
Extraction of ADP-Heptose and Kdo2-Lipid A from E. coli Deficient in the Heptosyltransferase I Gene


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Featured Application
Abstract
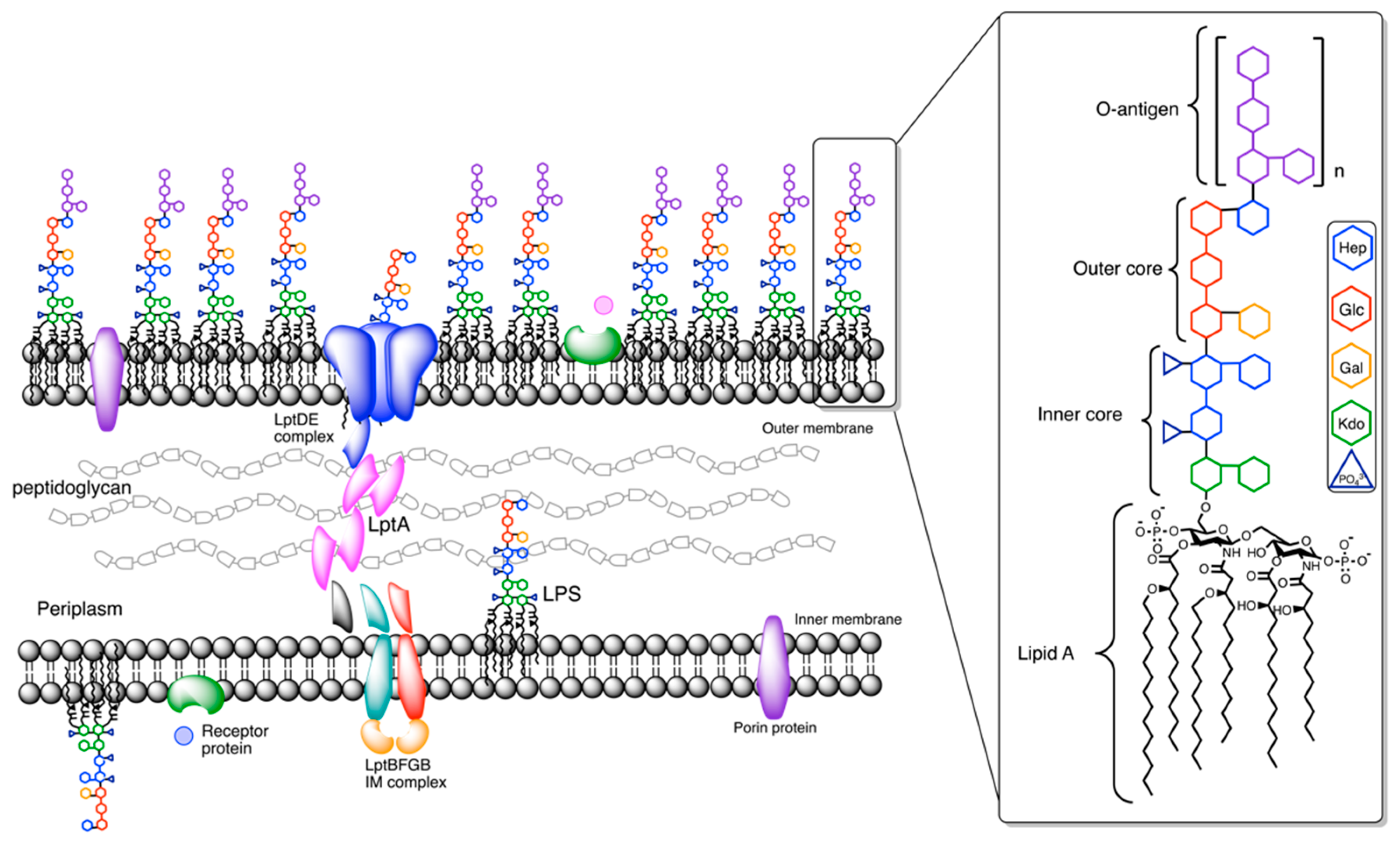
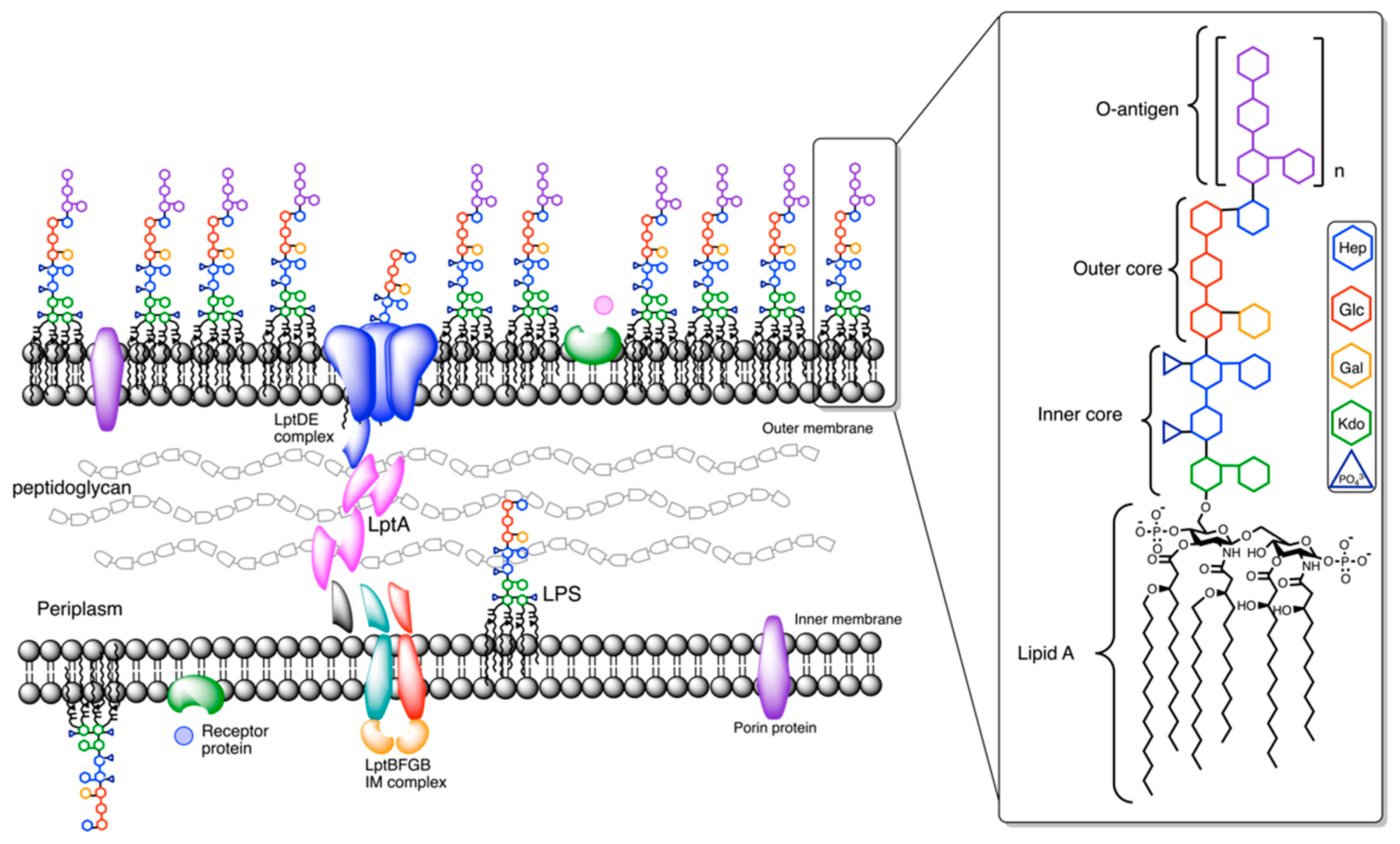
1. Introduction
2. Materials and Methods
2.1. General Materials and Equipment
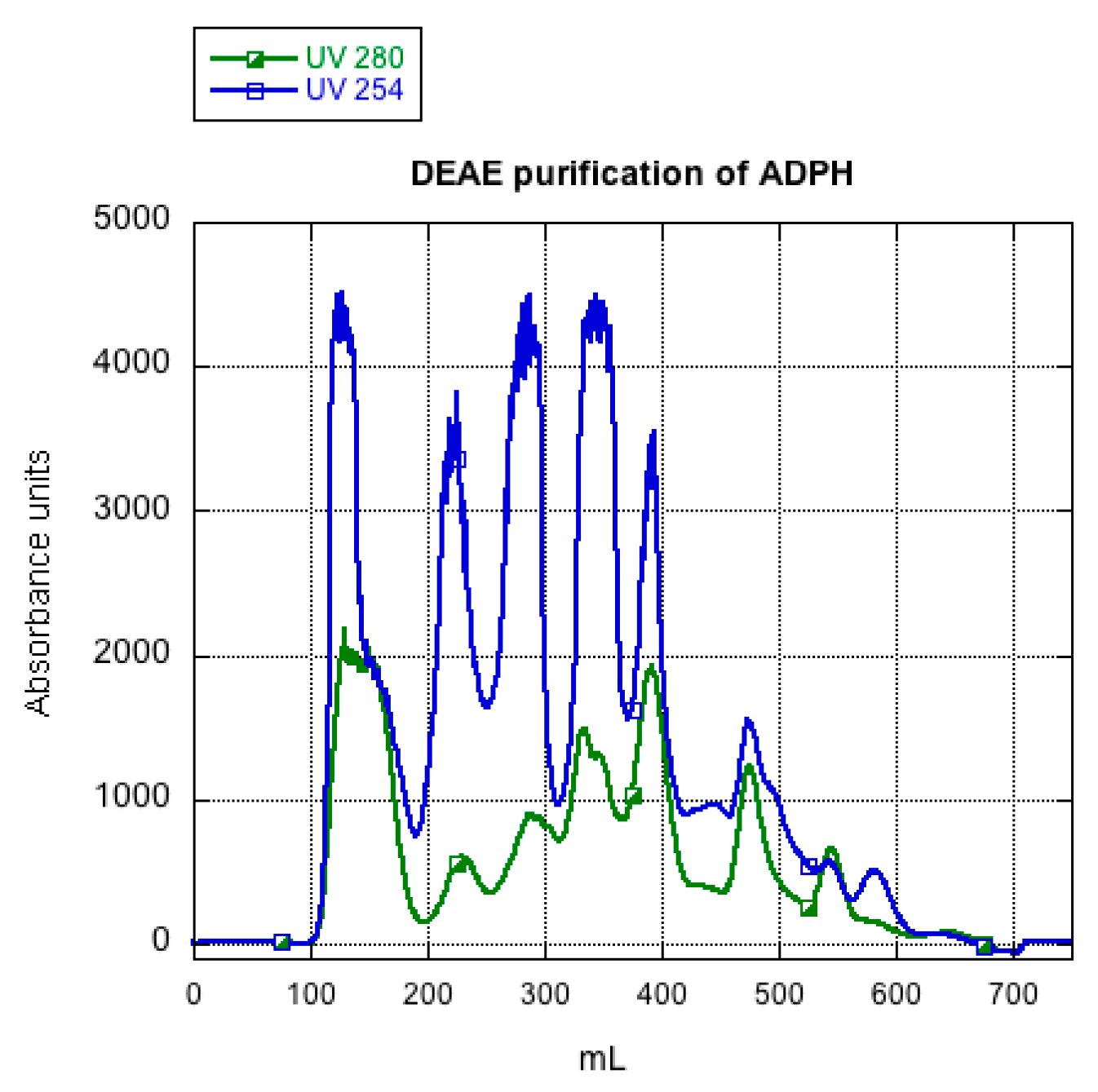
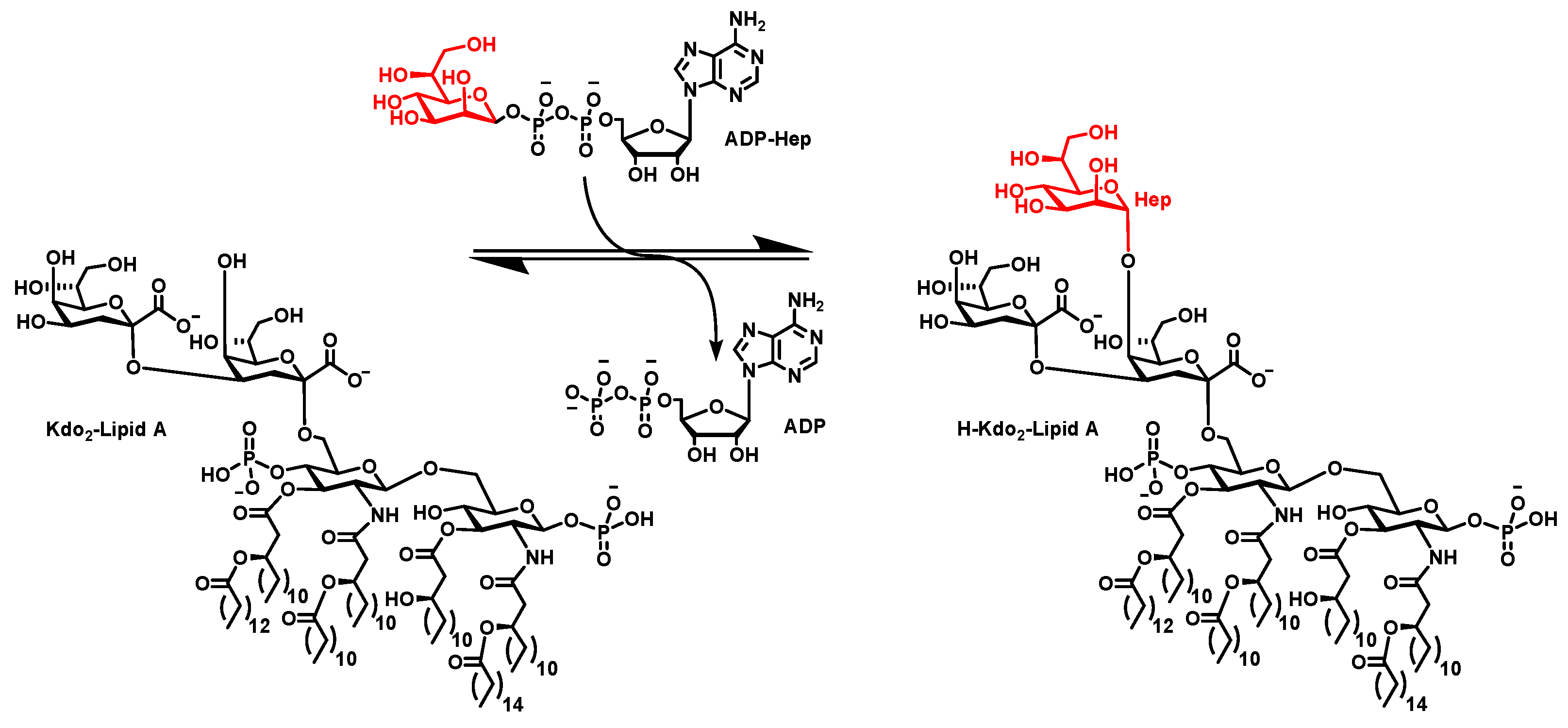
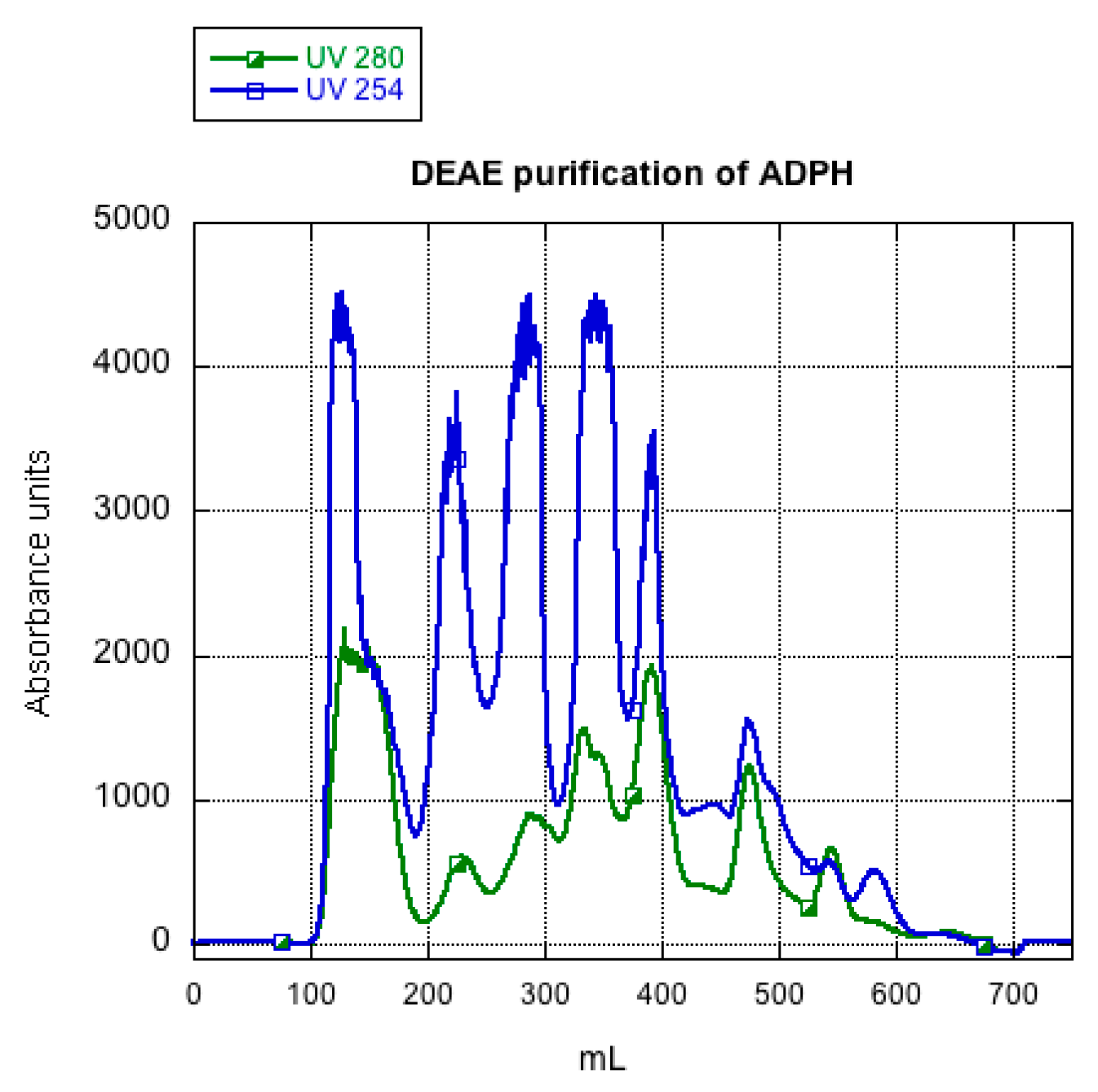
2.2. ADPH Isolation and Purification
2.3. Kdo2-Lipid A Isolation
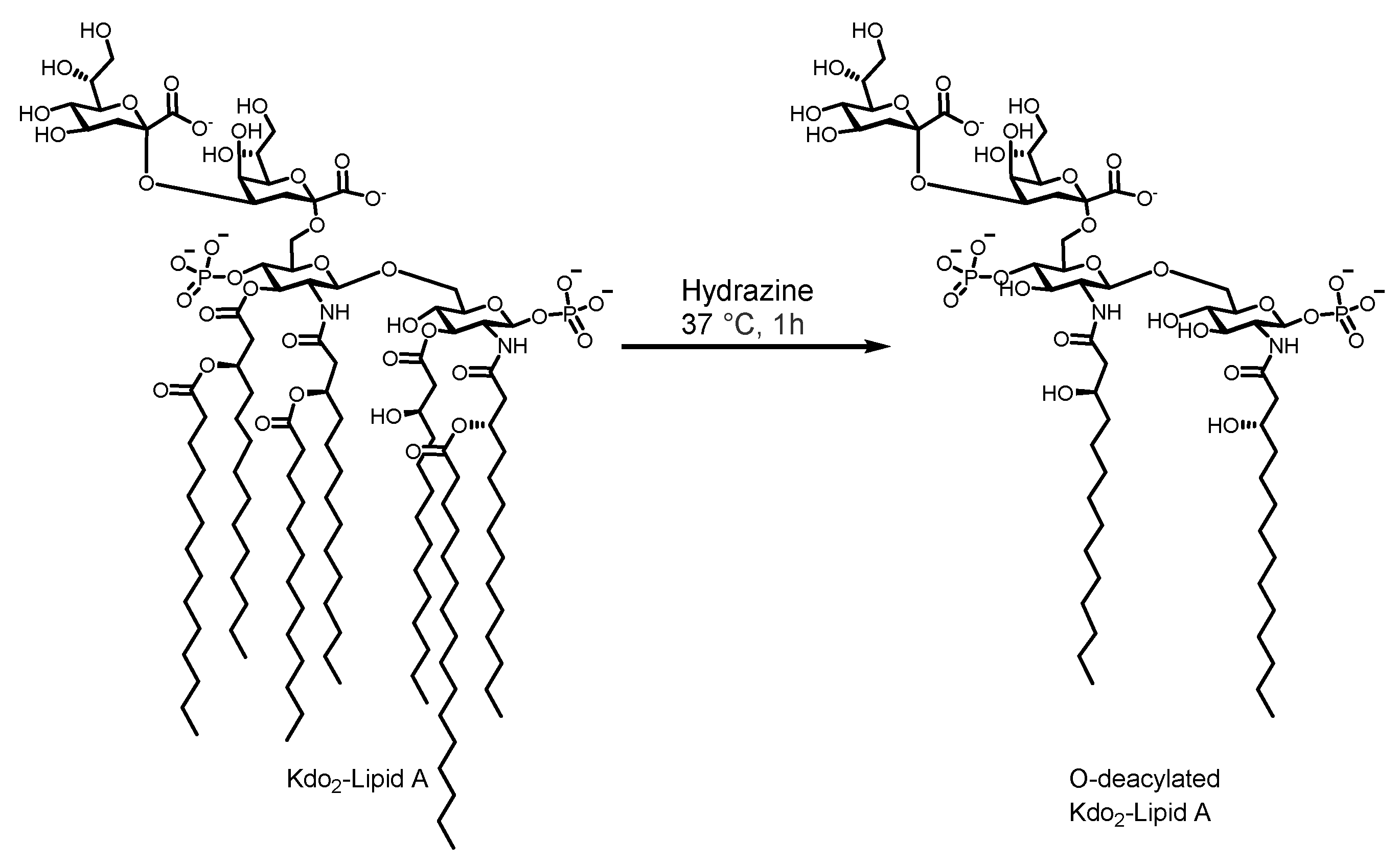
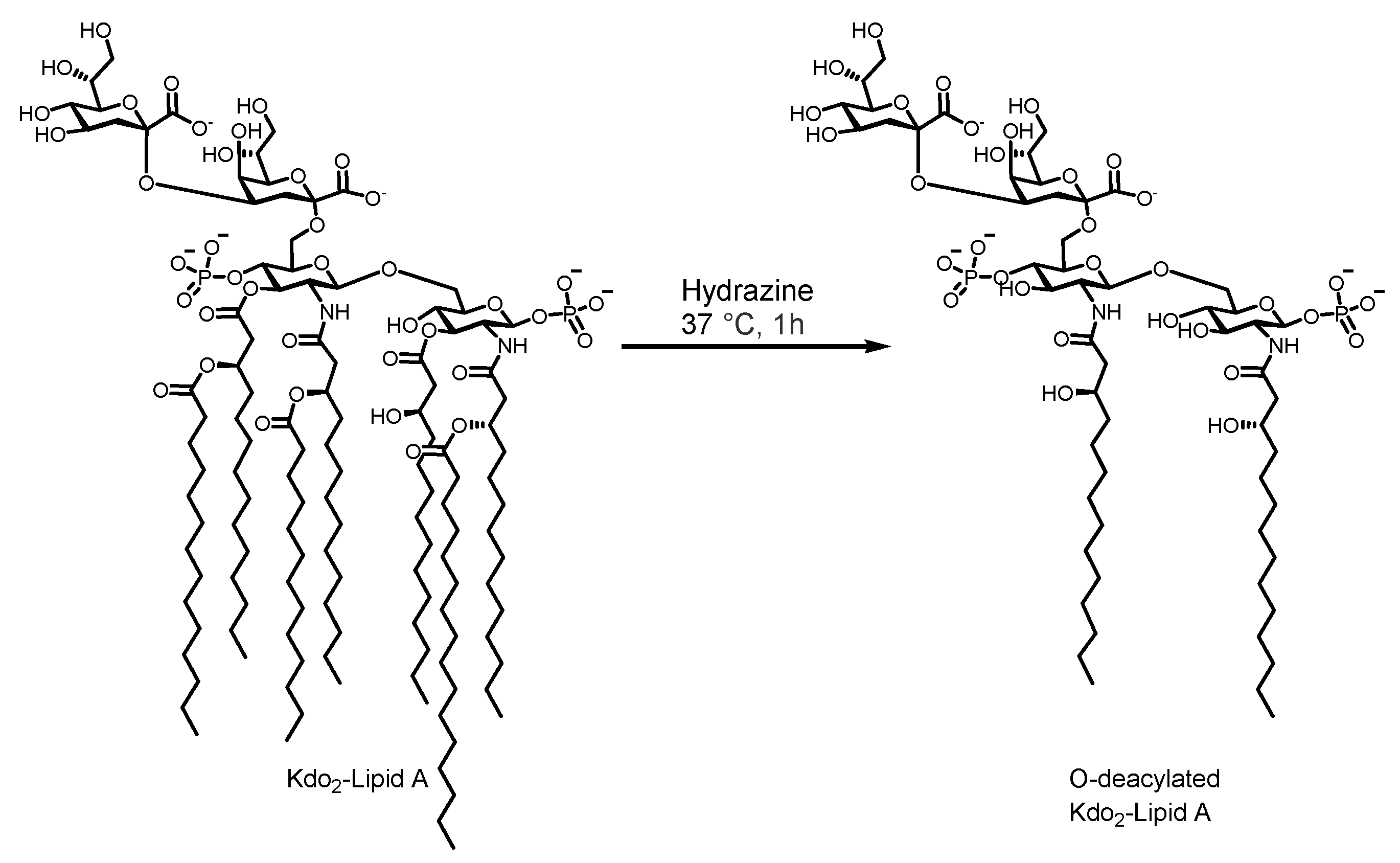
2.4. Kdo2-Lipid A Deacylation
3. Results
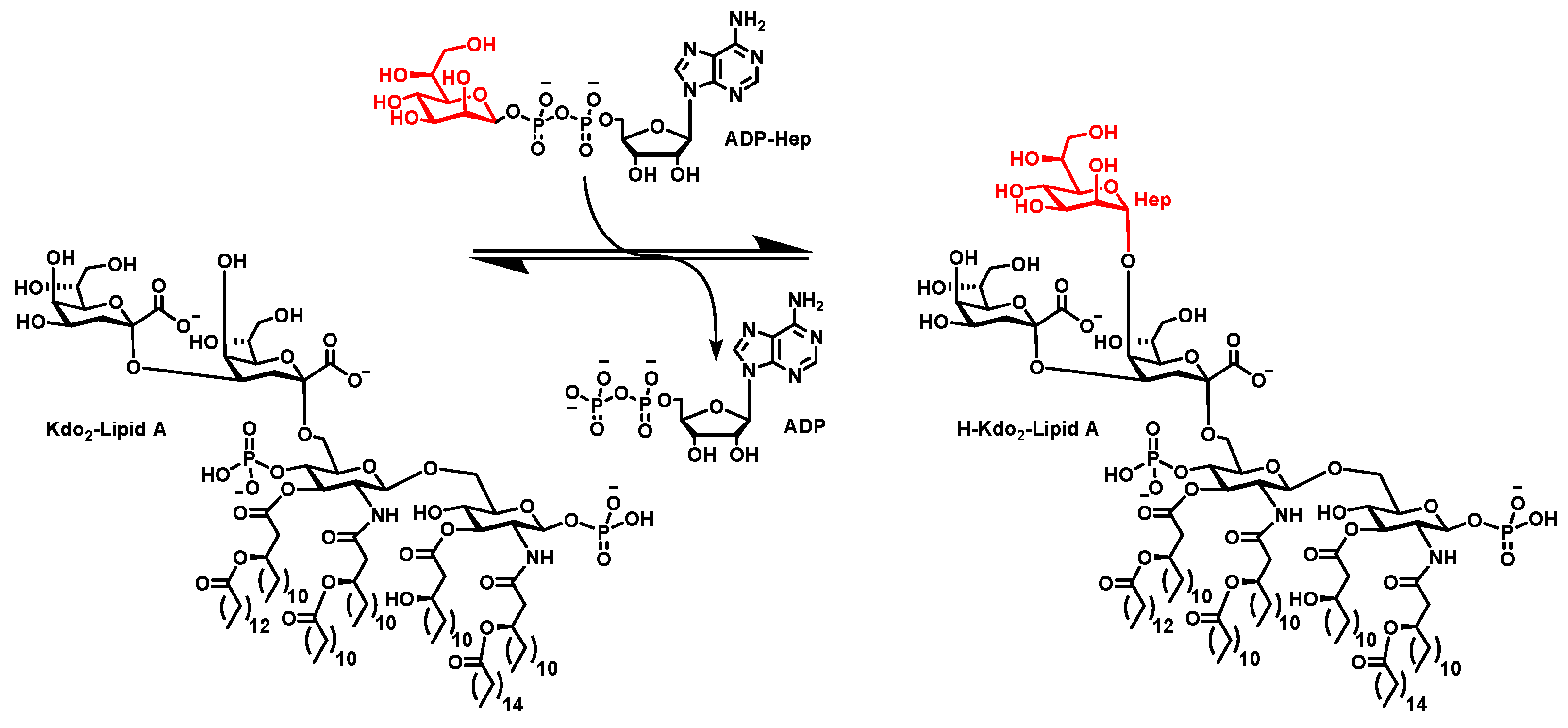
3.1. ADPH Extraction and Purification
3.2. Kdo2-Lipid A Extraction and Deacylation
4. Discussion
4.1. ADPH Extraction and Purification
4.2. Kdo2-Lipid A Extraction and Deacylation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakao, R.; Ramstedt, M.; Wai, S.N.; Uhlin, B.E. Enhanced Biofilm Formation by Escherichia coli LPS Mutants Defective in Hep Biosynthesis. PLoS ONE 2012, 7, e51241. [Google Scholar] [CrossRef] [Green Version]
- Cote, J.M.; Taylor, E.A. The Glycosyltransferases of LPS Core: A Review of Four Heptosyltransferase Enzymes in Context. Int. J. Mol. Sci. 2017, 18, 2256. [Google Scholar] [CrossRef] [Green Version]
- Alexander, C.; Rietschel, E.T. Invited review: Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef]
- Gunn, J.S. Bacterial modification of LPS and resistance to antimicrobial peptides. J. Endotoxin Res. 2001, 7, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Groisman, E.A.; Kayser, J.; Soncini, F.C. Regulation of polymyxin resistance and adaptation to low-Mg2+ environments. J. Bacteriol. 1997, 179, 7040–7045. [Google Scholar] [CrossRef] [Green Version]
- Nishino, K.; Hsu, F.-F.; Turk, J.; Cromie, M.J.; Wosten, M.; Groisman, E.A. Identification of the lipopolysaccharide modifications controlled by the Salmonella PmrA/PmrB system mediating resistance to Fe(III) and Al(III). Mol. Microbiol. 2006, 61, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Mamat, U.; Skurnik, M.; Bengoechea, J.A. Lipopolysaccharide Core Oligosaccharide Biosynthesis and Assembly. In Bacterial Lipopolysaccharides: Structure, Chemical Synthesis, Biogenesis and Interaction with Host Cells; Knirel, Y.A., Valvano, M.A., Eds.; Springer: Vienna, Austria, 2011; pp. 237–273. [Google Scholar] [CrossRef]
- Czyzyk, D.J.; Sawant, S.S.; Ramirez-Mondragon, C.A.; Hingorani, M.M.; Taylor, E.A. Escherichia coli Heptosyltransferase I: Investigation of Protein Dynamics of a GT-B Structural Enzyme. Biochemistry 2013, 52, 5158–5160. [Google Scholar] [CrossRef] [Green Version]
- Nkosana, N.K.; Czyzyk, D.J.; Siegel, Z.S.; Cote, J.M.; Taylor, E.A. Synthesis, kinetics and inhibition of Escherichia coli Heptosyltransferase I by monosaccharide analogues of Lipid A. Bioorg. Med. Chem. Lett. 2018, 28, 594–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, J.M.; Ramirez-Mondragon, C.A.; Siegel, Z.S.; Czyzyk, D.J.; Gao, J.; Sham, Y.Y.; Mukerji, I.; Taylor, E.A. The Stories Tryptophans Tell: Exploring Protein Dynamics of Heptosyltransferase I from Escherichia coli. Biochemistry 2017, 56, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Cote, J.M.; Hecht, C.J.S.; Patel, K.R.; Ramirez-Mondragon, C.A.; Sham, Y.Y.; Taylor, E.A. Opposites Attract: Escherichia coli Heptosyltransferase I Conformational Changes Induced by Interactions between the Substrate and Positively Charged Residues. Biochemistry 2020, 59, 3135–3147. [Google Scholar] [CrossRef]
- Hu, X.; Yang, C.; Wang, P.G.; Zhang, G.-L. ADP-Heptose: A new innate immune modulator. Carbohydr. Res. 2018, 473, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kneidinger, B.; Marolda, C.; Graninger, M.; Zamyatina, A.; McArthur, F.; Kosma, P.; Valvano, M.A.; Messner, P. Biosynthesis pathway of ADP-L-Glycero-Beta-D-Manno-Heptose in Escherichia coli. J. Bacteriol. 2002, 184, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfannkuch, L.; Hurwitz, R.; Trauisen, J.; Sigulla, J.; Poeschke, M.; Matzner, L.; Kosma, P.; Schmid, M.; Meyer, T.F. ADP heptose, a novel pathogen-associated molecular pattern identified in Helicobacter pylori. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 9087–9099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudapaka, J.; Taylor, E.A. Cloning and characterization of the Escherichia coli Heptosyltransferase III: Exploring substrate specificity in lipopolysaccharide core biosynthesis. FEBS Lett. 2015, 589, 1423–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durka, M.; Tikad, A.; Périon, R.; Bosco, M.; Andaloussi, M.; Floquet, S.; Malacain, E.; Moreau, F.; Oxoby, M.; Gerusz, V.; et al. Systematic Synthesis of Inhibitors of the Two First Enzymes of the Bacterial Heptose Biosynthetic Pathway: Towards Antivirulence Molecules Targeting Lipopolysaccharide Biosynthesis. Chem. A Eur. J. 2011, 17, 11305–11313. [Google Scholar] [CrossRef] [PubMed]
- Grizot, S.; Salem, M.; Vongsouthi, V.; Durand, L.; Moreau, F.; Dohi, H.; Vincent, S.; Escaich, S.; Ducruix, A. Structure of the Escherichia coli Heptosyltransferase WaaC: Binary Complexes with ADP AND ADP-2-deoxy-2-fluoro Heptose. J. Mol. Biol. 2006, 363, 383–394. [Google Scholar] [CrossRef]
- Balla, E.; Zamyatina, A.; Hofinger, A.; Kosma, P. Synthesis of a deoxy analogue of ADP l-glycero-d-manno-heptose. Carbohydr. Res. 2007, 342, 2537–2545. [Google Scholar] [CrossRef]
- Graziani, A.; Amer, H.; Zamyatina, A.; Hofinger, A.; Kosma, P. Synthesis of C-Glycosidically linked ADP glycero-β-d-manno-heptose analogues. Tetrahedron Asymmetry 2007, 18, 115–122. [Google Scholar] [CrossRef]
- Wang, X.; Quinn, P.J.; Yan, A. Kdo2-Lipid A: Structural diversity and impact on immunopharmacology. Biol. Rev. 2014, 90, 408–427. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.H.; Garrett, T.A.; Reynolds, C.M.; Shaw, W.A.; Moore, J.D.; Smith, D.C.; Ribeiro, A.A.; Murphy, R.C.; Ulevitch, R.J.; Fearns, C.; et al. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 2006, 47, 1097–1111. [Google Scholar] [CrossRef] [Green Version]
- Rutten, L.; Mannie, J.P.; Stead, C.M.; Raetz, C.R.; Reynolds, C.M.; Bonvin, A.M.; Tommassen, J.P.; Egmond, M.R.; Trent, M.S.; Gros, P. Active-site architecture and catalytic mechanism of the lipid A deacylase LpxR of Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 2009, 106, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Brabetz, W.; Muller-Loennies, S.; Holst, O.; Brade, H. Deletion of the Heptosyltransferase Genes rfaC and rfaF in Escherichia Coli K-12 Results in an Re-Type Lipopolysaccharide with a High Degree of 2-Aminoethanol Phosphate Substitution. JBIC J. Biol. Inorg. Chem. 1997, 247, 716–724. [Google Scholar] [CrossRef]
- Czyzyk, D.J.; Liu, C.; Taylor, E.A. Lipopolysaccharide Biosynthesis without the Lipids: Recognition Promiscuity of Escherichia coli Heptosyltransferase I. Biochemistry 2011, 50, 10570–10572. [Google Scholar] [CrossRef] [Green Version]
- Gronow, S.; Oertelt, C.; Ervelä, E.; Zamyatina, A.; Kosma, P.; Skurnik, M.; Holst, O. Characterization of the physiological substrate for lipopolysaccharide heptosyltransferases I and II. J. Endotoxin Res. 2001, 7, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Kanipes, M.I.; Lin, S.; Cotter, R.J.; Raetz, C.R.H. Ca2+-induced phosphoethanolamine transfer to the outer 3-deoxy-d-manno-octulosonic acid moiety of Escherichia coli lipopolysaccharide: A novel membrane enzyme dependent upon phosphatidylethanolamine. J. Biol. Chem. 2001, 276, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Hankins, J.V.; Madsen, J.A.; Needham, B.D.; Brodbelt, J.S.; Trent, M.S. The Outer Membrane of Gram-Negative Bacteria: Lipid A Isolation and Characterization. Methods Mol. Biol. 2012, 966, 239–258. [Google Scholar] [CrossRef] [Green Version]
- Galanos, C.; Luderitz, O.; Westphal, O. A New Method for the Extraction of R Lipopolysaccharides. JBIC J. Biol. Inorg. Chem. 1969, 9, 245–249. [Google Scholar] [CrossRef]
- Kneidinger, B.; Graninger, M.; Puchberger, M.; Kosma, P.; Messner, P. Biosynthesis of Nucleotide-activated-d-glycero-d-manno-Heptose. J. Biol. Chem. 2001, 276, 20935–20944. [Google Scholar] [CrossRef] [Green Version]
- García-Weber, D.; Dangeard, A.; Cornil, J.; Thai, L.; Rytter, H.; Zamyatina, A.; Mulard, L.A.; Arrieumerlou, C. ADP-heptose is a newly identified pathogen-associated molecular pattern of Shigella flexneri. EMBO Rep. 2018, 19, e46943. [Google Scholar] [CrossRef] [PubMed]
- Adekoya, I.A.; Guo, C.X.; Gray-Owen, S.D.; Cox, A.D.; Sauvageau, J. d-Glycero-β-d-Manno-Heptose 1-Phosphate and d-Glycero-β-d-Manno-Heptose 1,7-Biphosphate Are Both Innate Immune Agonists. Am. J. Immunol. 2018, 201, 2385–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muheim, C.; Bakali, A.; Engström, O.; Wieslander, Å.; Daley, D.O.; Widmalm, G. Identification of a Fragment-Based Scaffold that Inhibits the Glycosyltransferase WaaG from Escherichia coli. Antibiotics 2016, 5, 10. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milicaj, J.; Castro, C.D.; Jaunbocus, N.; Taylor, E.A. Extraction of ADP-Heptose and Kdo2-Lipid A from E. coli Deficient in the Heptosyltransferase I Gene. Appl. Sci. 2021, 11, 8314. https://doi.org/10.3390/app11188314
Milicaj J, Castro CD, Jaunbocus N, Taylor EA. Extraction of ADP-Heptose and Kdo2-Lipid A from E. coli Deficient in the Heptosyltransferase I Gene. Applied Sciences. 2021; 11(18):8314. https://doi.org/10.3390/app11188314
Chicago/Turabian StyleMilicaj, Jozafina, Colleen D. Castro, Nadiya Jaunbocus, and Erika A. Taylor. 2021. "Extraction of ADP-Heptose and Kdo2-Lipid A from E. coli Deficient in the Heptosyltransferase I Gene" Applied Sciences 11, no. 18: 8314. https://doi.org/10.3390/app11188314
APA StyleMilicaj, J., Castro, C. D., Jaunbocus, N., & Taylor, E. A. (2021). Extraction of ADP-Heptose and Kdo2-Lipid A from E. coli Deficient in the Heptosyltransferase I Gene. Applied Sciences, 11(18), 8314. https://doi.org/10.3390/app11188314