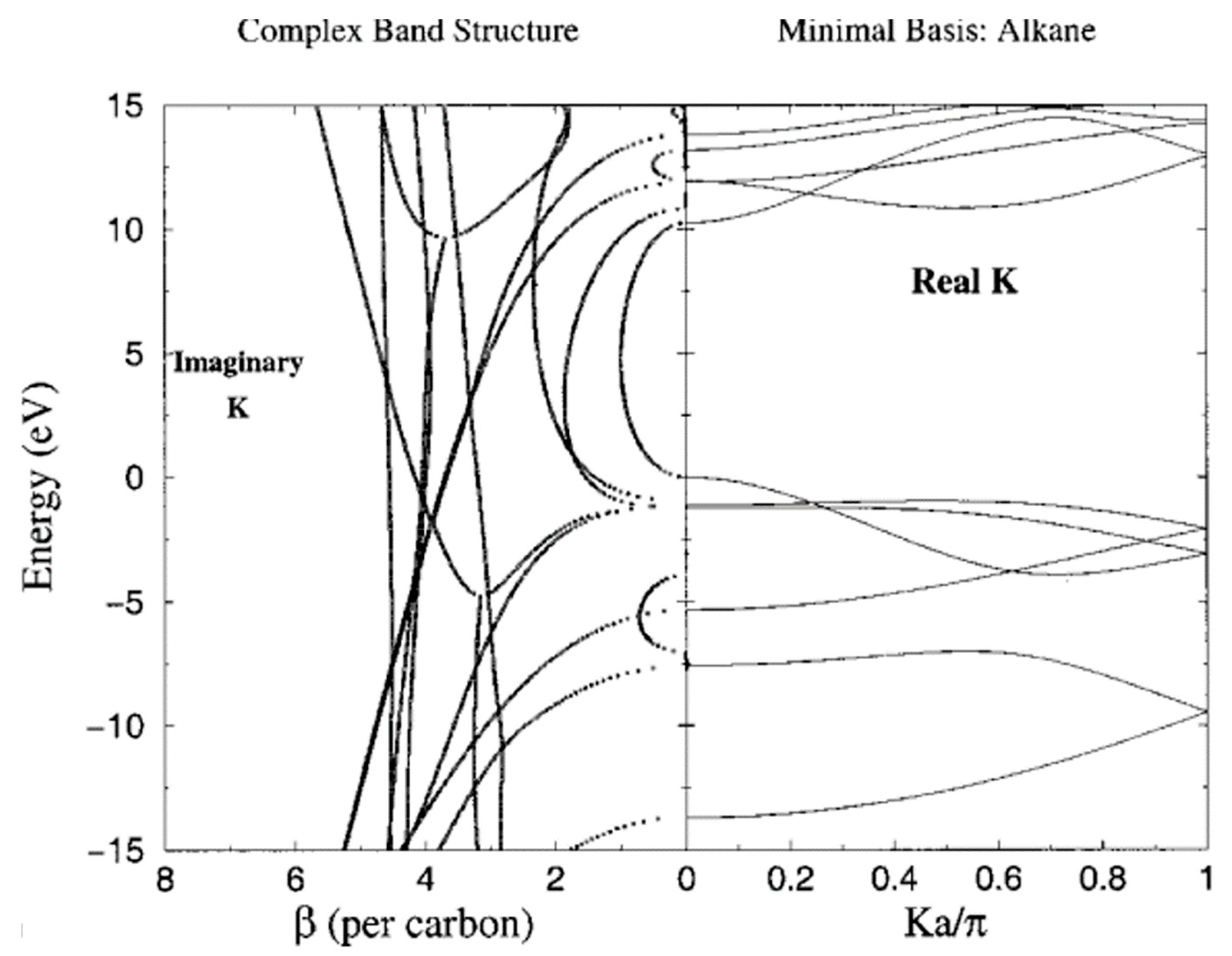
4. A Test Case: Alkane Chains
4.1. Role of the Anchoring Groups: Symmetric Junctions
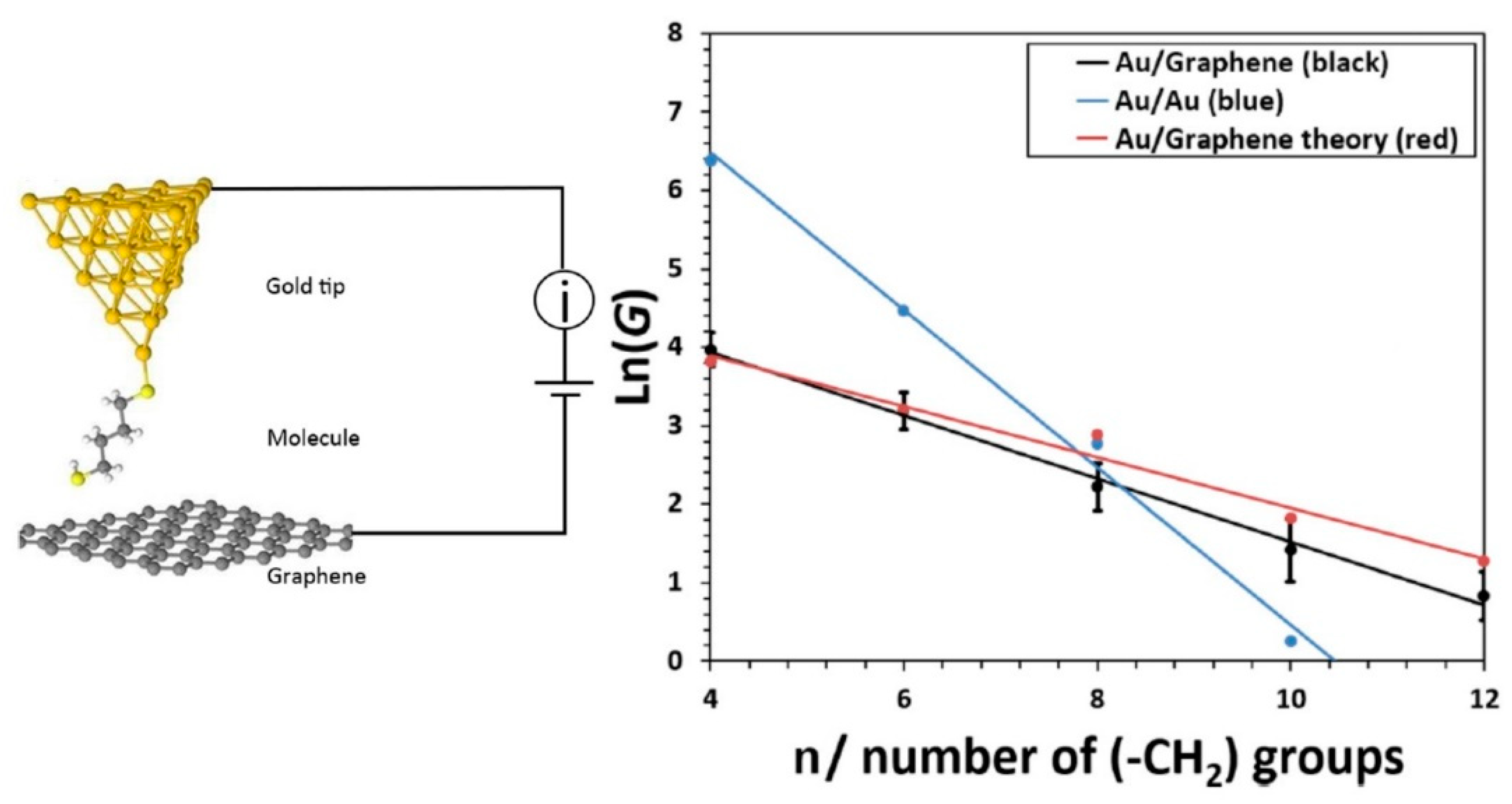
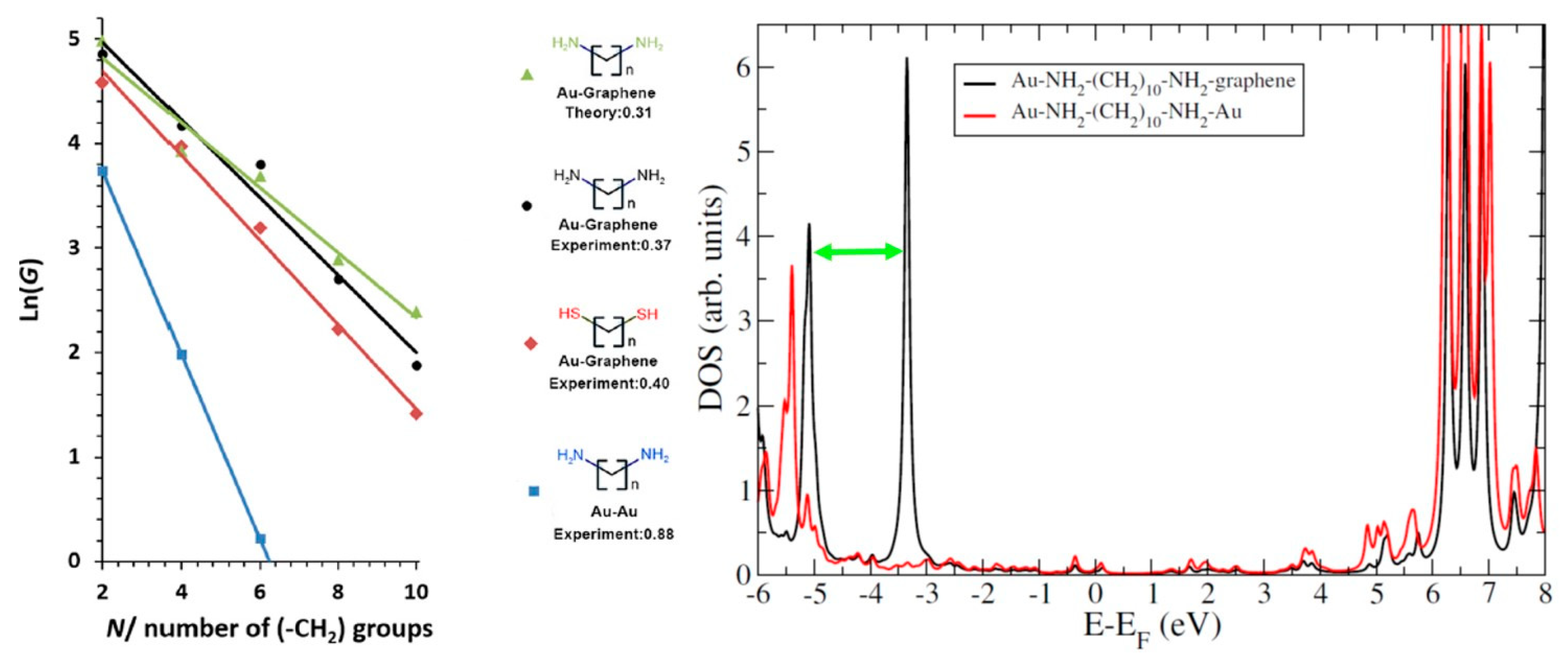
4.2. Role of the Electrodes: Hybrid Junctions Using a Graphene Electrode
4.3. The Case of Platinum–Graphene Hybrid Junctions
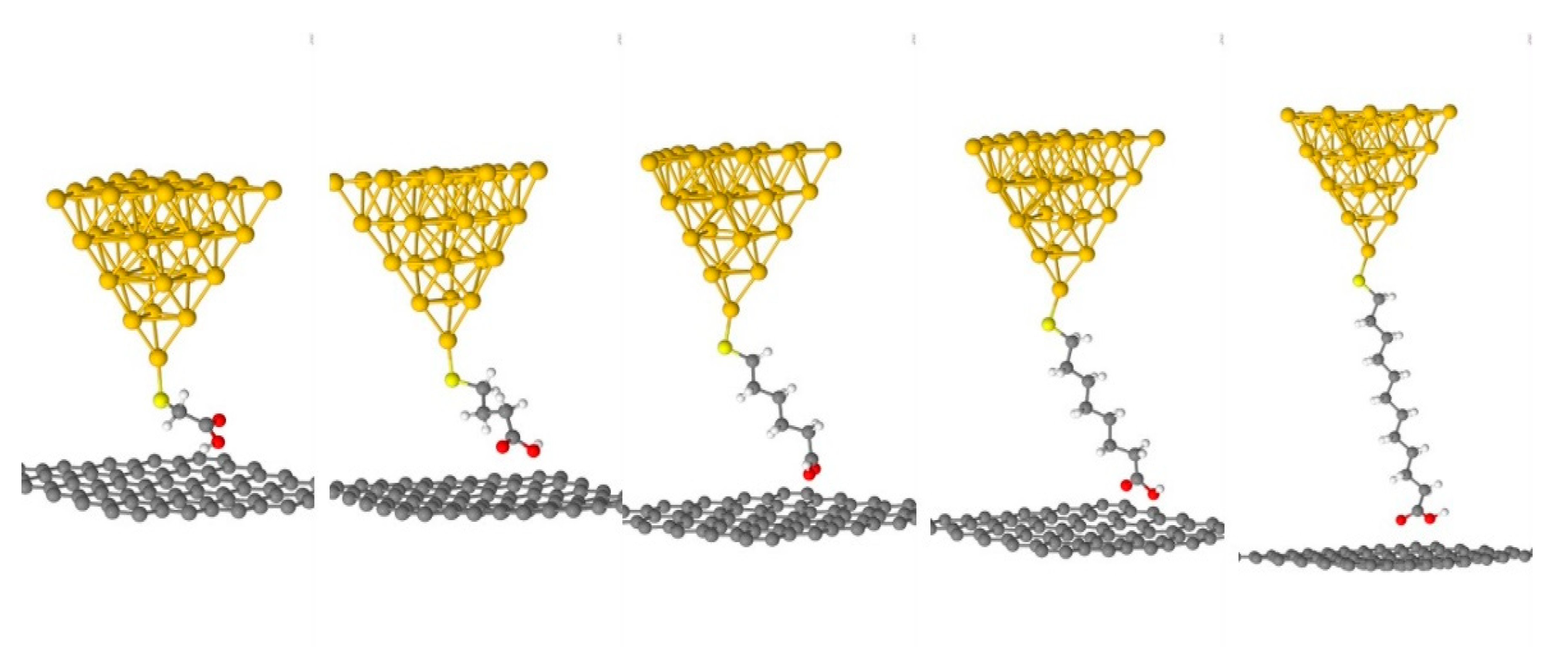
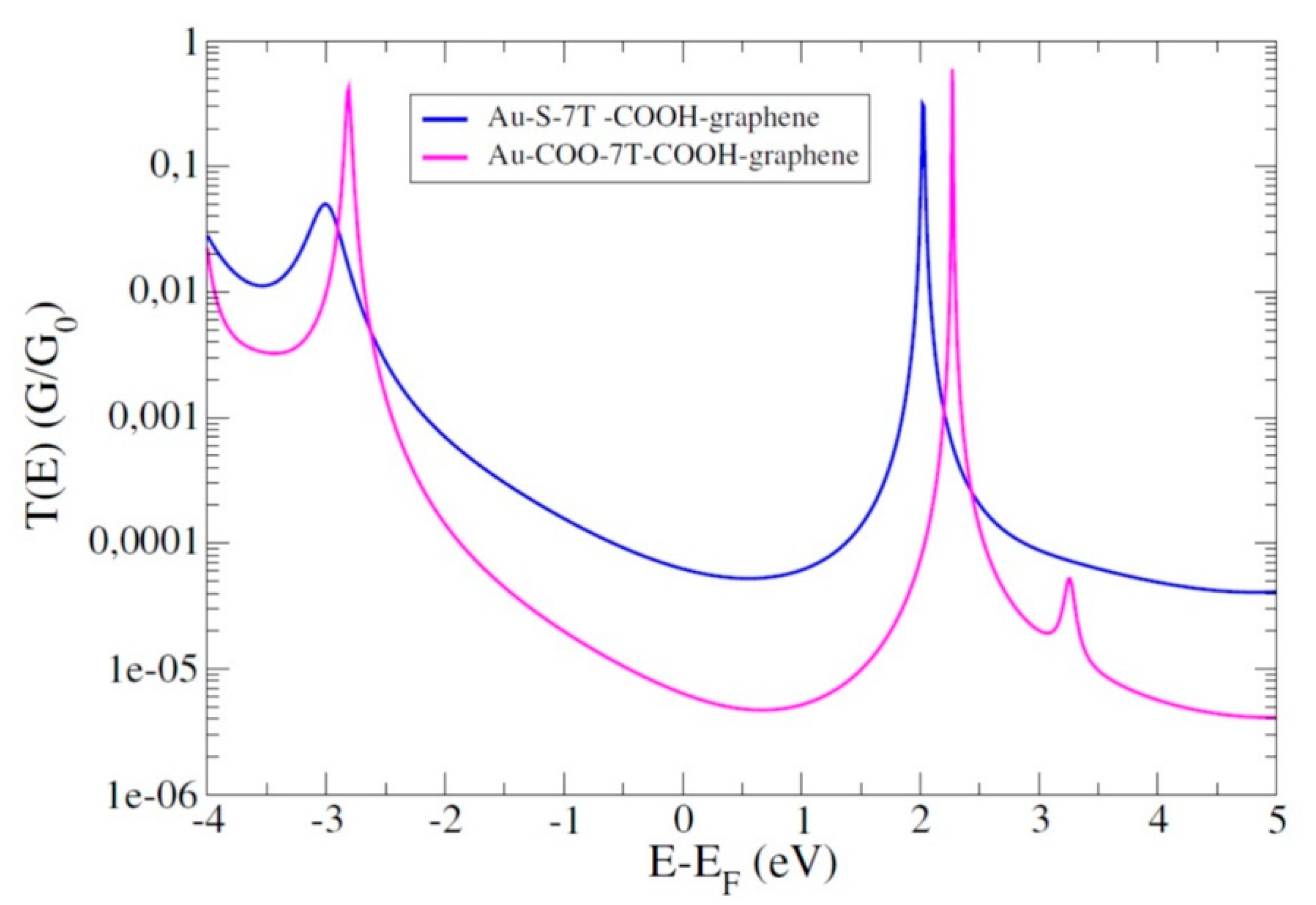
4.4. Role of the Anchoring Groups: Asymmetric Junctions
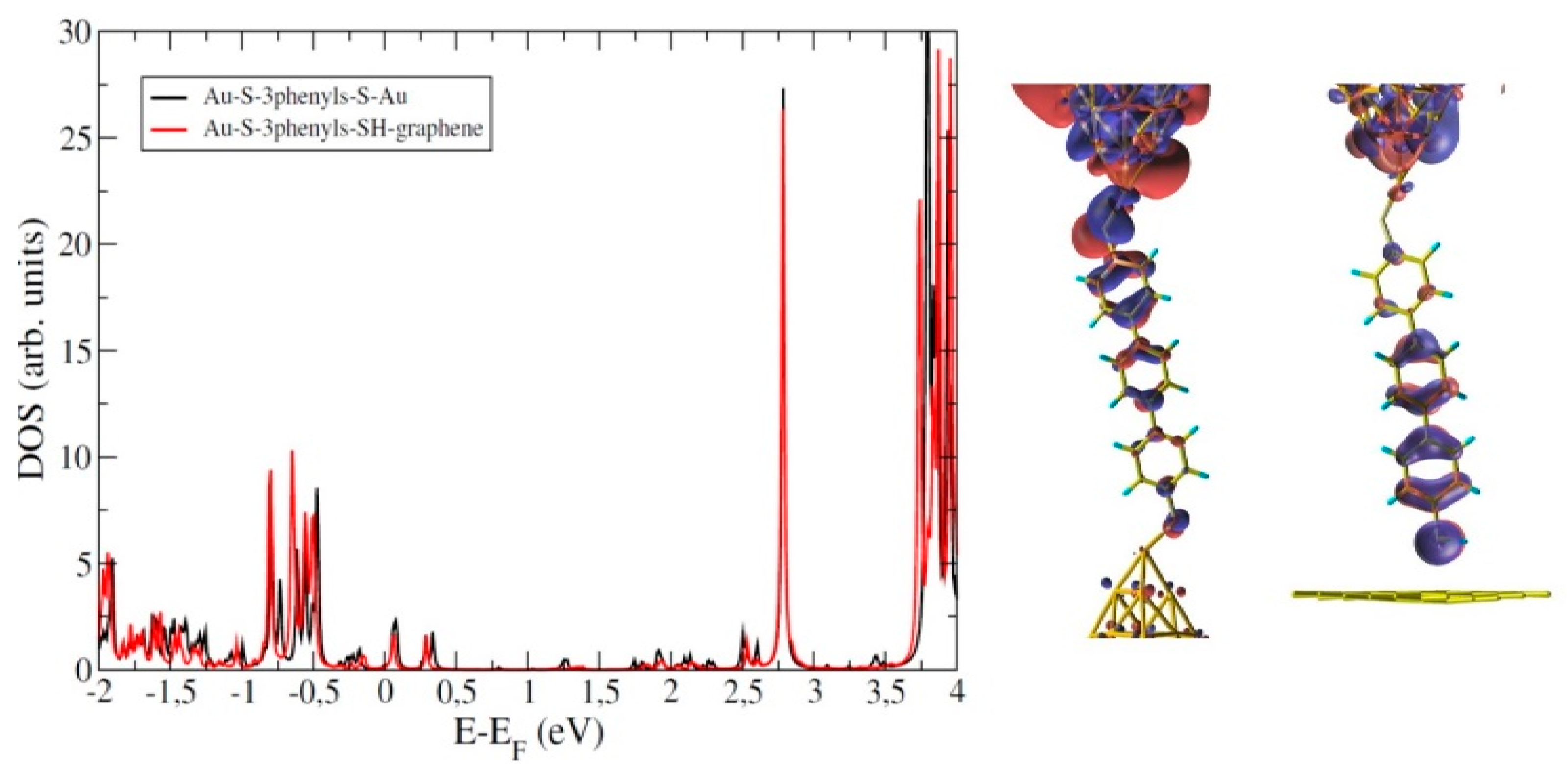
5. Importance of the Molecular Backbone: Conjugated Molecular Wires
6. Summary
Funding
Conflicts of Interest
References
- Cuevas, J.C.; Scheer, E. Molecular Electronics: An Introduction to Theory and Experiment; World Scientific: Singapore, 2010. [Google Scholar]
- Wang, W.; Lee, T.; Reed, M.A. Mechanism of electron conduction in self-assembled alkanethiol monolayer devices. Phys. Rev. B 2003, 68, 035416. [Google Scholar] [CrossRef]
- Sze, S.M. Physics of Semiconductor Devices, 2nd ed.; Wiley: New York, NY, USA, 1981. [Google Scholar]
- Hines, T.; Diez-Perez, I.; Hihath, J.; Liu, H.; Wang, Z.S.; Zhao, J.; Zhou, G.; Müllen, K.; Tao, N. Transition from Tunneling to Hopping in Single Molecular Junctions by Measuring Length and Temperature Dependence. J. Am. Chem. Soc. 2010, 132, 11658. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Nitzan, A.; Davis, W.B.; Wasielewski, M.R.; Ratner, M.A. Electron Transfer Rates in Bridged Molecular Systems 2. A Steady-State Analysis of Coherent Tunneling and Thermal Transitions. J. Phys. Chem. B 2000, 104, 3817. [Google Scholar] [CrossRef]
- Nitzan, A. The relationship between electron transfer rate and molecular conduction 2. The sequential hopping case. Isr. J. Chem. 2002, 42, 163. [Google Scholar] [CrossRef]
- Yan, H.; Bergren, A.J.; McCreery, R.; Luisa Della Rocca, M.; Martin, P.; Lafarge, P.H.; Lacroix, J.C. Activationless charge transport across 4.5 to 22 nm in molecular electronic junctions. Proc. Natl. Acad. Sci. USA 2013, 110, 5326. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, B.; Frisbie, C.D. Electrical Resistance of Long Conjugated Molecular Wires. Science 2008, 320, 1482. [Google Scholar] [CrossRef]
- Von Hippel, A.R. Molecular engineering. Science 1956, 123, 315. [Google Scholar] [CrossRef]
- Kamenetska, M.; Koentopp, M.; Whalley, A.C.; Park, Y.S.; Steigerwald, M.L.; Nuckolls, C.; Hybertsen, M.S.; Venkataraman, L. Formation and Evolution of Single-Molecule Junctions. Phys. Rev. Lett. 2009, 102, 126803. [Google Scholar] [CrossRef]
- Li, C.; Pobelov, I.; Wandlowski, T.; Bagrets, A.; Arnold, A.; Evers, F. Charge Transport in Single Au | Alkanedithiol | Au Junctions: Coordination Geometries and Conformational Degrees of Freedom. J. Am. Chem. Soc. 2008, 130, 318. [Google Scholar] [CrossRef]
- Li, X.; He, J.; Hihath, J.; Xu, B.; Lindsay, S.M.; Tao, N. Conductance of Single Alkanedithiols: Conduction Mechanism and Effect of Molecule-Electrode Contacts. J. Am. Chem. Soc. 2006, 128, 2138. [Google Scholar] [CrossRef]
- Klausen, R.S.; Widawsky, J.R.; Steigerwald, M.L.; Venkataraman, L.; Nuckolls, C. Conductive Molecular Silicon. J. Am. Chem. Soc. 2012, 134, 4541. [Google Scholar] [CrossRef] [PubMed]
- Su, T.A.; Li, H.; Zhang, V.; Neupane, M.; Batra, A.; Klausen, R.S.; Kumar, B.; Steigerwald, M.L.; Venkataraman, L.; Nuckolls, C. Single-Molecule Conductance in Atomically Precise Germanium Wires. J. Am. Chem. Soc. 2015, 137, 12400. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Garner, M.H.; Su, T.A.; Jensen, A.; Inkpen, M.S.; Steigerwald, M.L.; Venkataraman, L.; Solomon, G.C.; Nuckolls, C. Extreme Conductance Suppression in Molecular Siloxanes. J. Am. Chem. Soc. 2017, 139, 10212. [Google Scholar] [CrossRef] [PubMed]
- Bonifas, A.P.; McCreery, R.L. ‘Soft’ Au, Pt and Cu contacts for molecular junctions through surface-diffusion-mediated deposition. Nat. Nanotechnol. 2010, 5, 612. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, F.; Li, J.; Sankey, O.F.; Terazono, Y.; Herrero, C.; Gust, D.; Moore, T.A.; Moore, A.L.; Lindsay, S.M. Electronic Decay Constant of Carotenoid Polyenes from Single-Molecule Measurements. J. Am. Chem. Soc. 2005, 127, 1384. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, B.; Dell, E.J.; Berkelbach, T.C.; Reichman, D.R.; Venkataraman, L.; Campos, L.M. Length-Dependent Conductance of Oligothiophenes. J. Am. Chem. Soc. 2014, 136, 10486. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Hernangomez-Perez, D.; Davydenko, I.; Marder, S.; Evers, F.; Venkataraman, L. Near Length-Independent Conductance in Polymethine Molecular Wires. Nano Lett. 2018, 18, 6387. [Google Scholar] [CrossRef]
- Sedghi, G.; Garcia-Suarez, V.M.; Esdaile, L.J.; Anderson, H.L.; Lambert, C.J.; Martin, S.; Bethell, D.; Higgins, S.J.; Elliott, M.; Bennett, N.; et al. Long-range electron tunnelling in oligo-porphyrin molecular wires. Nat. Nano 2011, 6, 517. [Google Scholar] [CrossRef]
- Leary, E.; Limburg, B.; Alanazy, A.; Sangtarash, S.; Grace, I.; Swada, K.; Esdaile, L.J.; Noori, M.; Teresa Gonzalez, M.; Rubio-Bollinger, G.; et al. Bias-Driven Conductance Increase with Length in Porphyrin Tapes. J. Am. Chem. Soc. 2018, 140, 12877. [Google Scholar] [CrossRef]
- Brooke, C.; Vezzoli, A.; Higgins, S.J.; Zotti, L.A.; Palacios, J.J.; Nichols, R.J. Resonant transport and electrostatic effects in single-molecule electrical junctions. Phys. Rev. B 2015, 91, 195438. [Google Scholar] [CrossRef]
- Sayed, S.Y.; Fereiro, J.A.; Yan, H.; McCreery, R.L.; Bergren, A.J. Charge transport in molecular electronic junctions: Compression of the molecular tunnel barrier in the strong coupling regime. Proc. Natl. Acad. Sci. USA 2012, 109, 11498. [Google Scholar] [CrossRef] [PubMed]
- Fereiro, J.A.; McCreery, R.L.; Bergren, A.J. Direct Optical Determination of Interfacial Transport Barriers in Molecular Tunnel Junctions. J. Am. Chem. Soc. 2013, 135, 9584. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Manrique, D.Z.; Garcia-Suarez, V.M.; Haiss, W.; Higgins, S.J.; Lambert, C.J.; Nichols, R.J. Adverse effects of asymmetric contacts on single molecule conductances of HS(CH2)n COOH in nanoelectrical junctions. Nanotechnology 2009, 20, 125203. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhou, J.; Hamill, J.M.; Xu, B. Measurement and understanding of single-molecule break junction rectification caused by asymmetric contacts. J. Chem. Phys. 2014, 141, 054712. [Google Scholar] [CrossRef] [PubMed]
- Evers, F.; Korytar, R.; Tewari, S.; van Ruitenbeek, J.M. Advances and challenges in single-molecule electron transport. Rev. Mod. Phys. 2020, 92, 035001. [Google Scholar] [CrossRef]
- Simmons, J.G. Generalized Formula for the Electric Tunnel Effect between Similar Electrodes Separated by a Thin Insulating Film. J. Appl. Phys. 1963, 34, 1793. [Google Scholar] [CrossRef]
- Nitzan, A. Electron Transmission Through Molecules and Molecular Interfaces. Ann. Rev. Phys. Chem. 2001, 52, 681. [Google Scholar] [CrossRef]
- Tomfohr, J.K.; Sankey, O.F. Complex band structure, decay lengths, and Fermi level alignment in simple molecular electronic systems. Phys. Rev. B 2002, 65, 245105. [Google Scholar] [CrossRef]
- Picaud, F.; Smogunov, A.; Corso, A.D.; Tosatti, E. Complex band structures and decay length in polyethylene chains. J. Phys. Condens. Matter. 2003, 15, 3731. [Google Scholar] [CrossRef][Green Version]
- Desjonqueres, M.C.; Spanjaard, D. Concepts in surface physics. In Springer Series in Surface Science; Springer: Berlin, Germany, 1993; Volume 30. [Google Scholar]
- Kiguchi, M.; Kaneko, S. Single Molecule Bridging Between Metal Electrodes. Phys. Chem. Chem. Phys. 2013, 15, 2253–2267. [Google Scholar] [CrossRef]
- Quek, S.Y.; Choi, H.J.; Louie, S.G.; Neaton, J.B. Length Dependence of Conductance in Aromatic Single-Molecule Junctions. Nano Lett. 2009, 9, 3949–3953. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.L.; Chen, Z.B.; Zhou, X.Y.; Sun, Y.Y.; Liang, J.H.; Niu, Z.J.; Zhou, X.S.; Mao, B.W. Single Molecule Conductance of Carboxylic Acids Contacting Ag and Cu Electrodes. J. Phys. Chem. C 2012, 116, 21699–21705. [Google Scholar] [CrossRef]
- Kiguchi, M.; Miura, S.; Hara, K.; Sawamura, M.; Murakoshi, K. Conductance of a Single Molecule Anchored by an Isocyanide Substituent to Gold Electrodes. Appl. Phys. Lett. 2006, 89, 213104. [Google Scholar] [CrossRef]
- Park, Y.S.; Whalley, A.C.; Kamenetska, M.; Steigerwald, M.L.; Hybertsen, M.S.; Nuckolls, C.; Venkataraman, L. Contact Chemistry and Single-Molecule Conductance: A Comparison of Phosphines, Methyl Sulfides, and Amines. J. Am. Chem. Soc. 2007, 129, 15768–15769. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Li, X.; Hihath, J.; Huang, Z.; Tao, N. Effect of Anchoring Groups on Single-Molecule Conductance: Comparative Study of Thiol-, Amine-, and Carboxylic-Acid-Terminated Molecules. J. Am. Chem. Soc. 2006, 128, 15874–15881. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.A.; Zhou, C.; Muller, C.J.; Burgin, T.P.; Tour, J.M. Conductance of a Molecular Junction. Science 1997, 278, 252–254. [Google Scholar] [CrossRef]
- Xu, B.; Tao, N.J. Measurement of Single-Molecule Resistance by Repeated Formation of Molecular Junctions. Science 2003, 301, 1221–1223. [Google Scholar] [CrossRef]
- Cui, X.D.; Primak, A.; Zarate, X.; Tomfohr, J.; Sankey, O.F.; Moore, A.L.; Moore, T.A.; Gust, D.; Harris, G.; Lindsay, S.M. Reproducible Measurement of Single-Molecule Conductivity. Science 2001, 294, 571–574. [Google Scholar] [CrossRef]
- Haiss, W.; van Zalinge, H.; Higgins, S.J.; Bethell, D.; Höbenreich, H.; Schiffrin, D.J.; Nichols, R.J. Redox State Dependence of Single Molecule Conductivity. J. Am. Chem. Soc. 2003, 125, 15294. [Google Scholar] [CrossRef]
- Castellanos-Gomez, A.; Agraït, N.; Rubio-Bollinger, G. Carbon fibre tips for scanning probe microscopy based on quartz tuning fork force sensors. Nanotechnology 2010, 21, 145702. [Google Scholar] [CrossRef]
- Dappe, Y.J.; González, C.; Cuevas, J.C. Carbon tips for all-carbon single-molecule electronics. Nanoscale 2014, 6, 6953. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, L.; Tao, S.; Wang, C.; Zhao, C.; González, C.; Dappe, Y.J.; Nichols, R.J.; Yang, L. Graphene as a promising electrode for low current attenuation in asymmetric molecular junctions. Nano Lett. 2016, 16, 6534. [Google Scholar] [CrossRef]
- Zhang, Q.; Tao, S.; Yi, R.; He, C.; Zhao, C.; Su, W.; Smogunov, A.; Dappe, Y.J.; Nichols, R.J.; Yang, L. Symmetry Effects on Attenuation Factors in Graphene-Based Molecular Junctions. J. Phys. Chem. Lett. 2017, 8, 5987–5992. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, Q.; Gao, T.; Liu, C.; Chen, Z.; Zhao, C.; Zhao, C.; Nichols, R.J.; Dappe, Y.J.; Yang, L. Charge transport in hybrid platinum/molecule/graphene single molecule junctions. Phys. Chem. Chem. Phys. 2020, 22, 13498. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, Q.; Tao, S.; Zhao, C.Z.; Zhao, C.; Su, W.; Dappe, Y.J.; Nichols, R.J.; Yang, L. Carbon-contacted single molecule electrical junctions. Phys. Chem. Chem. Phys. 2018, 20, 24553. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhang, Q.; Fan, Y.; Zhao, C.Z.; Zhao, C.; Ye, J.; Dappe, Y.J.; Nichols, R.J.; Yang, L. Effect of Asymmetric Anchoring Groups on Electronic Transport in Hybrid Metal/Molecule/Graphene Single Molecule Junctions. Chem. Phys. Chem. 2019, 20, 1830. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Zhang, Q.; He, C.; Lin, X.; Xie, R.; Zhao, C.Z.; Zhao, C.; Smogunov, A.; Dappe, Y.J.; Nichols, R.J.; et al. Graphene-Contacted Single Molecular Junctions with Conjugated Molecular Wires. ACS Appl. Nano Mater. 2019, 2, 12. [Google Scholar] [CrossRef]








© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dappe, Y.J. Attenuation Factors in Molecular Electronics: Some Theoretical Concepts. Appl. Sci. 2020, 10, 6162. https://doi.org/10.3390/app10186162
Dappe YJ. Attenuation Factors in Molecular Electronics: Some Theoretical Concepts. Applied Sciences. 2020; 10(18):6162. https://doi.org/10.3390/app10186162
Chicago/Turabian StyleDappe, Yannick J. 2020. "Attenuation Factors in Molecular Electronics: Some Theoretical Concepts" Applied Sciences 10, no. 18: 6162. https://doi.org/10.3390/app10186162
APA StyleDappe, Y. J. (2020). Attenuation Factors in Molecular Electronics: Some Theoretical Concepts. Applied Sciences, 10(18), 6162. https://doi.org/10.3390/app10186162