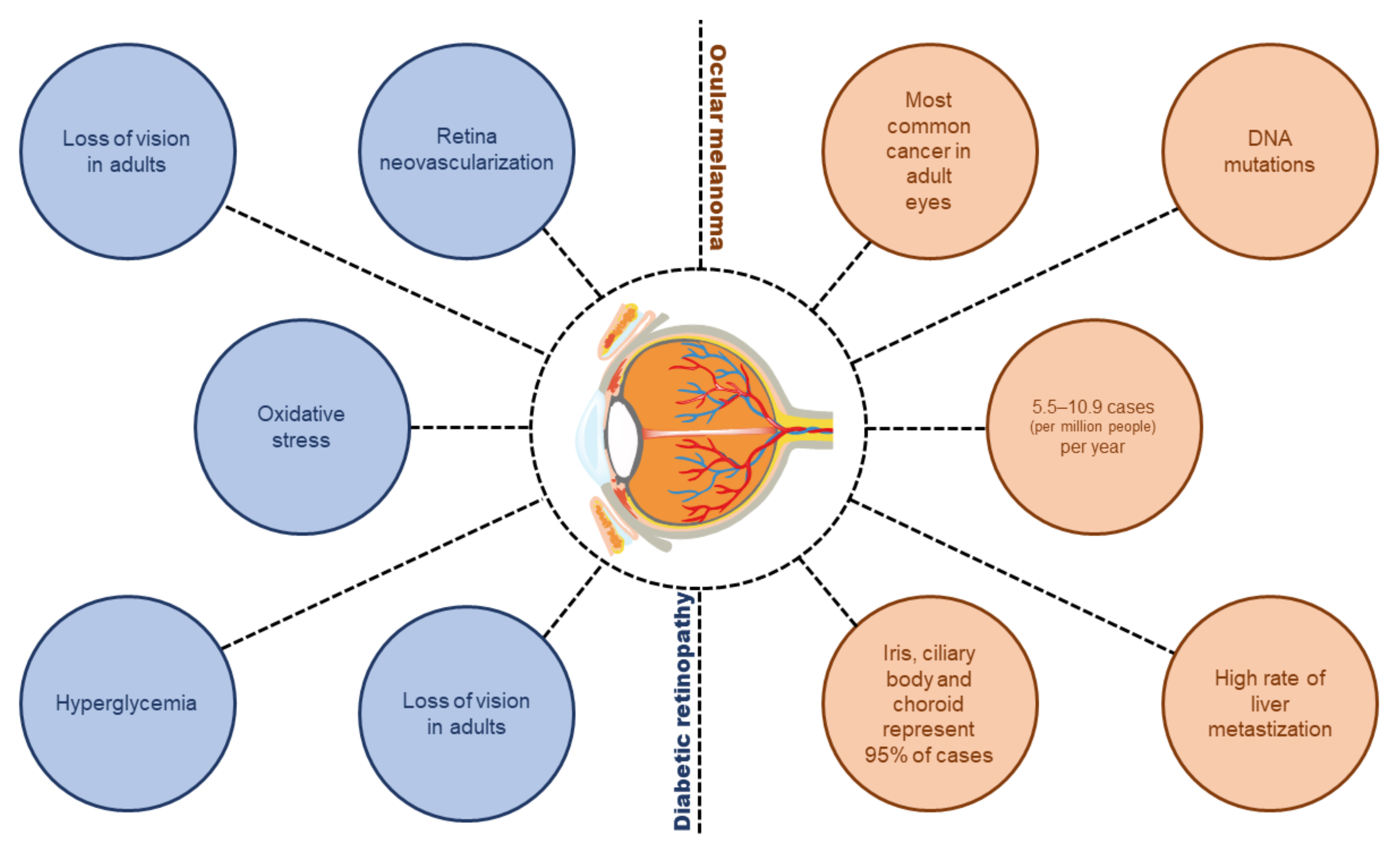
Diabetic Retinopathy and Ocular Melanoma: How Far We Are?

 , , ,
, , ,  ,
, 
 , , ,
, , ,  and
and
Abstract
1. Introduction
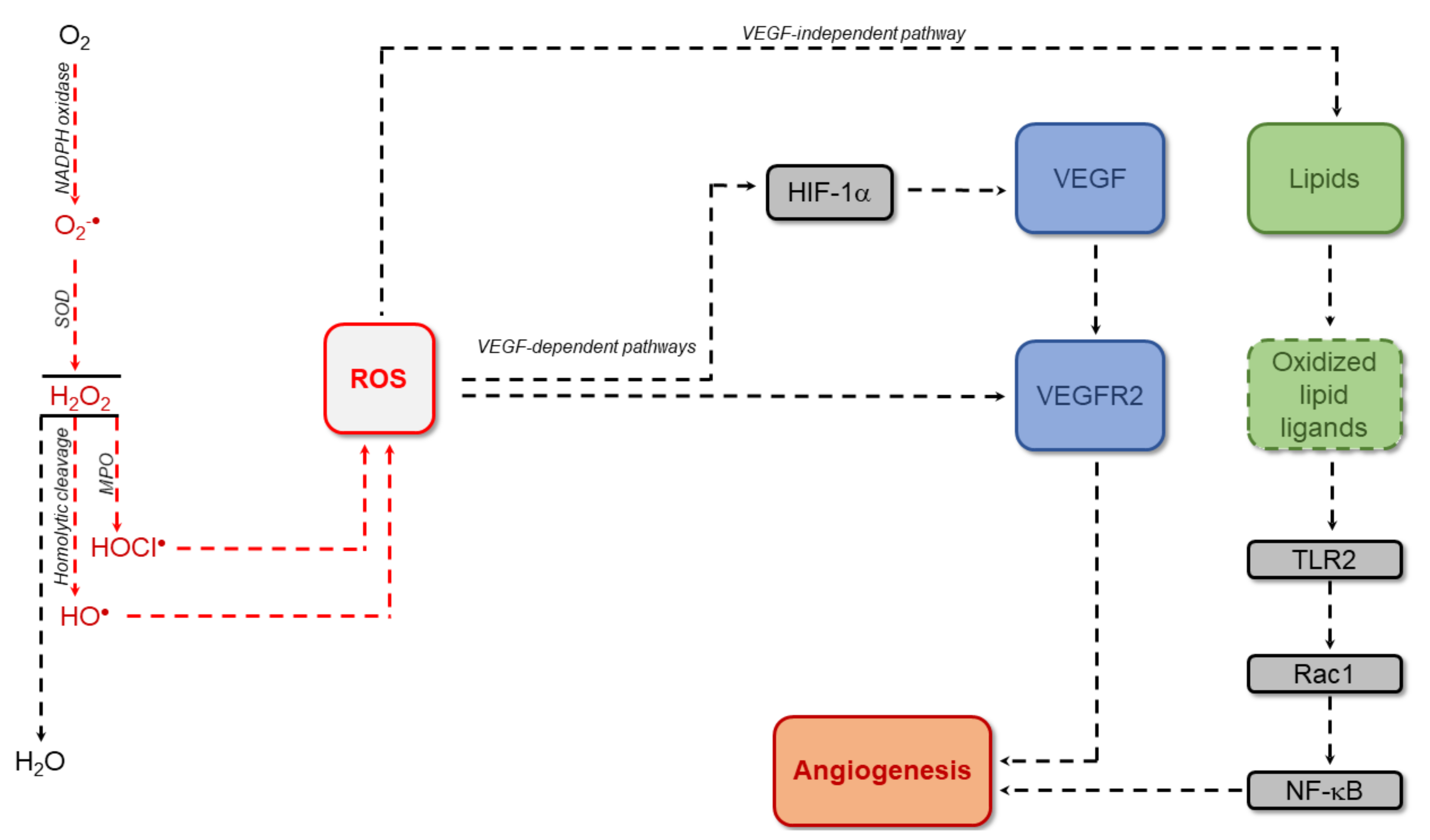
2. Angiogenesis and Anti-Angiogenic Therapy
3. Diabetic Retinopathy, Angiogenesis and Anti-Angiogenic Therapy
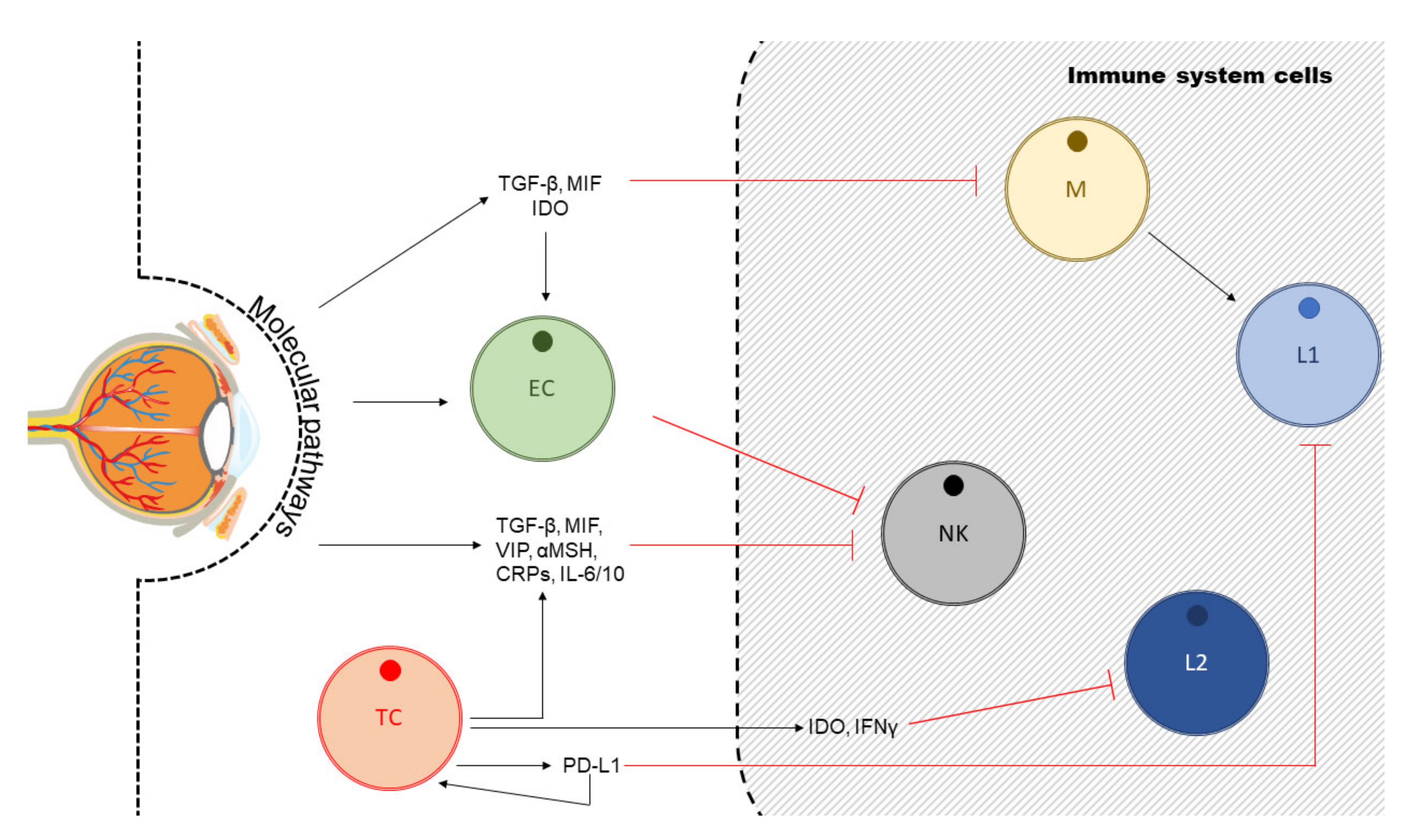
4. Uveal Melanoma, Angiogenesis and Anti-Angiogenic Therapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMD | Age-related macular degeneration |
| Ang-2 | Angiopoietin-2 |
| bFGF | basic fibroblast growth factor |
| BRB | Blood-retinal barriers |
| CNV | Choroidal neovascularization |
| DME | Diabetic macular edema |
| DR | Diabetic retinopathy |
| GFAP | Glial fibrillary acidic protein |
| HMGB1 | High mobility group protein B1 |
| HOS | Hyperosmolar stress |
| IGF-1R | Insulin growth factor- 1 receptor |
| IL | Interleukins |
| IL6 | Interleukin 6 |
| IL8 | Interleukin 8 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MGCs | Müller glial cells |
| MGSA | Melanoma growth stimulatory activity |
| MIP1 | Macrophage inflammatory protein 1 |
| MMPs | Matrix metalloproteinases |
| NAP2 | Neutrophil activating protein 2 |
| NFAT5 | Nuclear factor of activated T-cells 5 |
| NPDR | Non-proliferative DR |
| PDGF | Platelet-derived growth factor-BB |
| PDR | Proliferation DR |
| PIGF | Placental growth factor |
| RGCs | Retinal ganglion cells |
| ROS | Reactive oxygen species |
| RPE | Retinal pigmented epithelial cells |
| RT | Radiotherapy |
| SOD | Superoxide dismutase |
| TLRs | Toll-like receptors |
| TNFα | Tumour necrosis alpha |
| TonEBP | Transcription factor tonicity-responsive binding-protein |
| UV | Ultraviolet |
| VEGF | Vascular endothelial growth factor |
| VEGR | Vascular endothelial growth receptor |
References
- Souto, E.B.; Zielinska, A.; Luis, M.; Carbone, C.; Martins-Gomes, C.; Souto, S.B.; Silva, A.M. Uveal melanoma: Physiopathology and new in situ-specific therapies. Cancer Chemother. Pharmacol. 2019, 84, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Balne, P.K.; Meissner, K.E.; Barathi, V.A.; Schmetterer, L.; Agrawal, R. Assessment of flow dynamics in retinal and choroidal microcirculation. Surv. Ophthalmol. 2018, 63, 646–664. [Google Scholar] [CrossRef]
- Willermain, F.; Scifo, L.; Weber, C.; Caspers, L.; Perret, J.; Delporte, C. Potential Interplay between Hyperosmolarity and Inflammation on Retinal Pigmented Epithelium in Pathogenesis of Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 1056. [Google Scholar] [CrossRef]
- Fangueiro, J.F.; Silva, A.M.; Garcia, M.L.; Souto, E.B. Current nanotechnology approaches for the treatment and management of diabetic retinopathy. Eur. J. Pharm. Biopharm. 2014. [Google Scholar] [CrossRef]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef]
- Willermain, F.; Libert, S.; Motulsky, E.; Salik, D.; Caspers, L.; Perret, J.; Delporte, C. Origins and consequences of hyperosmolar stress in retinal pigmented epithelial cells. Front. Physiol. 2014, 5, 199. [Google Scholar] [CrossRef]
- Madonna, R.; Giovannelli, G.; Confalone, P.; Renna, F.V.; Geng, Y.-J.; De Caterina, R. High glucose-induced hyperosmolarity contributes to COX-2 expression and angiogenesis: Implications for diabetic retinopathy. Cardiovasc. Diabetol. 2016, 15, 18. [Google Scholar] [CrossRef]
- Bringmann, A.; Hollborn, M.; Kohen, L.; Wiedemann, P. Intake of dietary salt and drinking water: Implications for the development of age-related macular degeneration. Mol. Vis. 2016, 22, 1437. [Google Scholar] [PubMed]
- Mashayekhi, A.; Schönbach, E.; Shields, C.L.; Shields, J.A. Early Subclinical Macular Edema in Eyes with Uveal Melanoma: Association with Future Cystoid Macular Edema. Ophthalmology 2015. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Struyf, S.; Verbeke, H.; Van Damme, J.; Geboes, K. Circulating bone-marrow-derived endothelial precursor cells contribute to neovascularization in diabetic epiretinal membranes. Acta Ophthalmol. 2011, 89, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.X.; Wang, Y.S.; Shi, Y.Y.; Hou, H.Y.; Zhang, C.; Cai, Y.; Dou, G.R.; Yao, L.B.; Li, F.Y. Hypoxia specific SDF-1 expression by retinal pigment epithelium initiates bone marrow-derived cells to participate in Choroidal neovascularization in a laser-induced mouse model. Curr. Eye Res. 2011, 36, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Xue, Y.; Hedlund, E.M.; Zhong, Z.; Tritsaris, K.; Tondelli, B.; Lucchini, F.; Zhu, Z.; Dissing, S.; Cao, Y. VEGFR1-mediated pericyte ablation links VEGF and PlGF to cancer-associated retinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Cao, Y. Cancer-associated retinopathy: A new mechanistic insight on vascular remodeling. Cell Cycle 2010, 9, 1882–1885. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, L.; Tonissi, F.; Torta, I.; Gianello, L.; Russi, E.; Milano, G.; Merlano, M.; Lo Nigro, C. Role of IL-8 induced angiogenesis in uveal melanoma. Investig. New Drugs 2013, 31, 1107–1114. [Google Scholar] [CrossRef]
- Jovanovic, P.; Mihajlovic, M.; Djordjevic-Jocic, J.; Vlajkovic, S.; Cekic, S.; Stefanovic, V. Ocular melanoma: An overview of the current status. Int J Clin Exp Pathol 2013, 6, 1230–1244. [Google Scholar]
- Logan, P.; Burnier, J.; Burnier, M.N., Jr. Vascular endothelial growth factor expression and inhibition in uveal melanoma cell lines. Ecancermedicalscience 2013, 7, 336. [Google Scholar] [CrossRef]
- Leyvraz, S.; Keilholz, U. Ocular melanoma: What’s new? Curr. Opin. Oncol. 2012, 24, 162–169. [Google Scholar] [CrossRef]
- Grajewski, R.S.; Schuler-Thurner, B.; Mauch, C.; Kreuzberg, N.; Koch, K.R.; Bergua, A.; Cursiefen, C.; Heindl, L.M. Ocular diseases in metastatic cutaneous melanoma: Review of 108 consecutive patients in two German tertiary centers. Graefe’s Arch. Clin. Exp. Ophthalmol. 2014, 252, 679–685. [Google Scholar] [CrossRef]
- Bishop, K.D.; Olszewski, A.J. Epidemiology and survival outcomes of ocular and mucosal melanomas: A population-based analysis. Int. J. Cancer 2014, 134, 2961–2971. [Google Scholar] [CrossRef]
- Damato, B.; Eleuteri, A.; Taktak, A.F.; Coupland, S.E. Estimating prognosis for survival after treatment of choroidal melanoma. Prog. Retin. Eye Res. 2011, 30, 285–295. [Google Scholar] [CrossRef]
- Harbour, J.W.; Chao, D.L. A molecular revolution in uveal melanoma: Implications for patient care and targeted therapy. Ophthalmology 2014, 121, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.; Dopierala, J.A.; Coupland, S.E. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin. Cancer Res. 2010, 16, 6083–6092. [Google Scholar] [CrossRef] [PubMed]
- Bianciotto, C.; Shields, C.L.; Pirondini, C.; Mashayekhi, A.; Furuta, M.; Shields, J.A. Proliferative radiation retinopathy after plaque radiotherapy for uveal melanoma. Ophthalmology 2010, 117, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Ettcheto, M.; Chang, J.H.; Barroso, E.; Espina, M.; Kuhne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J. Control Release 2019, 301, 62–75. [Google Scholar] [CrossRef]
- Souto, E.B.; Souto, S.B.; Campos, J.R.; Severino, P.; Pashirova, T.N.; Zakharova, L.Y.; Silva, A.M.; Durazzo, A.; Lucarini, M.; Izzo, A.A.; et al. Nanoparticle Delivery Systems in the Treatment of Diabetes Complications. Molecules 2019, 24, 4209. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Masood, R.; Cai, J.; Zheng, T.; Smith, D.L.; Hinton, D.R.; Gill, P.S. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor–positive human tumors. Blood 2001, 98, 1904–1913. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist 2004, 9, 2–10. [Google Scholar] [CrossRef]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, M.; Hendgen-Cotta, U.B.; Kelm, M.; Rassaf, T. Crosstalk between nitrite, myoglobin and reactive oxygen species to regulate vasodilation under hypoxia. PLoS ONE 2014, 9, e105951. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-Inducible Factor-1 Mediates Activation of Cultured Vascular Endothelial Cells by Inducing Multiple Angiogenic Factors. Circ. Res. 2003, 93, 664–673. [Google Scholar] [CrossRef]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix Metalloproteinases: Inflammatory Regulators of Cell Behaviors in Vascular Formation and Remodeling. Mediat. Inflamm. 2013, 2013, 14. [Google Scholar] [CrossRef]
- Motiejunaite, R.; Kazlauskas, A. Pericytes and ocular diseases. Exp. Eye Res. 2008, 86, 171–177. [Google Scholar] [CrossRef]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149. [Google Scholar] [CrossRef]
- Criscuolo, G.R.; Merrill, M.J.; Oldfield, E.H. Further characterization of malignant glioma-derived vascular permeability factor. J. Neurosurg. 1988, 69, 254–262. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669. [Google Scholar] [CrossRef]
- el Filali, M.; Van der Velden, P.A.; Luyten, G.P.; Jager, M.J. Anti-angiogenic therapy in uveal melanoma. In Current Concepts in Uveal Melanoma; Karger Publishers: Basel, Switzerland, 2012; Volume 49, pp. 117–136. [Google Scholar]
- Vieira, R.; Souto, S.B.; Sanchez-Lopez, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Silva, A.M.; Fortuna, A.; Garcia, M.L.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome-Strategies for In Vivo Administration: Part-II. J. Clin. Med. 2019, 8, 1332. [Google Scholar] [CrossRef]
- Vieira, R.; Souto, S.B.; Sanchez-Lopez, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; Garcia, M.L.; Silva, A.M.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome-Review of Classical and New Compounds: Part-I. Pharmaceuticals (Basel) 2019, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Souto, S.B.; Souto, E.B.; Braga, D.C.; Medina, J.L. Prevention and current onset delay approaches of type 2 diabetes mellitus (T2DM). Eur. J. Clin. Pharmacol. 2011, 67, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Reports on Diabetes; World Health Organization: Geneva, Switzerland, 2018; Available online: http://www.who.int.org (accessed on 17 January 2020).
- Daliu, P.; Santini, A.; Novellino, E. A decade of nutraceutical patents: Where are we now in 2018? Expert Opin. Ther. Pat. 2018, 28, 875–882. [Google Scholar] [CrossRef]
- Santini, A.; Novellino, E. To Nutraceuticals and Back: Rethinking a Concept. Foods 2017, 6, 74. [Google Scholar] [CrossRef]
- Santini, A.; Novellino, E. Nutraceuticals—Shedding light on the grey area between pharmaceuticals and food. Expert Rev. Clin. Pharmacol. 2018, 11, 545–547. [Google Scholar] [CrossRef]
- Daliu, P.; Santini, A.; Novellino, E. From pharmaceuticals to nutraceuticals: Bridging disease prevention and management. Expert Rev. Clin. Pharmacol. 2019, 12, 1–7. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A concise overview on the chemistry, occurrence, and human health. Phytother. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef]
- Durazzo, A.; D’Addezio, L.; Camilli, E.; Piccinelli, R.; Turrini, A.; Marletta, L.; Marconi, S.; Lucarini, M.; Lisciani, S.; Gabrielli, P.; et al. From Plant Compounds to Botanicals and Back: A Current Snapshot. Molecules 2018, 23, 1844. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M.; Novellino, E.; Souto, E.B.; Daliu, P.; Santini, A. Abelmoschus esculentus (L.): Bioactive Components’ Beneficial Properties-Focused on Antidiabetic Role-For Sustainable Health Applications. Molecules 2018, 24, 38. [Google Scholar] [CrossRef]
- Daliu, P.; Annunziata, G.; Tenore, G.C.; Santini, A. Abscisic acid identification in Okra, Abelmoschus esculentus L. (Moench): Perspective nutraceutical use for the treatment of diabetes. Nat. Prod. Res. 2020, 34, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Ata, A.; Anil Kumar, V.N.; Sharopov, F.; Ramírez-Alarcón, K.; Ruiz-Ortega, A.; Abdulmajid Ayatollahi, S.; Tsouh Fokou, P.V.; Kobarfard, F.; Amiruddin Zakaria, Z.; et al. Antidiabetic potential of medicinal plants and their active components. Biomolecules 2019, 9, 551. (in press) [CrossRef] [PubMed]
- Frank, R.N. Diabetic retinopathy and systemic factors. Middle E. Afr. J. Ophthalmol. 2015, 22, 151. [Google Scholar] [CrossRef] [PubMed]
- Shah, C. Diabetic retinopathy: A comprehensive review. Indian J. Med Sci. 2008, 62, 500. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.; Al-Shabrawey, M.; Caldwell, R.W.; Caldwell, R.B. Inflammation and Diabetic Retinal Microvascular Complications; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- BURDITT, A.F.; Caird, F.; Draper, G. The natural history of diabetic retinopathy. QJM Int. J. Med. 1968, 37, 303–317. [Google Scholar]
- Lu, L.; Jiang, Y.; Jaganathan, R.; Hao, Y. Current Advances in Pharmacotherapy and Technology for Diabetic Retinopathy: A Systematic Review. J. Ophthalmol. 2018, 2018, 1694187. [Google Scholar]
- Lorenzi, M.; Gerhardinger, C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia 2001, 44, 791–804. [Google Scholar] [CrossRef]
- Lieth, E.; Gardner, T.W.; Barber, A.J.; Antonetti, D.A. Retinal neurodegeneration: Early pathology in diabetes. Clin. Exp. Ophthalmol. 2000, 28, 3–8. [Google Scholar] [CrossRef]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Kern, T.S.; Barber, A.J. Retinal ganglion cells in diabetes. J. Physiol. 2008, 586, 4401–4408. [Google Scholar] [CrossRef]
- Park, S.-H.; Park, J.-W.; Park, S.-J.; Kim, K.-Y.; Chung, J.-W.; Chun, M.-H.; Oh, S.-J. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia 2003, 46, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Énzsöly, A.; Szabó, A.; Kántor, O.; Dávid, C.; Szalay, P.; Szabó, K.; Szél, Á.; Németh, J.; Lukáts, Á. Pathologic alterations of the outer retina in streptozotocin-induced diabetes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3686–3699. [Google Scholar] [CrossRef] [PubMed]
- Ostroy, S.E.; Frede, S.M.; Wagner, E.F.; Gaitatzes, C.G.; Janle, E.M. Decreased rhodopsin regeneration in diabetic mouse eyes. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3905–3909. [Google Scholar]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Müller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef]
- El-Asrar, A.M.A.; Dralands, L.; Missotten, L.; Al-Jadaan, I.A.; Geboes, K. Expression of apoptosis markers in the retinas of human subjects with diabetes. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2760–2766. [Google Scholar] [CrossRef]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345. [Google Scholar] [CrossRef]
- Cunha-Vaz, J. Mechanisms of Retinal Fluid Accumulation and Blood-Retinal Barrier Breakdown. In Macular Edema; Karger Publishers: Basel, Switzerland, 2017; Volume 58, pp. 11–20. [Google Scholar]
- Eshaq, R.S.; Aldalati, A.M.; Alexander, J.S.; Harris, N.R. Diabetic retinopathy: Breaking the barrier. Pathophysiology 2017, 24, 229–241. [Google Scholar] [CrossRef]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. J. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Schmidt, D.; Kirste, G.; Schrader, W. Progressive proliferative diabetic retinopathy after transplantation of the pancreas. Acta Ophthalmol. 1994, 72, 743–751. [Google Scholar] [CrossRef]
- Brownlee, M.D.M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, e93751. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.-P. Diabetic retinopathy: Hyperglycaemia, oxidative stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Shamsul Ola, M.; S Alhomida, A.; M Ferrario, C.; Ahmad, S. Role of Tissue Renin-angiotensin System and the Chymase/angiotensin-(1-12) Axis in the Pathogenesis of Diabetic Retinopathy. Curr. Med. Chem. 2017, 24, 3104–3114. [Google Scholar]
- Khullar, M.; Cheema, B.S.; Raut, S.K. emerging evidence of epigenetic Modifications in vascular Complication of Diabetes. Front. Endocrinol. 2017, 8, 237. [Google Scholar] [CrossRef]
- Kowluru, R.A. Diabetic retinopathy, metabolic memory and epigenetic modifications. Vis. Res. 2017, 139, 30–38. [Google Scholar] [CrossRef]
- Paccola, L.; Costa, R.A.; Folgosa, M.S.; Barbosa, J.C.; Scott, I.U.; Jorge, R. Intravitreal triamcinolone versus bevacizumab for treatment of refractory diabetic macular oedema (IBEME study). Br. J. Ophthalmol. 2008, 92, 76–80. [Google Scholar] [CrossRef]
- Shimura, M.; Nakazawa, T.; Yasuda, K.; Shiono, T.; Iida, T.; Sakamoto, T.; Nishida, K. Comparative therapy evaluation of intravitreal bevacizumab and triamcinolone acetonide on persistent diffuse diabetic macular edema. Am. J. Ophthalmol. 2008, 145, 854–861. [Google Scholar] [CrossRef]
- Miyamoto, K.; Khosrof, S.; Bursell, S.-E.; Moromizato, Y.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am. J. Pathol. 2000, 156, 1733–1739. [Google Scholar] [CrossRef]
- Das, A.; McGuire, P.G.; Monickaraj, F. Novel pharmacotherapies in diabetic retinopathy: Current status and what’s in the horizon? Indian J. Ophthalmol. 2016, 64, 4. [Google Scholar] [CrossRef]
- Amadio, M.; Bucolo, C.; Leggio, G.; Drago, F.; Govoni, S.; Pascale, A. The PKCβ/HuR/VEGF pathway in diabetic retinopathy. Biochem. Pharmacol. 2010, 80, 1230–1237. [Google Scholar] [CrossRef]
- Lupo, G.; Motta, C.; Giurdanella, G.; Anfuso, C.D.; Alberghina, M.; Drago, F.; Salomone, S.; Bucolo, C. Role of phospholipases A2 in diabetic retinopathy: In vitro and in vivo studies. Biochem. Pharmacol. 2013, 86, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Pascale, A.; Cupri, S.; Pignatello, R.; Osera, C.; Leggio, G.M.; Ruozi, B.; Govoni, S.; Drago, F.; Bucolo, C. Nanosystems based on siRNA silencing HuR expression counteract diabetic retinopathy in rat. Pharmacol. Res. 2016, 111, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Sone, H.; Okuda, Y.; Kawakami, Y.; Hanatani, M.; Suzuki, H.; Kozawa, T.; Honmura, S.; Yamashita, K. Vascular endothelial growth factor level in aqueous humor of diabetic patients with rubeotic glaucoma is markedly elevated. Diabetes Care 1996, 19, 1306–1307. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Bamonte, G.; Cifariello, F.; Romano, M.R.; Costagliola, C. Vitreous mediators in retinal hypoxic diseases. Mediat. Inflamm. 2013, 2013, 935301. [Google Scholar]
- Caldwell, R.B.; Bartoli, M.; Behzadian, M.A.; El-Remessy, A.E.; Al-Shabrawey, M.; Platt, D.H.; Caldwell, R.W. Vascular endothelial growth factor and diabetic retinopathy: Pathophysiological mechanisms and treatment perspectives. Diabetes/Metab. Res. Rev. 2003, 19, 442–455. [Google Scholar] [CrossRef]
- Ponnalagu, M.; Subramani, M.; Jayadev, C.; Shetty, R.; Das, D. Retinal pigment epithelium-secretome: A diabetic retinopathy perspective. Cytokine 2017, 95, 126–135. [Google Scholar] [CrossRef]
- Funatsu, H.; Noma, H.; Mimura, T.; Eguchi, S.; Hori, S. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology 2009, 116, 73–79. [Google Scholar] [CrossRef]
- Hong, Y.R.; Kim, Y.H.; Kim, S.Y.; Nam, G.Y.; Cheon, H.J.; Lee, S.J. Plasma concentrations of vascular endothelial growth factor in retinopathy of prematurity after intravitreal bevacizumab injection. Retina 2015, 35, 1772–1777. [Google Scholar] [CrossRef]
- Rangasamy, S.; McGuire, P.G.; Nitta, C.F.; Monickaraj, F.; Oruganti, S.R.; Das, A. Chemokine mediated monocyte trafficking into the retina: Role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS ONE 2014, 9, e108508. [Google Scholar] [CrossRef]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFα is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P.; Grigoropoulos, V.; Emfietzoglou, I.; Theodossiadis, G.; Tentolouris, N.; Delicha, E.; Katsiari, C.; Alexiadou, K.; Hatziagelaki, E.; Theodossiadis, P.G. Infliximab for diabetic macular edema refractory to laser photocoagulation: A randomized, double-blind, placebo-controlled, crossover, 32-week study. Diabetes Care 2010, 33, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.-Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Mocan, M.C.; Kadayifcilar, S.; Eldem, B. Elevated intravitreal interleukin-6 levels in patients with proliferative diabetic retinopathy. Can. J. Ophthalmol. 2006, 41, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Yamashita, H.; Noma, H.; Mimura, T.; Nakamura, S.; Sakata, K.; Hori, S. Aqueous humor levels of cytokines are related to vitreous levels and progression of diabetic retinopathy in diabetic patients. Graefe’s Arch. Clin. Exp. Ophthalmol. 2005, 243, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Cancarini, A.; Rezzola, S.; Romano, M.; Costagliola, C. Diabetic retinopathy: Vascular and inflammatory disease. J. Diabetes Res. 2015, 2015, 582060. [Google Scholar] [CrossRef]
- Elner, S.G.; Elner, V.M.; Jaffe, G.J.; Stuart, A.; Kunkel, S.L.; Strieter, R.M. Cytokines in proliferative diabetic retinopathy and proliferative vitreoretinopathy. Curr. Eye Res. 1995, 14, 1045–1053. [Google Scholar] [CrossRef]
- Rosenbaum, J.T.; Samples, J.R.; Hefeneider, S.H.; Howes, E.L. Ocular inflammatory effects of intravitreal interleukin 1. Arch. Ophthalmol. 1987, 105, 1117–1120. [Google Scholar] [CrossRef]
- Vincent, J.A.; Mohr, S. Inhibition of caspase-1/interleukin-1β signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 2007, 56, 224–230. [Google Scholar] [CrossRef]
- Xu, W.-Q.; Wang, Y.-S. The role of Toll-like receptors in retinal ischemic diseases. Int. J. Ophthalmol. 2016, 9, 1343. [Google Scholar]
- Xu, H.; Chen, M. Diabetic retinopathy and dysregulated innate immunity. Vis. Res. 2017, 139, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Simó-Servat, O.; Hernández, C.; Simó, R. Usefulness of the vitreous fluid analysis in the translational research of diabetic retinopathy. Mediat. Inflamm. 2012, 2012, 872978. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Ki-i, Y.; Hashiguchi, T.; Kawahara, K.-i.; Biswas, K.K.; Nakamura, M.; Sonoda, Y.; Yamakiri, K.; Okubo, A.; Sakamoto, T. Intraocular expression and release of high-mobility group box 1 protein in retinal detachment. Lab. Investig. 2009, 89, 278. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Keino, H.; Sato, Y.; Kudo, A.; Kawakami, H.; Okada, A.A. High mobility group box protein-1 in experimental autoimmune uveoretinitis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2283–2290. [Google Scholar] [CrossRef] [PubMed]
- Jakuš, V.; Rietbrock, N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol. Res. 2004, 53, 131–142. [Google Scholar] [PubMed]
- Santos, A.R.C.; Dvoriantchikova, G.; Li, Y.; Mohammad, G.; El-Asrar, A.M.A.; Wen, R.; Ivanov, D. Cellular mechanisms of high mobility group 1 (HMGB-1) protein action in the diabetic retinopathy. PLoS ONE 2014, 9, e87574. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Zhang, X.-D.; Li, Y.-Y.; Chen, X.-M.; Tang, D.-R.; Ran, R.-J. Involvement of HMGB1 mediated signalling pathway in diabetic retinopathy: Evidence from type 2 diabetic rats and ARPE-19 cells under diabetic condition. Br. J. Ophthalmol. 2013, 97, 1598–1603. [Google Scholar] [CrossRef]
- Rajamani, U.; Jialal, I. Hyperglycemia induces Toll-like receptor-2 and-4 expression and activity in human microvascular retinal endothelial cells: Implications for diabetic retinopathy. J. Diabetes Res. 2014, 2014, 790902. [Google Scholar] [CrossRef]
- Liao, Y.-R.; Li, Z.-J.; Zeng, P.; Lan, Y.-Q. TLR7 deficiency contributes to attenuated diabetic retinopathy via inhibition of inflammatory response. Biochem. Biophys. Res. Commun. 2017, 493, 1136–1142. [Google Scholar] [CrossRef]
- Platania, C.B.M.; Giurdanella, G.; Di Paola, L.; Leggio, G.M.; Drago, F.; Salomone, S.; Bucolo, C. P2X7 receptor antagonism: Implications in diabetic retinopathy. Biochem. Pharmacol. 2017, 138, 130–139. [Google Scholar] [CrossRef]
- Miller, J.W.; Adamis, A.P.; Shima, D.T.; D’Amore, P.A.; Moulton, R.S.; O’Reilly, M.S.; Folkman, J.; Dvorak, H.F.; Brown, L.F.; Berse, B. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am. J. Pathol. 1994, 145, 574. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.A.; Kermany, D.S.; Waters, J.; Jansen, M.E.; Tyler, L. Diabetic macular edema: It is more than just VEGF. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.W.; Abcouwer, S.F.; Barber, A.J.; Jackson, G.R. An integrated approach to diabetic retinopathy research. Arch. Ophthalmol. 2011, 129, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Murata, T.; Tsujikawa, A.; Kirchhof, B.; Bursell, S.-E.; Adamis, A.P. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am. J. Pathol. 2001, 158, 147–152. [Google Scholar] [CrossRef]
- Powell, E.U.; Field, R. Diabetic retinopathy and rheumatoid arthritis. Lancet 1964, 284, 17–18. [Google Scholar] [CrossRef]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef]
- Mitchell, P.; Wong, T.Y. Management paradigms for diabetic macular edema. Am. J. Ophthalmol. 2014, 157, 505–513. [Google Scholar] [CrossRef]
- Stewart, M.W.; Flynn Jr, H.W.; Schwartz, S.G.; Scott, I.U. Extended duration strategies for the pharmacologic treatment of diabetic retinopathy: Current status and future prospects. Expert Opin. Drug Deliv. 2016, 13, 1277–1287. [Google Scholar] [CrossRef]
- Kumar, B.; Gupta, S.; Saxena, R.; Srivastava, S. Current trends in the pharmacotherapy of diabetic retinopathy. J. Postgrad. Med. 2012, 58, 132. [Google Scholar]
- Kompella, U.B.; Kadam, R.S.; Lee, V.H. Recent advances in ophthalmic drug delivery. Ther. Deliv. 2010, 1, 435–456. [Google Scholar] [CrossRef] [PubMed]
- G Schwartz, S.; W Flynn, H. Fluocinolone acetonide implantable device for diabetic retinopathy. Curr. Pharm. Biotechnol. 2011, 12, 347–351. [Google Scholar] [CrossRef]
- Śniegocka, M.; Podgórska, E.; Płonka, P.M.; Elas, M.; Romanowska-Dixon, B.; Szczygieł, M.; Żmijewski, M.A.; Cichorek, M.; Markiewicz, A.; Brożyna, A.A. Transplantable Melanomas in Hamsters and Gerbils as Models for Human Melanoma. Sensitization in Melanoma Radiotherapy—From Animal Models to Clinical Trials. Int. J. Mol. Sci. 2018, 19, 1048. [Google Scholar] [CrossRef] [PubMed]
- Reichstein, D.; Karan, K. Plaque brachytherapy for posterior uveal melanoma in 2018: Improved techniques and expanded indications. Curr. Opin. Ophthalmol. 2018, 29, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Urteaga, B.O.; Pack, G.T. On the antiquity of melanoma. Cancer 1966, 19, 607–610. [Google Scholar] [CrossRef]
- Slominski, R.M.; Zmijewski, M.A.; Slominski, A.T. The role of melanin pigment in melanoma. Exp. Dermatol. 2015, 24, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Damato, B. Ocular treatment of choroidal melanoma in relation to the outcome of metastatic death–A personal view. Prog. Retin. Eye Res. 2018, 66, 187–199. [Google Scholar] [CrossRef]
- Peinado, H.; Lavotshkin, S.; Lyden, D. The secreted factors responsible for pre-metastatic niche formation: Old sayings and new thoughts. Semin. Cancer Biol. 2011, 21, 139–146. [Google Scholar] [CrossRef]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332. [Google Scholar] [CrossRef]
- el Filali, M.; van der Velden, P.A.; Luyten, G.P.; Jager, M.J. Anti-angiogenic therapy in uveal melanoma. Dev. Ophthalmol. 2012, 49, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Proniewska-Skretek, E.; Mariak, Z. Tumor angiogensis in uveal melanoma. Role of vascular endothelial growth factor (VEGF). Klin. Ocz. 2004, 106, 682–685. [Google Scholar]
- Folberg, R.; Pe’er, J.; Gruman, L.M.; Woolson, R.F.; Jeng, G.; Montague, P.R.; Moninger, T.O.; Yi, H.; Moore, K.C. The morphologic characteristics of tumor blood vessels as a marker of tumor progression in primary human uveal melanoma: A matched case-control study. Hum. Pathol. 1992, 23, 1298–1305. [Google Scholar] [CrossRef]
- Notting, I.C.; Missotten, G.S.O.A.; Sijmons, B.; Boonman, Z.F.H.M.; Keunen, J.E.E.; van der Pluijm, G. Angiogenic Profile of Uveal Melanoma. Curr. Eye Res. 2006, 31, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Noma, H.; Mimura, T.; Yasuda, K.; Shimura, M. Role of soluble vascular endothelial growth factor receptor signaling and other factors or cytokines in central retinal vein occlusion with macular edema. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Funk, M.; Kriechbaum, K.; Prager, F.; Benesch, T.; Georgopoulos, M.; Zlabinger, G.J.; Schmidt-Erfurth, U. Intraocular concentrations of growth factors and cytokines in retinal vein occlusion and the effect of therapy with bevacizumab. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1025–1032. [Google Scholar] [CrossRef]
- Guyer, D.R.; Mukai, S.; Egan, K.M.; Seddon, J.M.; Walsh, S.M.; Gragoudas, E.S. Radiation Maculopathy after Proton Beam Irradiation for Choroidal Melanoma. Ophthalmology 1992, 99, 1278–1285. [Google Scholar] [CrossRef]
- Boyd, S.R.; Tan, D.; Bunce, C.; Gittos, A.; Neale, M.H.; Hungerford, J.L.; Charnock-Jones, S.; Cree, I.A. Vascular endothelial growth factor is elevated in ocular fluids of eyes harbouring uveal melanoma: Identification of a potential therapeutic window. Br. J. Ophthalmol. 2002, 86, 448–452. [Google Scholar] [CrossRef]
- Koch, K.R.; Refaian, N.; Hos, D.; Schlereth, S.L.; Bosch, J.J.; Cursiefen, C.; Heindl, L.M. Autocrine impact of VEGF-A on uveal melanoma cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2697–2704. [Google Scholar] [CrossRef]
- Sahin, A.; Kiratli, H.; Soylemezoglu, F.; Tezel, G.G.; Bilgic, S.; Saracbasi, O. Expression of vascular endothelial growth factor-A, matrix metalloproteinase-9, and extravascular matrix patterns and their correlations with clinicopathologic parameters in posterior uveal melanomas. Jpn. J. Ophthalmol. 2007, 51, 325–331. [Google Scholar] [CrossRef]
- Jászai, J.; Schmidt, M.H.H. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef] [PubMed]
- Witmer, A.N.; van Blijswijk, B.C.; van Noorden, C.J.; Vrensen, G.F.; Schlingemann, R.O. In vivo angiogenic phenotype of endothelial cells and pericytes induced by vascular endothelial growth factor-A. J. Histochem. Cytochem. 2004, 52, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular mediators of angiogenesis. J. Burn Care Res. 2010, 31, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Flores, L.; Gutierrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Martin-Vasallo, P.; Diaz-Flores, L., Jr. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol. Histopathol. 2009, 24, 909–969. [Google Scholar] [PubMed]
- Ozerdem, U. Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic Res. 2006, 38, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 593–598. [Google Scholar] [CrossRef]
- Murakami, M. Signaling Required for Blood Vessel Maintenance: Molecular Basis and Pathological Manifestations. Int. J. Vasc. Med. 2012, 2012, 15. [Google Scholar] [CrossRef]
- Kumar, B.; Chile, S.A.; Ray, K.B.; Reddy, G.E.; Addepalli, M.K.; Kumar, A.S.; Ramana, V.; Rajagopal, V. VEGF-C differentially regulates VEGF-A expression in ocular and cancer cells; promotes angiogenesis via RhoA mediated pathway. Angiogenesis 2011, 14, 371–380. [Google Scholar] [CrossRef]
- Cao, R.; Ji, H.; Feng, N.; Zhang, Y.; Yang, X.; Andersson, P.; Sun, Y.; Tritsaris, K.; Hansen, A.J.; Dissing, S.; et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. of Sci. USA 2012, 109, 15894–15899. [Google Scholar] [CrossRef]
- Hirakawa, S.; Brown, L.F.; Kodama, S.; Paavonen, K.; Alitalo, K.; Detmar, M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood 2007, 109, 1010–1017. [Google Scholar] [CrossRef]
- Jehs, T.; Faber, C.; Juel, H.B.; Bronkhorst, I.H.; Jager, M.J.; Nissen, M.H. Inflammation-induced chemokine expression in uveal melanoma cell lines stimulates monocyte chemotaxis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5169–5175. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ma, A.; Cai, J.; Boulton, M. VEGF-A regulates the expression of VEGF-C in human retinal pigment epithelial cells. Br. J. Ophthalmol. 2006, 90, 1052–1059. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, B.; Smith, G.; Cai, J.; Ma, A.; Boulton, M. Vascular endothelial growth factor C promotes survival of retinal vascular endothelial cells via vascular endothelial growth factor receptor-2. Br. J. Ophthalmol. 2007, 91, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci. Signal. 2009, 2, re1. [Google Scholar] [CrossRef]
- Barak, V.; Pe’er, J.; Kalickman, I.; Frenkel, S. VEGF as a biomarker for metastatic uveal melanoma in humans. Curr. Eye Res. 2011, 36, 386–390. [Google Scholar] [CrossRef]
- el Filali, M.; Missotten, G.S.; Maat, W.; Ly, L.V.; Luyten, G.P.; van der Velden, P.A.; Jager, M.J. Regulation of VEGF-A in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2329–2337. [Google Scholar] [CrossRef]
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. Am. J. Physiol.-Cell Physiol. 2002, 282, C947–C970. [Google Scholar] [CrossRef]
- Yang, H.; Jager, M.J.; Grossniklaus, H.E. Bevacizumab suppression of establishment of micrometastases in experimental ocular melanoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2835–2842. [Google Scholar] [CrossRef]
- Melo, F.H.M.d.; Molognoni, F.; Jasiulionis, M.G. The Role of Oxidative Stress in Melanoma Development, Progression and Treatment. In Recent Advances in the Biology, Therapy and Management of Melanoma; InTechOpen: London, UK, 2013; p. 42738. [Google Scholar]
- Murphy, M.P.; Holmgren, A.; Larsson, N.-G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the Biological Roles of Reactive Oxygen Species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef]
- Govindarajan, B.; Sligh, J.E.; Vincent, B.J.; Li, M.; Canter, J.A.; Nickoloff, B.J.; Rodenburg, R.J.; Smeitink, J.A.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 2007, 117, 719–729. [Google Scholar] [CrossRef]
- Lin, J.; Yu, Y.; Shigdar, S.; Fang, D.Z.; Du, J.R.; Wei, M.Q.; Danks, A.; Liu, K.; Duan, W. Enhanced antitumor efficacy and reduced systemic toxicity of sulfatide-containing nanoliposomal Doxorubicin in a xenograft model of colorectal cancer. PLoS ONE 2012, 7, e49277. [Google Scholar] [CrossRef] [PubMed]
- Wittgen, H.G.; van Kempen, L.C. Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Res. 2007, 17, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Galeotti, T.; Chiarugi, P. Metastasis: Cancer cell’s escape from oxidative stress. Cancer Metastasis Rev. 2010, 29, 351–378. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; McNulty, S.E.; Buckmeier, J.A.; Tohidian, N.B.; Spillane, T.J.; Kahlon, R.S.; Gonzalez, R.I. Aberrant redox regulation in human metastatic melanoma cells compared to normal melanocytes. Free Radic. Biol. Med. 2001, 31, 799–808. [Google Scholar] [CrossRef]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Chau, H.V.; Tohidian, N.; Buckmeier, J. Luminol-enhanced chemiluminescent response of human melanocytes and melanoma cells to hydrogen peroxide stress. Pigment Cell Res. 1997, 10, 184–189. [Google Scholar] [CrossRef]
- Blasi, M.A.; Maresca, V.; Roccella, M.; Roccella, F.; Sansolini, T.; Grammatico, P.; Balestrazzi, E.; Picardo, M. Antioxidant Pattern in Uveal Melanocytes and Melanoma Cell Cultures. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3012–3016. [Google Scholar]
- Picardo, M.; Maresca, V.; Eibenschutz, L.; De Bernardo, C.; Rinaldi, R.; Grammatico, P. Correlation between antioxidants and phototypes in melanocytes cultures. A possible link of physiologic and pathologic relevance. J. Investig. Dermatol. 1999, 113, 424–425. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, S.; Clement, M.V. Superoxide anion: Oncogenic reactive oxygen species? Int. J. Biochem. Cell Biol. 2007, 39, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Pokrzywniak, A.; Jockel, K.H.; Bornfeld, N.; Sauerwein, W.; Stang, A. Positive interaction between light iris color and ultraviolet radiation in relation to the risk of uveal melanoma: A case-control study. Ophthalmology 2009, 116, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Moan, J.E.; Micu, E.; Porojnicu, A.C.; Juzeniene, A. Similarities in solar ultraviolet irradiance and other environmental factors may explain much of the family link between uveal melanoma and other cancers. Fam. Cancer 2010, 9, 659–660. [Google Scholar] [CrossRef]
- Coupland, S.E.; Campbell, I.; Damato, B. Routes of extraocular extension of uveal melanoma: Risk factors and influence on survival probability. Ophthalmology 2008, 115, 1778–1785. [Google Scholar] [CrossRef]
- Ly, L.V.; Odish, O.F.; de Wolff-Rouendaal, D.; Missotten, G.S.; Luyten, G.P.; Jager, M.J. Intravascular presence of tumor cells as prognostic parameter in uveal melanoma: A 35-year survey. Investig. Ophthalmol. Vis. Sci. 2010, 51, 658–665. [Google Scholar] [CrossRef]
- Augsburger, J.J.; Corrêa, Z.M.; Freire, J.; Brady, L.W. Long-term survival in choroidal and ciliary body melanoma after enucleation versus plaque radiation therapy. Ophthalmology 1998, 105, 1670–1678. [Google Scholar] [CrossRef]
- Kujala, E.; Makitie, T.; Kivela, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef]
- Economou, M.A.; All-Ericsson, C.; Bykov, V.; Girnita, L.; Bartolazzi, A.; Larsson, O.; Seregard, S. Receptors for the liver synthesized growth factors IGF-1 and HGF/SF in uveal melanoma: Intercorrelation and prognostic implications. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4372–4375. [Google Scholar] [CrossRef]
- Baserga, R. The insulin-like growth factor I receptor: A key to tumor growth? Cancer Res. 1995, 55, 249–252. [Google Scholar]
- Slomiany, M.G.; Rosenzweig, S.A. IGF-1–induced VEGF and IGFBP-3 secretion correlates with increased HIF-1α expression and activity in retinal pigment epithelial cell line D407. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Benjamin, L.E. Angiogenesis: Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401. [Google Scholar] [CrossRef] [PubMed]
- Witmer, A.; Vrensen, G.; Van Noorden, C.; Schlingemann, R. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 2003, 22, 1–29. [Google Scholar] [CrossRef]
- Boyd, S.; Tan, D.; De Souza, L.; Neale, M.; Myatt, N.; Alexander, R.; Robb, M.; Hungerford, J.; Cree, I. Uveal melanomas express vascular endothelial growth factor and basic fibroblast growth factor and support endothelial cell growth. Br. J. Ophthalmol. 2002, 86, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Ijland, S.; Jager, M.; Heijdra, B.; Westphal, J.; Peek, R. Expression of angiogenic and immunosuppressive factors by uveal melanoma cell lines. Melanoma Res. 1999, 9, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Sheidow, T.G.; Hooper, P.L.; Crukley, C.; Young, J.; Heathcote, J.G. Expression of vascular endothelial growth factor in uveal melanoma and its correlation with metastasis. Br. J. Ophthalmol. 2000, 84, 750–756. [Google Scholar] [CrossRef][Green Version]
- Ang, W.J.; Zunaina, E.; Norfadzillah, A.J.; Raja-Norliza, R.O.; Julieana, M.; Ab-Hamid, S.A.; Mahaneem, M. Evaluation of vascular endothelial growth factor levels in tears and serum among diabetic patients. PLoS ONE 2019, 14, e0221481. [Google Scholar] [CrossRef]
- Missotten, G.S.; Notting, I.C.; Schlingemann, R.O.; Zijlmans, H.J.; Lau, C.; Eilers, P.H.; Keunen, J.E.; Jager, M.J. Vascular endothelial growth factor a in eyes with uveal melanoma. Arch. Ophthalmol. 2006, 124, 1428–1434. [Google Scholar] [CrossRef]
- Pötgens, A.; Lubsen, N.H.; van Altena, M.C.; Schoenmakers, J.; Ruiter, D.J.; De Waal, R. Vascular permeability factor expression influences tumor angiogenesis in human melanoma lines xenografted to nude mice. Am. J. Pathol. 1995, 146, 197. [Google Scholar]
- Claffey, K.P.; Brown, L.F.; del Aguila, L.F.; Tognazzi, K.; Yeo, K.-T.; Manseau, E.J.; Dvorak, H.F. Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis, and experimental metastasis. Cancer Res. 1996, 56, 172–181. [Google Scholar]
- Bande Rodríguez, M.F.; Fernandez Marta, B.; Lago Baameiro, N.; Santiago-Varela, M.; Silva-Rodríguez, P.; Blanco-Teijeiro, M.J.; Pardo Perez, M.; Piñeiro Ces, A. Blood Biomarkers of Uveal Melanoma: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.) 2020, 14, 157–169. [Google Scholar] [CrossRef]
- Crosby, M.B.; Yang, H.; Gao, W.; Zhang, L.; Grossniklaus, H.E. Serum vascular endothelial growth factor (VEGF) levels correlate with number and location of micrometastases in a murine model of uveal melanoma. Br. J. Ophthalmol. 2010. [Google Scholar] [CrossRef]
- Sudaka, A.; Susini, A.; Nigro, C.L.; Fischel, J.-L.; Toussan, N.; Formento, P.; Tonissi, F.; Lattanzio, L.; Russi, E.; Etienne-Grimaldi, M.-C. Combination of bevacizumab and irradiation on uveal melanoma: An in vitro and in vivo preclinical study. Investig. New Drugs 2013, 31, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Hovinga, K.; Stalpers, L.; Van Bree, C.; Donker, M.; Verhoeff, J.; Rodermond, H.; Bosch, D.; van Furth, W. Radiation-enhanced vascular endothelial growth factor (VEGF) secretion in glioblastoma multiforme cell lines–a clue to radioresistance? J. Neuro-Oncol. 2005, 74, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.E.; Lee, J.; Kuang, W.-J.; Rice, G.C.; Wood, W.I. Structure and functional expression of a human interleukin-8 receptor. Science 1991, 253, 1278–1280. [Google Scholar] [CrossRef]
- Singh, S.; Nannuru, K.; Sadanandam, A.; Varney, M.; Singh, R. CXCR1 and CXCR2 enhances human melanoma tumourigenesis, growth and invasion. Br. J. Cancer 2009, 100, 1638. [Google Scholar] [CrossRef]
- Martin, D.; Galisteo, R.; Gutkind, J.S. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFκB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem. 2009, 284, 6038–6042. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.-N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef]
- Nagarkatti-Gude, N.; Bronkhorst, I.H.; van Duinen, S.G.; Luyten, G.P.; Jager, M.J. Cytokines and chemokines in the vitreous fluid of eyes with uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6748–6755. [Google Scholar] [CrossRef]
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye (London, England) 2017, 31, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Ono, M. Molecular links between tumor angiogenesis and inflammation: Inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008, 99, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Dhawahir, A.; Sum, E.Y.; Urosevic, J.; Lechuga, C.G.; Esteban, L.M.; Castellano, E.; Guerra, C.; Santos, E.; Barbacid, M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010, 29, 1091–1104. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef]
- McArthur, G.A. The coming of age of MEK. Lancet Oncol. 2012, 13, 744–745. [Google Scholar] [CrossRef]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T. Survival in BRAF V600–mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A. MEK1/2 inhibition delays progression of uveal melanoma. Lancet Oncol. 2014, 15, e366. [Google Scholar] [CrossRef]
- Shields, C.L.; Cater, J.; Shields, J.A.; Chao, A.; Krema, H.; Materin, M.; Brady, L.W. Combined plaque radiotherapy and transpupillary thermotherapy for choroidal melanoma: Tumor control and treatment complications in 270 consecutive patients. Arch. Ophthalmol. 2002, 120, 933–940. [Google Scholar] [CrossRef]
- Jampol, L.M.; Moy, C.S.; Murray, T.G.; Reynolds, S.M.; Albert, D.M.; Schachat, A.P.; Diddie, K.R.; Engstrom Jr, R.E.; Finger, P.T.; Hovland, K.R. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: IV. Local treatment failure and enucleation in the first 5 years after brachytherapy. COMS report no. 19. Ophthalmology 2002, 109, 2197–2206. [Google Scholar] [CrossRef]
- Melia, B.; Abramson, D.; Albert, D.; Boldt, H.; Earle, J.; Hanson, W.; Montague, P.; Moy, C.; Schachat, A.; Simpson, E. Collaborative ocular melanoma study (COMS) randomized trial of I-125 brachytherapy for medium choroidal melanoma. I. Visual acuity after 3 years COMS report no. 16. Ophthalmology 2001, 108, 348–366. [Google Scholar]
- Gragoudas, E.S.; Lane, A.M.; Munzenrider, J.; Egan, K.M.; Li, W. Long-term risk of local failure after proton therapy for choroidal/ciliary body melanoma. Trans. Am. Ophthalmol. Soc. 2002, 100, 43. [Google Scholar]
- Mangiameli, D.P.; Blansfield, J.A.; Kachala, S.; Lorang, D.; Schafer, P.H.; Muller, G.W.; Stirling, D.I.; Libutti, S.K. Combination therapy targeting the tumor microenvironment is effective in a model of human ocular melanoma. J. Transl. Med. 2007, 5, 38. [Google Scholar] [CrossRef]
- Eisen, T.; Ahmad, T.; Flaherty, K.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer 2006, 95, 581. [Google Scholar] [CrossRef]
- Chow, L.Q.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef]
- Routy, J.-P.; Routy, B.; Graziani, G.M.; Mehraj, V. The kynurenine pathway is a double-edged sword in immune-privileged sites and in cancer: Implications for immunotherapy. Int. J. Tryptophan Res. 2016, 9, IJTR–S38355. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.N.; McCormick, S.A.; Roberts, J.E. Effects of melatonin, its precursors and derivatives on the growth of cultured human uveal melanoma cells. Melanoma Res. 1998, 8, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.W.; Mellon, J.K.; Mayhew, E.; Wang, S.; He, Y.G.; Hogan, N.; Niederkorn, J.Y. Uveal melanoma expression of indoleamine 2, 3-deoxygenase: Establishment of an immune privileged environment by tryptophan depletion. Exp. Eye Res. 2007, 85, 617–625. [Google Scholar] [CrossRef]
- Terai, M.; Londin, E.; Rochani, A.; Link, E.; Lam, B.; Kaushal, G.; Bhushan, A.; Orloff, M.; Sato, T. Expression of Tryptophan 2, 3-Dioxygenase in Metastatic Uveal Melanoma. Cancers 2020, 12, 405. [Google Scholar] [CrossRef]
- Liang, C.; Peng, L.; Zeng, S.; Zhao, Q.; Tang, L.; Jiang, X.; Zhang, J.; Yan, N.; Chen, Y. Investigation of indoleamine 2, 3-dioxygenase 1 expression in uveal melanoma. Exp. Eye Res. 2019, 181, 112–119. [Google Scholar] [CrossRef]
- European Medicines Agency: Avastin®. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/avastin (accessed on 17 January 2020).
- US Food & Drugs (FDA): Avastin®. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=125085 (accessed on 17 January 2020).
- Shah, S.U.; Shields, C.L.; Bianciotto, C.G.; Iturralde, J.; Al-Dahmash, S.A.; Say, E.A.T.; Badal, J.; Mashayekhi, A.; Shields, J.A. Intravitreal bevacizumab at 4-month intervals for prevention of macular edema after plaque radiotherapy of uveal melanoma. Ophthalmology 2014, 121, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Vazzana, M.; Andreani, T.; Fangueiro, J.; Faggio, C.; Silva, C.; Santini, A.; Garcia, M.L.; Silva, A.M.; Souto, E.B. Tramadol hydrochloride: Pharmacokinetics, pharmacodynamics, adverse side effects, co-administration of drugs and new drug delivery systems. Biomed. Pharmacother. 2015, 70, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.; Severino, P.; Nalone, L.A.; Souto, S.B.; Silva, A.M.; Lucarini, M.; Durazzo, A.; Santini, A.; Souto, E.B. Sucupira Oil-Loaded Nanostructured Lipid Carriers (NLC): Lipid Screening, Factorial Design, Release Profile and Cytotoxicity. Molecules 2020, 25, 685. [Google Scholar] [CrossRef] [PubMed]
- Danielli, R.; Ridolfi, R.; Chiarion-Sileni, V.; Queirolo, P.; Testori, A.; Plummer, R.; Boitano, M.; Calabrò, L.; De Rossi, C.; Giacomo, A.M.D.; et al. Ipilimumab in pretreated patients with metastatic uveal melanoma: Safety and clinical efficacy. Cancer Immunol. Immunother. 2012, 61, 41–48. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency: Lucentis®. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/lucentis (accessed on 17 January 2020).
- Jose, S.; Cinu, T.A.; Sebastian, R.; Shoja, M.H.; Aleykutty, N.A.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B. Transferrin-Conjugated Docetaxel-PLGA Nanoparticles for Tumor Targeting: Influence on MCF-7 Cell Cycle. Polymers 2019, 11, 905. [Google Scholar] [CrossRef]
- Li, J.; Cui, Y.; Wang, Q.; Guo, D.; Pan, X.; Wang, X.; Bi, H.; Chen, W.; Liu, Z.; Zhao, S. The proliferation of malignant melanoma cells could be inhibited by ranibizumab via antagonizing VEGF through VEGFR1. Mol. Vis. 2014, 20, 649. [Google Scholar] [PubMed]
- Zielinska, A.; Ferreira, N.R.; Durazzo, A.; Lucarini, M.; Cicero, N.; Mamouni, S.E.; Silva, A.M.; Nowak, I.; Santini, A.; Souto, E.B. Development and Optimization of Alpha-Pinene-Loaded Solid Lipid Nanoparticles (SLN) Using Experimental Factorial Design and Dispersion Analysis. Molecules 2019, 24, 2683. [Google Scholar] [CrossRef] [PubMed]
- Kottschade, L.A.; McWilliams, R.R.; Markovic, S.N.; Block, M.S.; Bisneto, J.V.; Pham, A.Q.; Esplin, B.L.; Dronca, R.S. The use of pembrolizumab for the treatment of metastatic uveal melanoma. Melanoma Res. 2016, 26, 300–303. [Google Scholar] [CrossRef] [PubMed]
- US Food & Drugs (FDA): Sutent®. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021938s036lbl.pdf (accessed on 17 January 2020).
- European Medicines Agency: Sutent®. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/sutent (accessed on 17 January 2020).
- Mahipal, A.; Tijani, L.; Chan, K.; Laudadio, M.; Mastrangelo, M.J.; Sato, T. A pilot study of sunitinib malate in patients with metastatic uveal melanoma. Melanoma Res. 2012, 22, 440–446. [Google Scholar] [CrossRef]
- De Ceu Teixeira, M.; Sanchez-Lopez, E.; Espina, M.; Garcia, M.L.; Durazzo, A.; Lucarini, M.; Novellino, E.; Souto, S.B.; Santini, A.; Souto, E.B. Sirtuins and SIRT6 in Carcinogenesis and in Diet. Int. J. Mol. Sci. 2019, 20, 4945. [Google Scholar] [CrossRef]
- US Food & Drugs (FDA): Nevaxar®. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/021923lbl.pdf (accessed on 17 January 2020).
- Kaempgen, E.; Schmid, M.; Erdmann, M.; Keikavoussi, P.; Strobel, D.; Schuler-Thurner, B.; Schuler, G. Predictable clinical responses to sorafenib in stage IV uveal melanoma. J. Clin. Oncol. 2012, 30, e19032. [Google Scholar] [CrossRef]
- Mouriaux, F.; Servois, V.; Piperno-Neumann, S.; Lesimple, T.; Thyss, A.; Jouary, T.; Neidhardt, E.-M.; Penel, N.; Delcambre, C.; Parienti, J.-J.; et al. O-mel-sora: A national multicenter phase II trial of sorafenib in metastatic uveal melanoma. J. Clin. Oncol. 2014, 32, e20004. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug | Studies/Observations |
|---|---|
| Bevacizumab | Yang et al. proved that in ocular melanoma bevacizumab allows suppression of primary tumour growth, reduction of liver micrometastases and decreased VEGF levels [162]. Filali et al. observed that in vitro uveal melanoma cell proliferation did not occur, but in vivo, the use of bevacizumab caused greater intraocular tumour growth, especially under hypoxic conditions. With treatment, anterior chamber and tumour bleedings were observed in the eyes, increasing microvascular permeability due to induced VEGF expression. It is thought to be a consequence of an adaptive or evasive tumour response [40]. |
| Sorafenib | Sorafenib inhibits VEGFR. Mangiameli et al. proved that after sorafenib therapy there is an inhibition of tumour growth and metastasis [223]. In patients with metastatic cutaneous melanoma, sorafenib monotherapy has not shown significant antitumour activity [224]. Filali et al. compared the treatment of patients with metastatic melanoma (not including uvea) with carboplatin, paclitaxel and sorafenib or placebo, where the results of the phase 3 study showed no relevance to overall survival [40]. |
| Sunitinib | Sunitinib also inhibits VEGFR [225]. A preclinical phase 2 study demonstrated the benefit of sunitinib monotherapy in patients with advanced metastatic melanoma [40]. |
| Product | Type | Target | Status | Ref. |
|---|---|---|---|---|
| Bevacizumab sold as Avastin® | Monoclonal antibody Angiogenesis inhibitor | VEGF | Authorized for the treatment of various tumour types in EU and USA | [231,232,233] |
| Ipilimumab sold as Yervoy® | Monoclonal antibody Immune system modulator | CTLA-4 | Authorized for melanoma and carcinoma treatment in EU and USA | [234,235,236] |
| Ranibizumab sold as Lucentis® | Monoclonal antibody fragment Angiogenesis inhibitor | VEGF-A | Authorized for macular edema and diabetic retinopathy treatment in EU and USA | [237,238,239] |
| Pembrolizumab sold as Keytruda® | Monoclonal antibody Immune system modulator | PD-1 | Authorized for the treatment of melanoma and other tumour types in EU and USA | [41,240,241] |
| Sunitinib sold as Keytruda® | Multiple receptor tyrosine kinases inhibitor Angiogenesis inhibitor | c-KIT VEGFR PDGFR M-CSFR | Authorized for the treatment of various tumour types in EU and USA | [242,243,244] |
| Sorafenib sold as Nexavar® | Synthetic drug Multikinase inhibitor Angiogenesis inhibitor | Protein kinase | Authorized for the treatment of various carcinoma types in EU and USA | [245,246,247,248] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souto, E.B.; Campos, J.R.; Da Ana, R.; Fangueiro, J.F.; Martins-Gomes, C.; Durazzo, A.; Lucarini, M.; Sánchez López, E.; Espina, M.; García, M.L.; et al. Diabetic Retinopathy and Ocular Melanoma: How Far We Are? Appl. Sci. 2020, 10, 2777. https://doi.org/10.3390/app10082777
Souto EB, Campos JR, Da Ana R, Fangueiro JF, Martins-Gomes C, Durazzo A, Lucarini M, Sánchez López E, Espina M, García ML, et al. Diabetic Retinopathy and Ocular Melanoma: How Far We Are? Applied Sciences. 2020; 10(8):2777. https://doi.org/10.3390/app10082777
Chicago/Turabian StyleSouto, Eliana B., Joana R. Campos, Raquel Da Ana, Joana F. Fangueiro, Carlos Martins-Gomes, Alessandra Durazzo, Massimo Lucarini, Elena Sánchez López, Marta Espina, Maria Luisa García, and et al. 2020. "Diabetic Retinopathy and Ocular Melanoma: How Far We Are?" Applied Sciences 10, no. 8: 2777. https://doi.org/10.3390/app10082777
APA StyleSouto, E. B., Campos, J. R., Da Ana, R., Fangueiro, J. F., Martins-Gomes, C., Durazzo, A., Lucarini, M., Sánchez López, E., Espina, M., García, M. L., Silva, A. M., Mendonça, F., Santini, A., & Souto, S. B. (2020). Diabetic Retinopathy and Ocular Melanoma: How Far We Are? Applied Sciences, 10(8), 2777. https://doi.org/10.3390/app10082777