Author Contributions
Conceptualization, F.G., T.A., M.A., Q.S, and T.F.; Methodology, F.G., T.A., M.A., Q.S., T.V.; software, T.V., and F.G., validation, T.V., and F.G.; formal analysis, F.G; resources, M.H., and T.F.; writing—original draft preparation, F.G.; writing—review and editing, F.G., T.A., M.A., Q.S, T.V., and T.F.; supervision, T.F., M.H., and M.A.; project administration, M.H.; funding acquisition, M.H., and T.F. All authors have read and agreed to the published version of the manuscript.
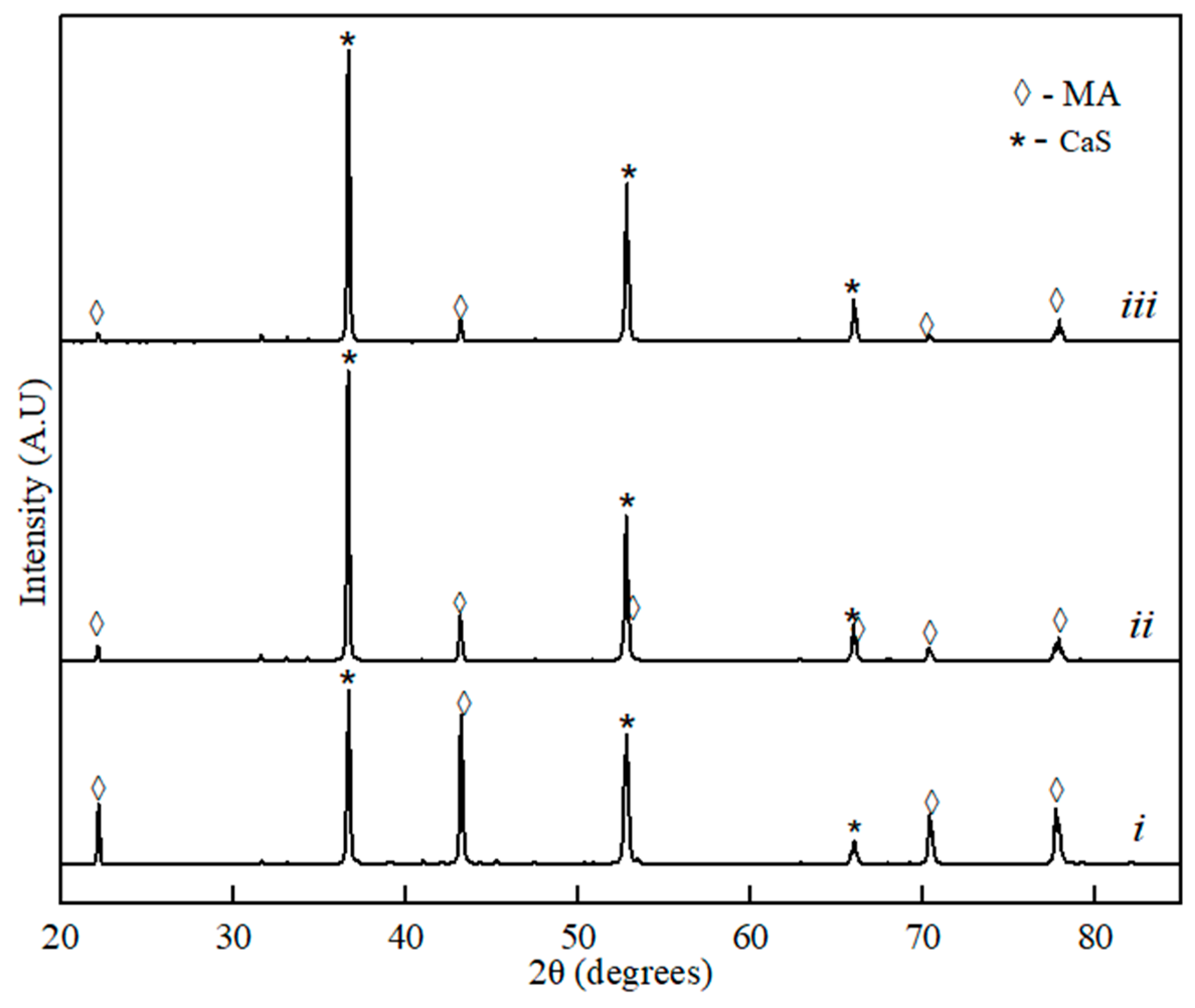
Figure 1.
XRD spectra for the binary sample of MgO∙Al2O3 (MA)–CaS: (i) 20% of CaS, (ii) 60% of CaS, and (iii) 80% of the CaS phase component.
Figure 1.
XRD spectra for the binary sample of MgO∙Al2O3 (MA)–CaS: (i) 20% of CaS, (ii) 60% of CaS, and (iii) 80% of the CaS phase component.
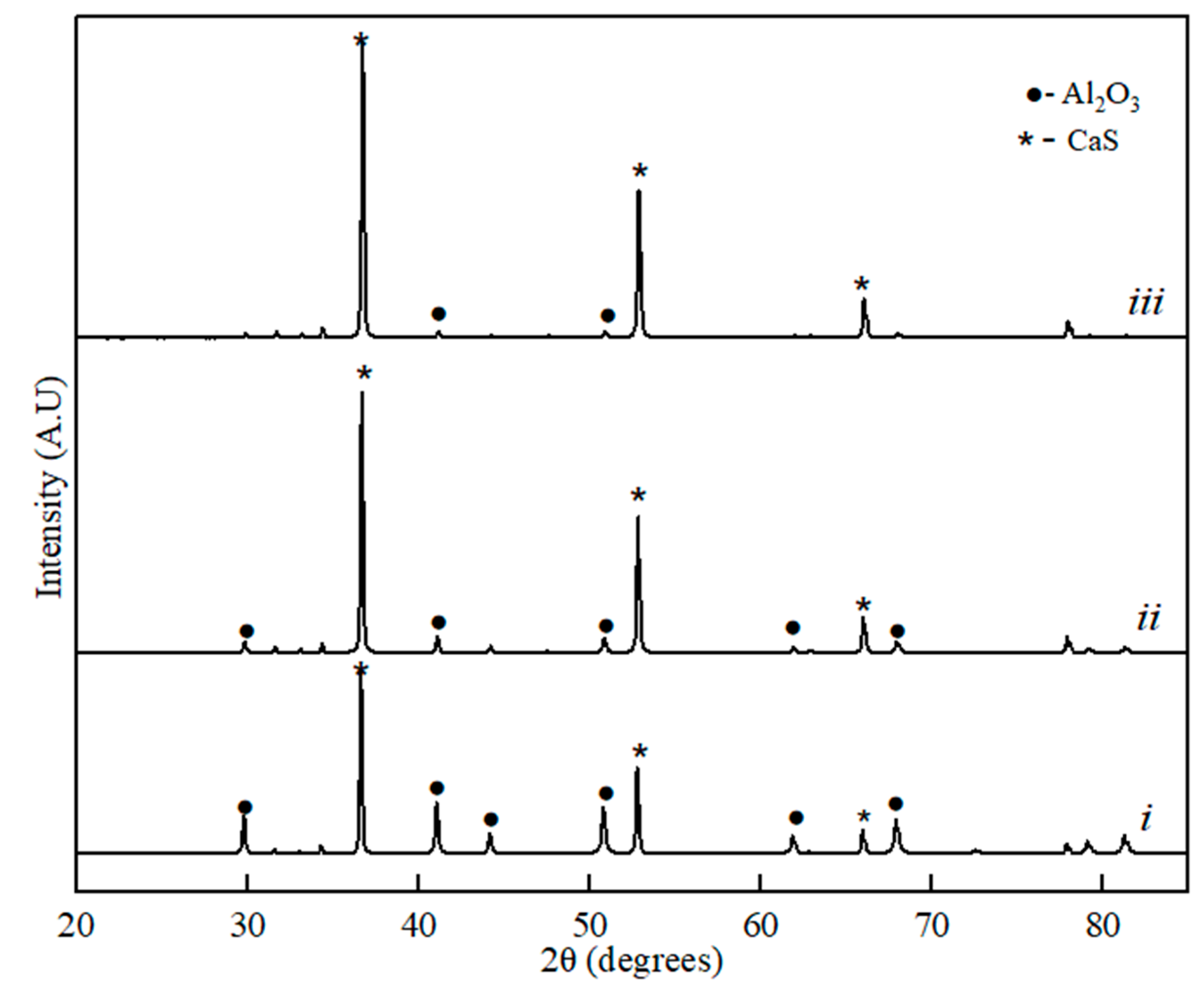
Figure 2.
XRD spectra for the binary sample of CaS-Al2O3: (i) 20% of CaS, (ii) 60% of CaS, and (iii) 80% of the CaS phase component.
Figure 2.
XRD spectra for the binary sample of CaS-Al2O3: (i) 20% of CaS, (ii) 60% of CaS, and (iii) 80% of the CaS phase component.
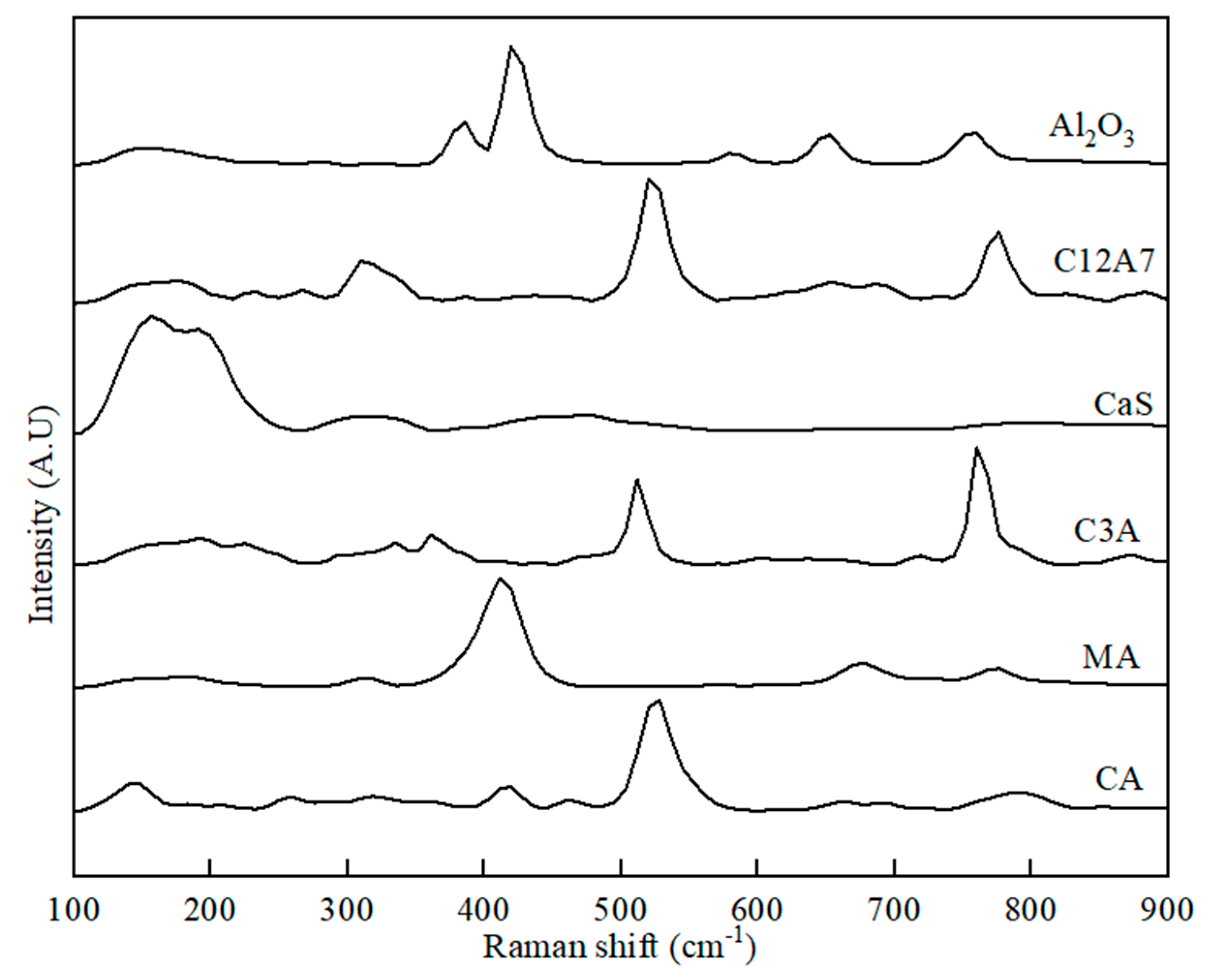
Figure 3.
Raman spectra for initial phases of Al2O3, C12A7, CaS, C3A, MA, and CA used for preparing the binary phase samples.
Figure 3.
Raman spectra for initial phases of Al2O3, C12A7, CaS, C3A, MA, and CA used for preparing the binary phase samples.
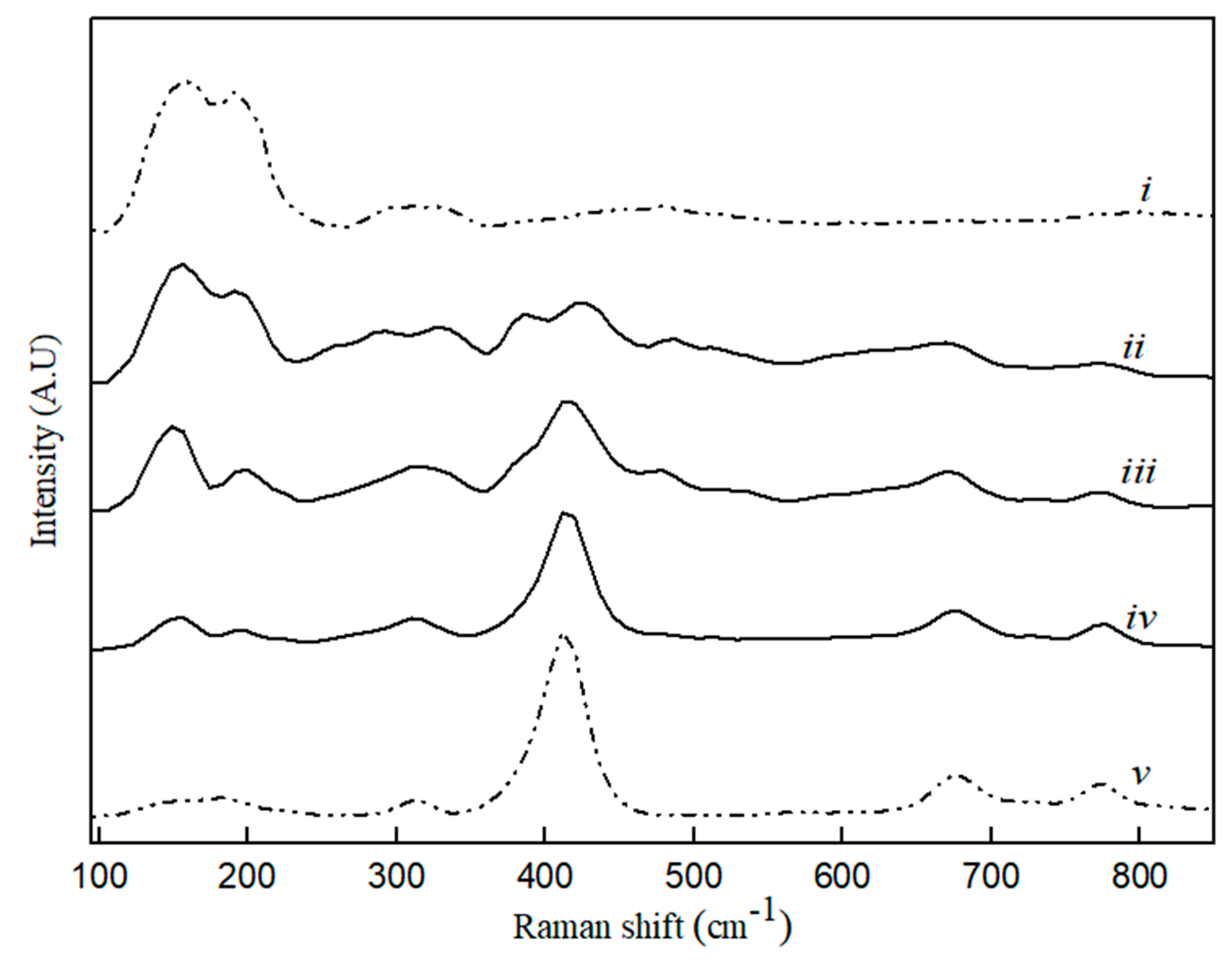
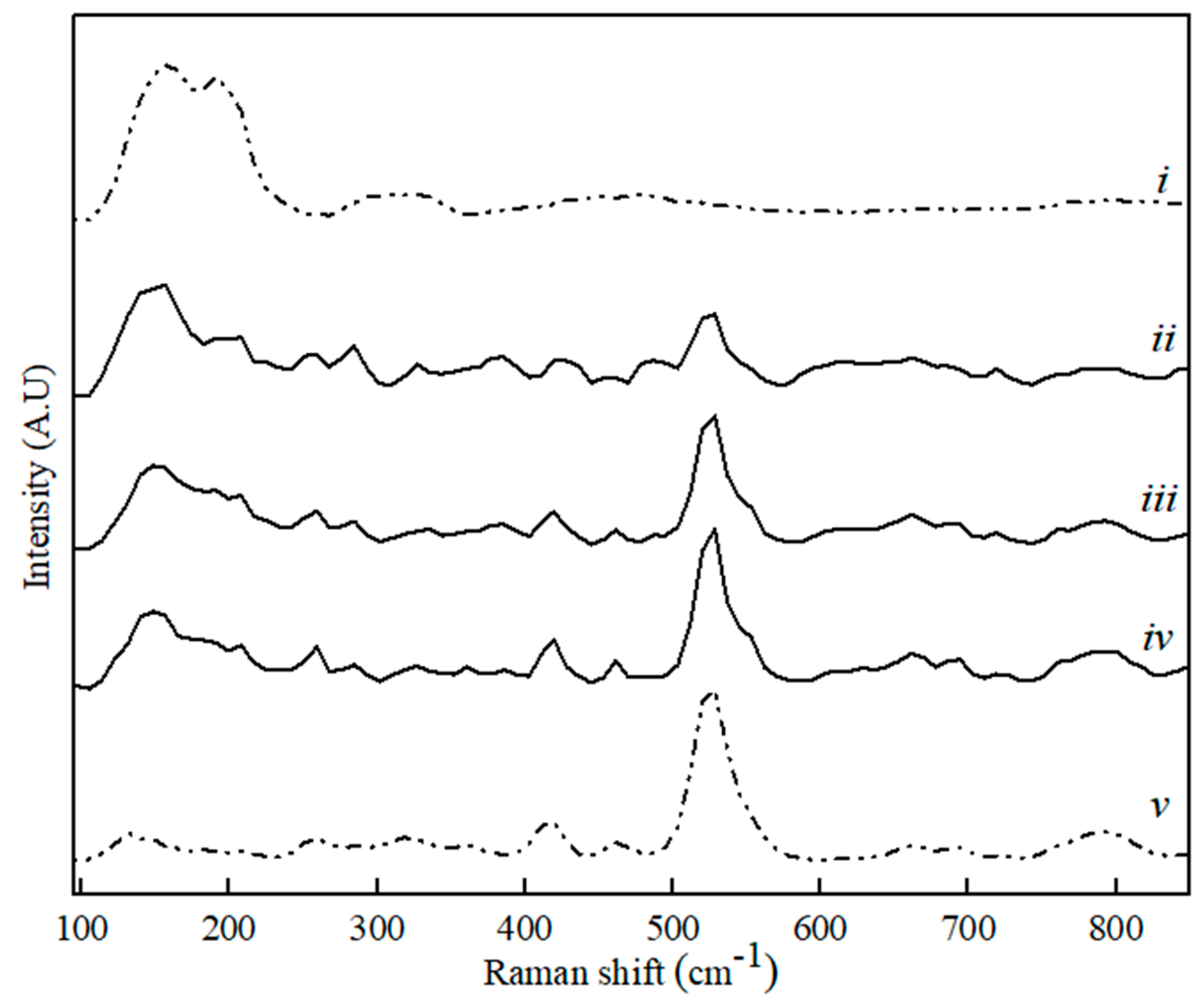
Figure 4.
Raman spectra for CaS (i), MA (v) and binary MA–CaS phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
Figure 4.
Raman spectra for CaS (i), MA (v) and binary MA–CaS phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
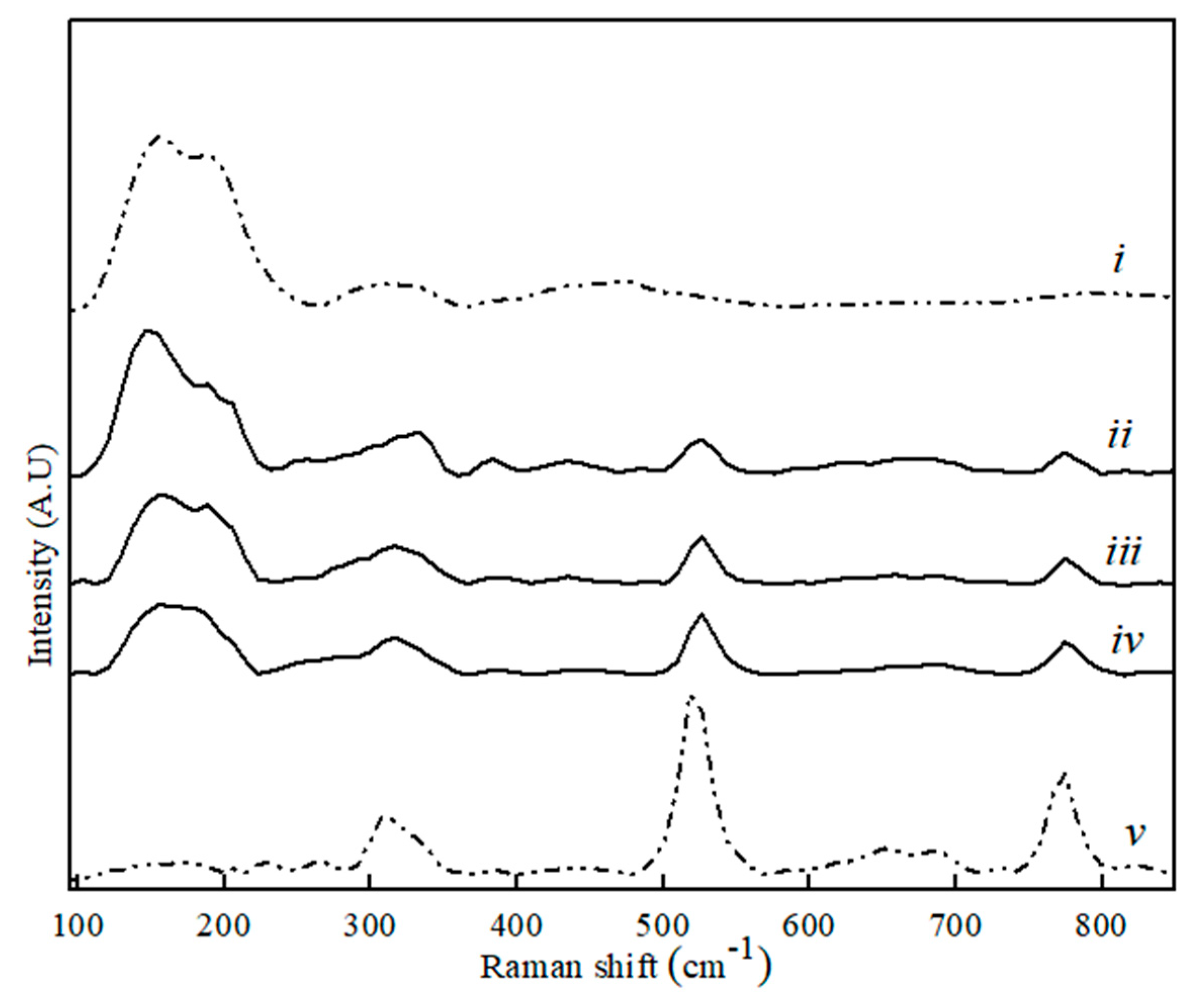
Figure 5.
Raman spectra for CaS (i), CA (v) and binary CA–CaS phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
Figure 5.
Raman spectra for CaS (i), CA (v) and binary CA–CaS phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
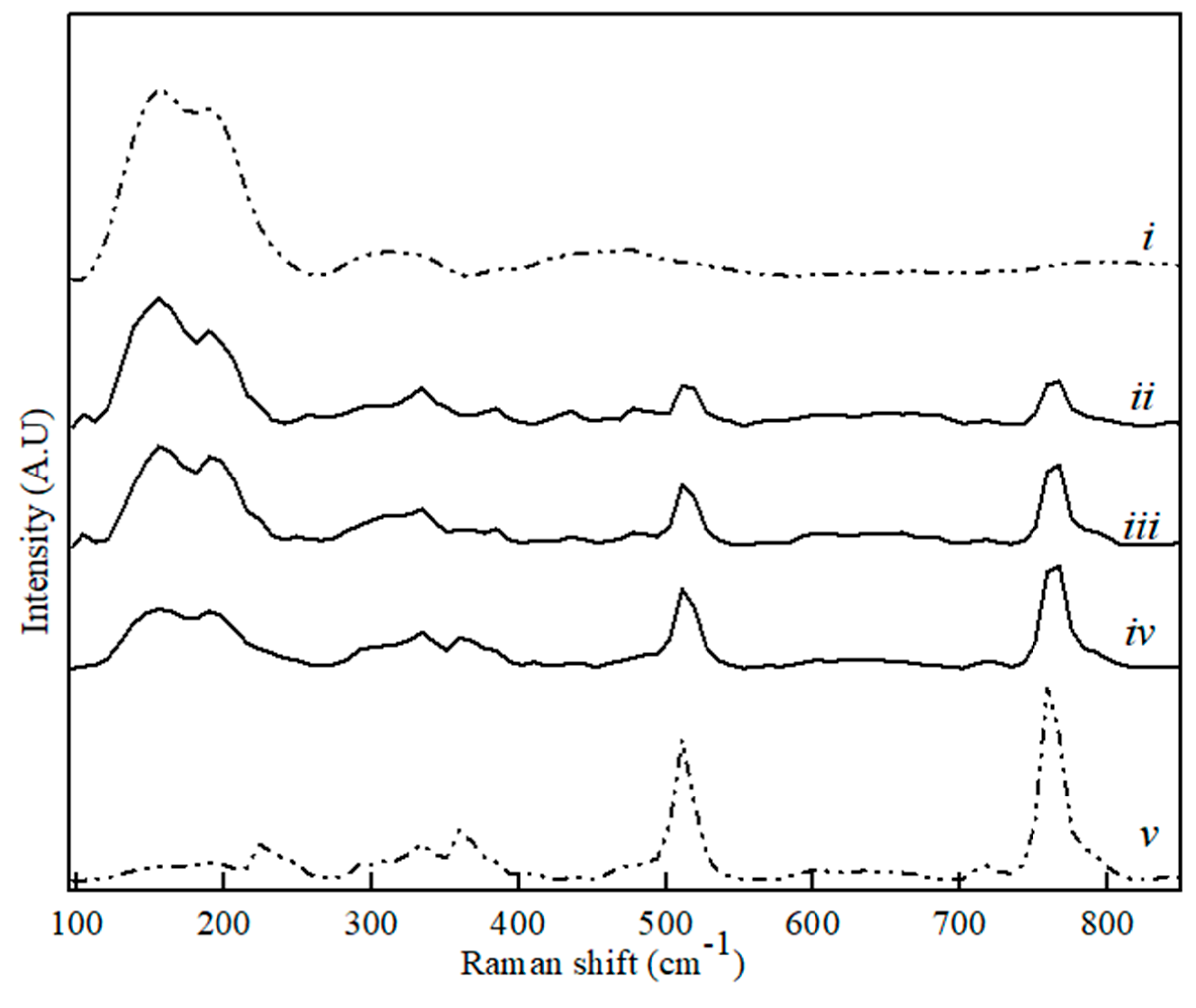
Figure 6.
Raman spectra for CaS (i), C12A7 (v) and binary CaS–C12A7 phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
Figure 6.
Raman spectra for CaS (i), C12A7 (v) and binary CaS–C12A7 phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
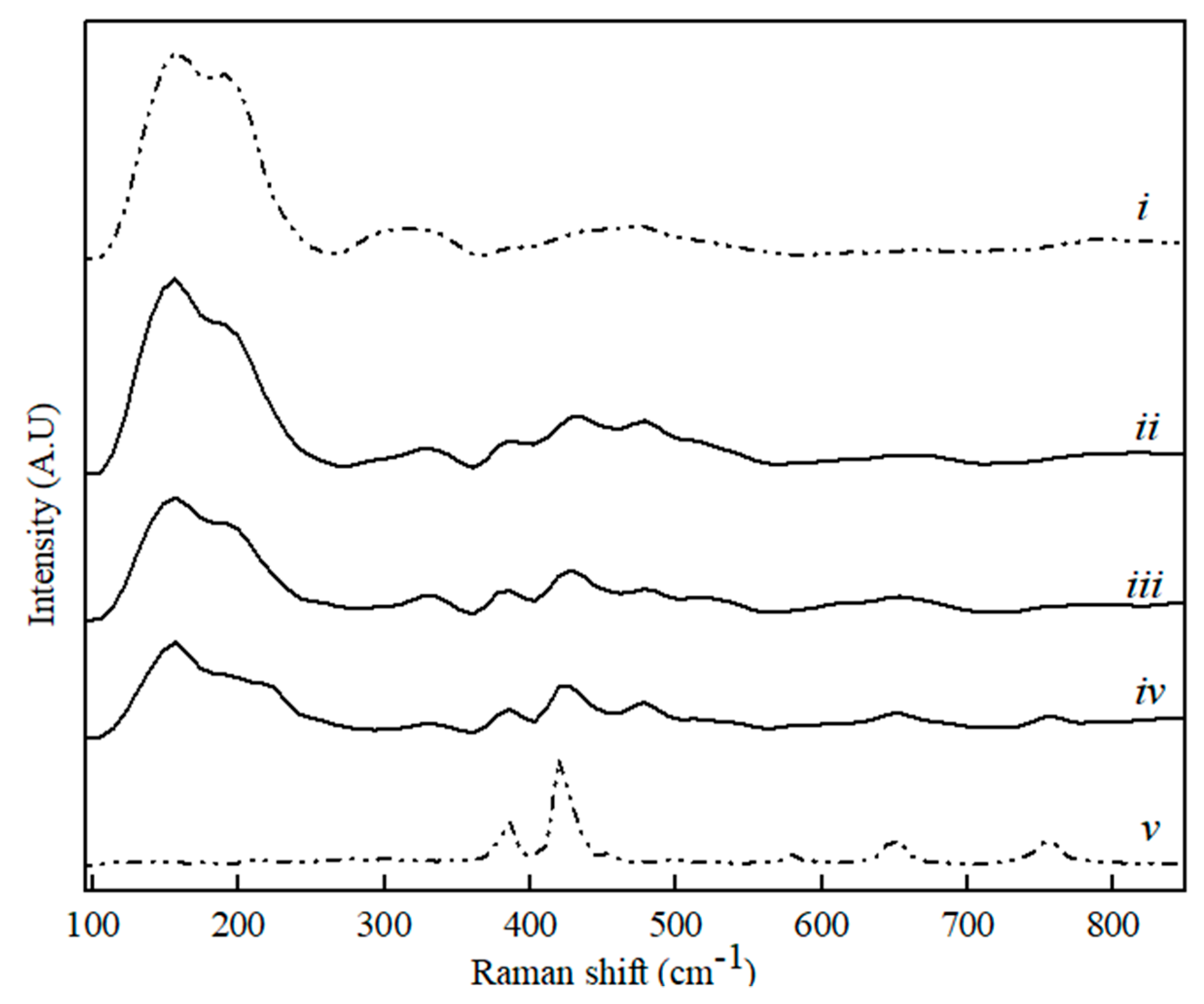
Figure 7.
Raman spectra for CaS (i), C3A (v) and binary CaS–C3A phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
Figure 7.
Raman spectra for CaS (i), C3A (v) and binary CaS–C3A phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
Figure 8.
Raman spectra for CaS (i), Al2O3 (v) and binary CaS–Al2O3 phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
Figure 8.
Raman spectra for CaS (i), Al2O3 (v) and binary CaS–Al2O3 phase sample with CaS content, where (ii) 80 wt. %, (iii) 60 wt. %, and (iv) 20 wt. %.
Figure 9.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for CaS–CA.
Figure 9.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for CaS–CA.
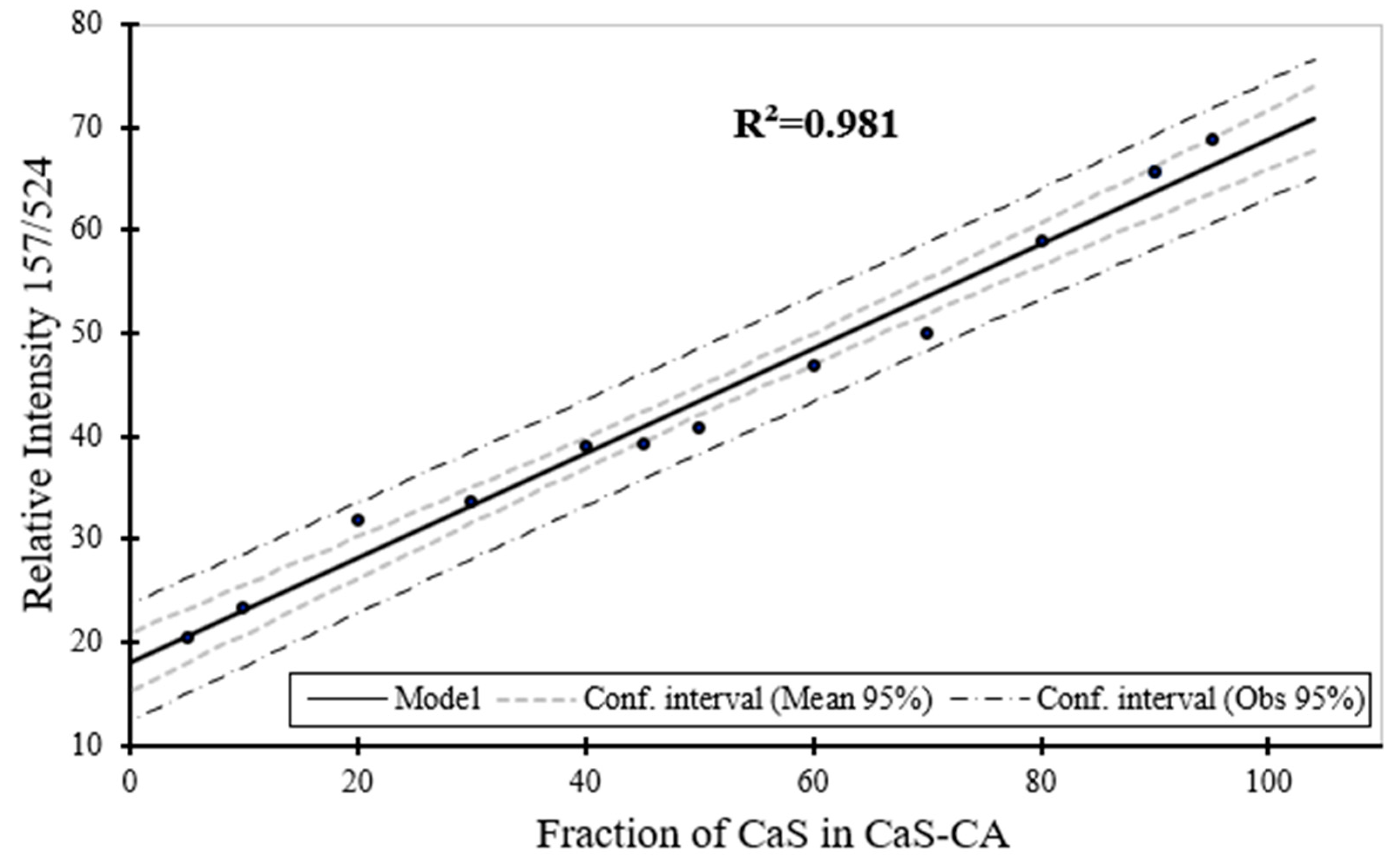
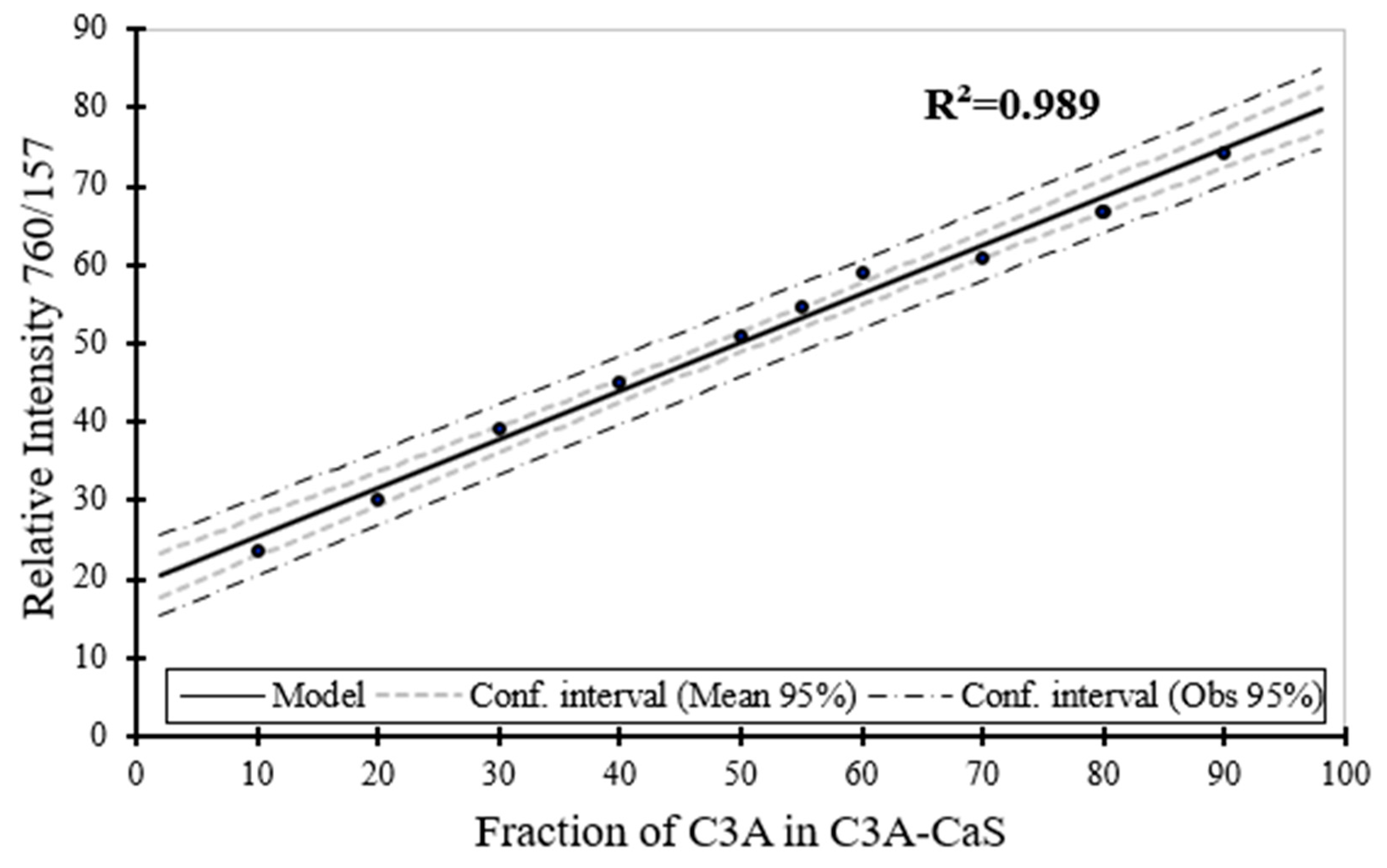
Figure 10.
Raman spectroscopy estimation for the phase content (wt. %) for CaS as a function of the phase content of CaS in CaS–CA sample.
Figure 10.
Raman spectroscopy estimation for the phase content (wt. %) for CaS as a function of the phase content of CaS in CaS–CA sample.
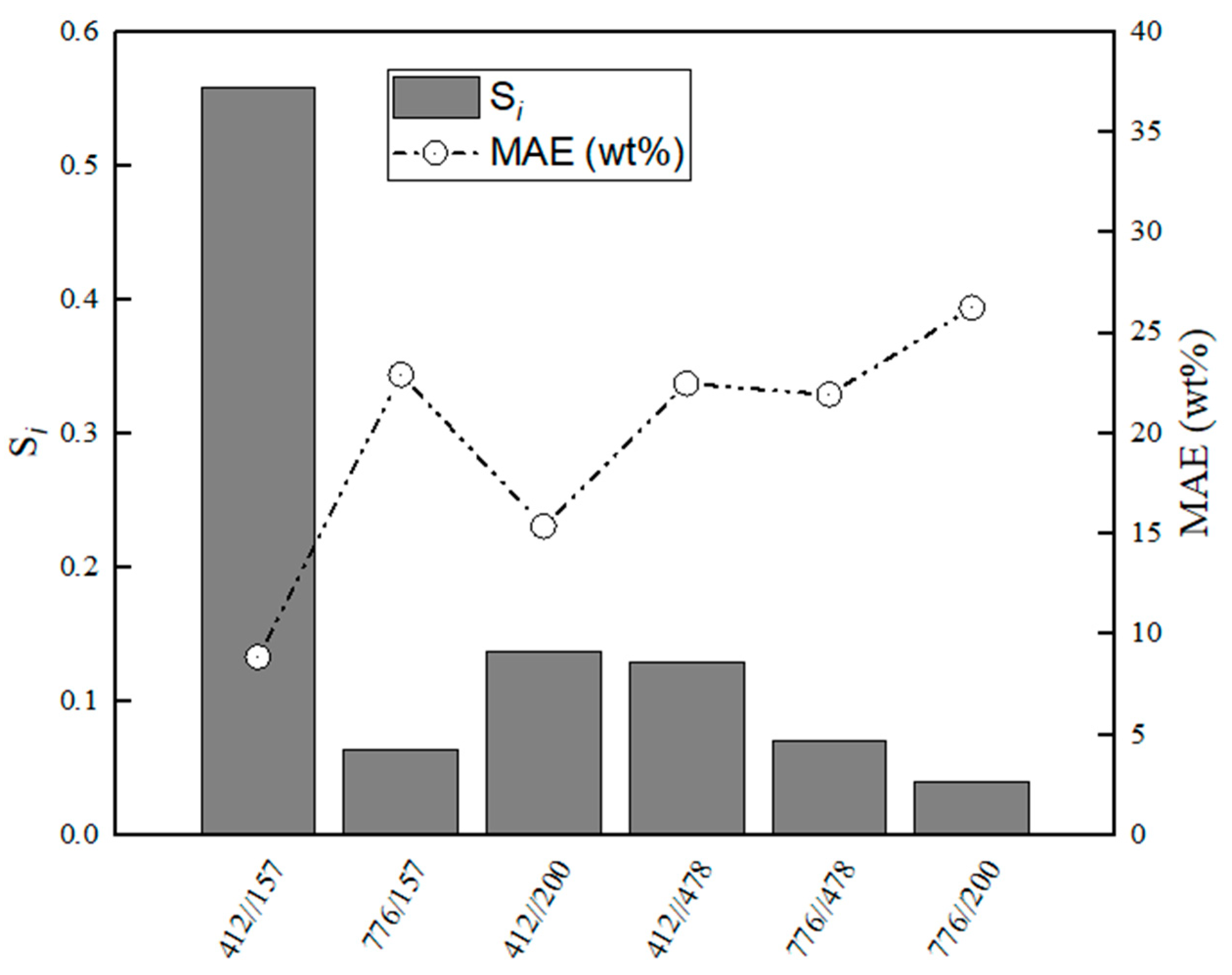
Figure 11.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for MA–CaS.
Figure 11.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for MA–CaS.
Figure 12.
Raman spectroscopy estimation for the phase content (wt. %) for MA as a function of the phase content of MA in MA–CaS sample.
Figure 12.
Raman spectroscopy estimation for the phase content (wt. %) for MA as a function of the phase content of MA in MA–CaS sample.
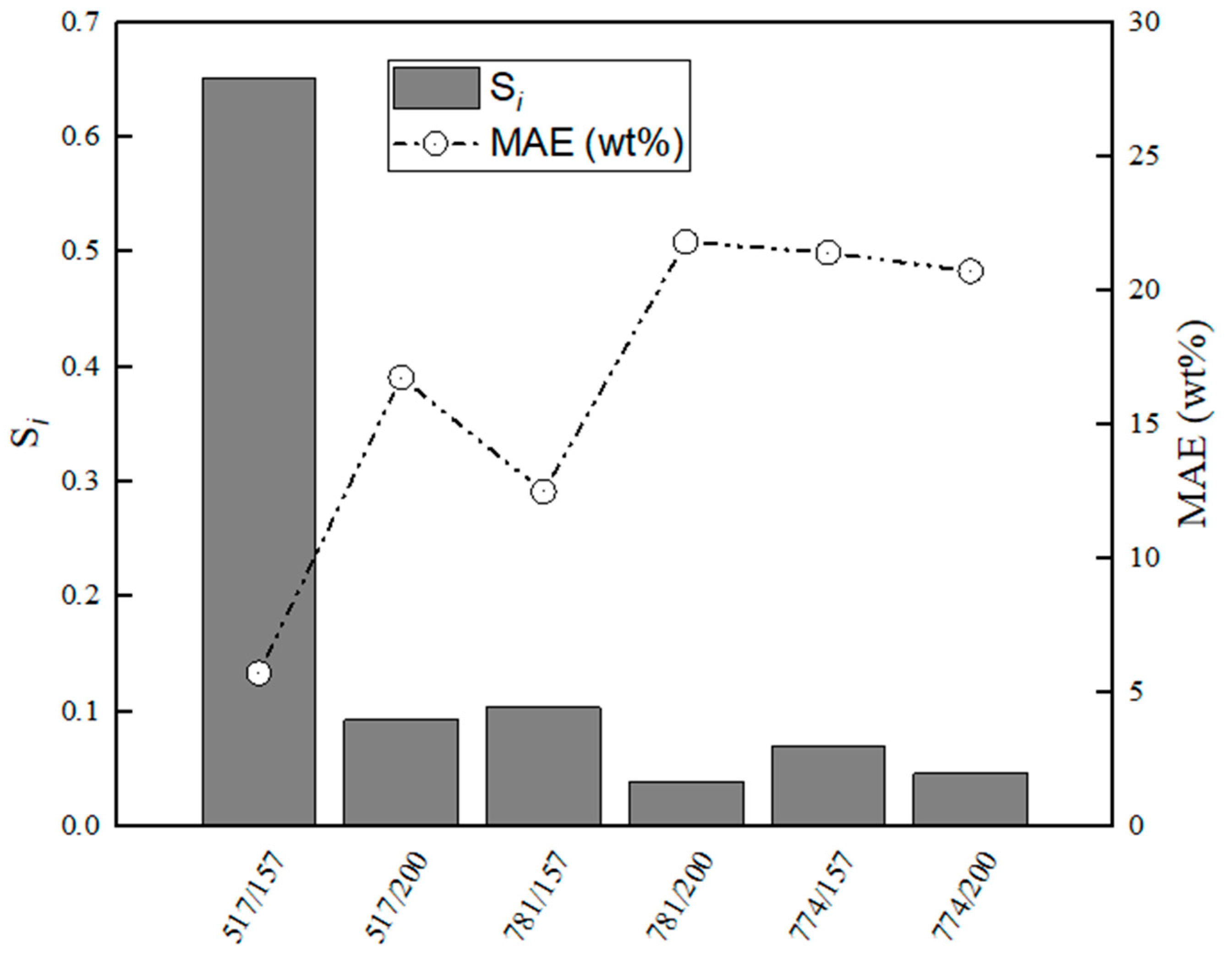
Figure 13.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for C12A7–CaS.
Figure 13.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for C12A7–CaS.
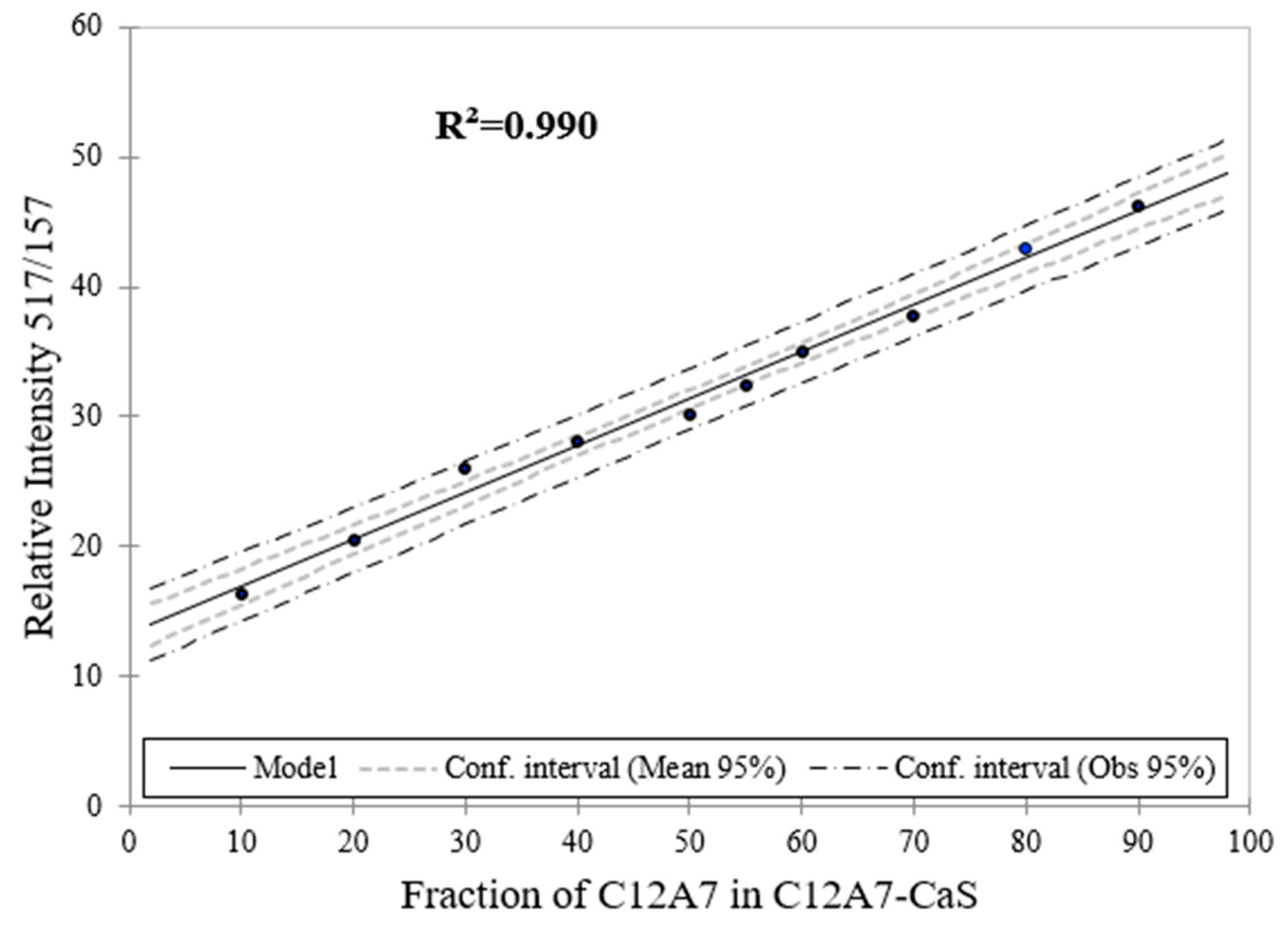
Figure 14.
Raman spectroscopy estimation for the phase content (wt. %) for C12A7 as a function of the phase content of C12A7 in C12A7–CaS sample.
Figure 14.
Raman spectroscopy estimation for the phase content (wt. %) for C12A7 as a function of the phase content of C12A7 in C12A7–CaS sample.
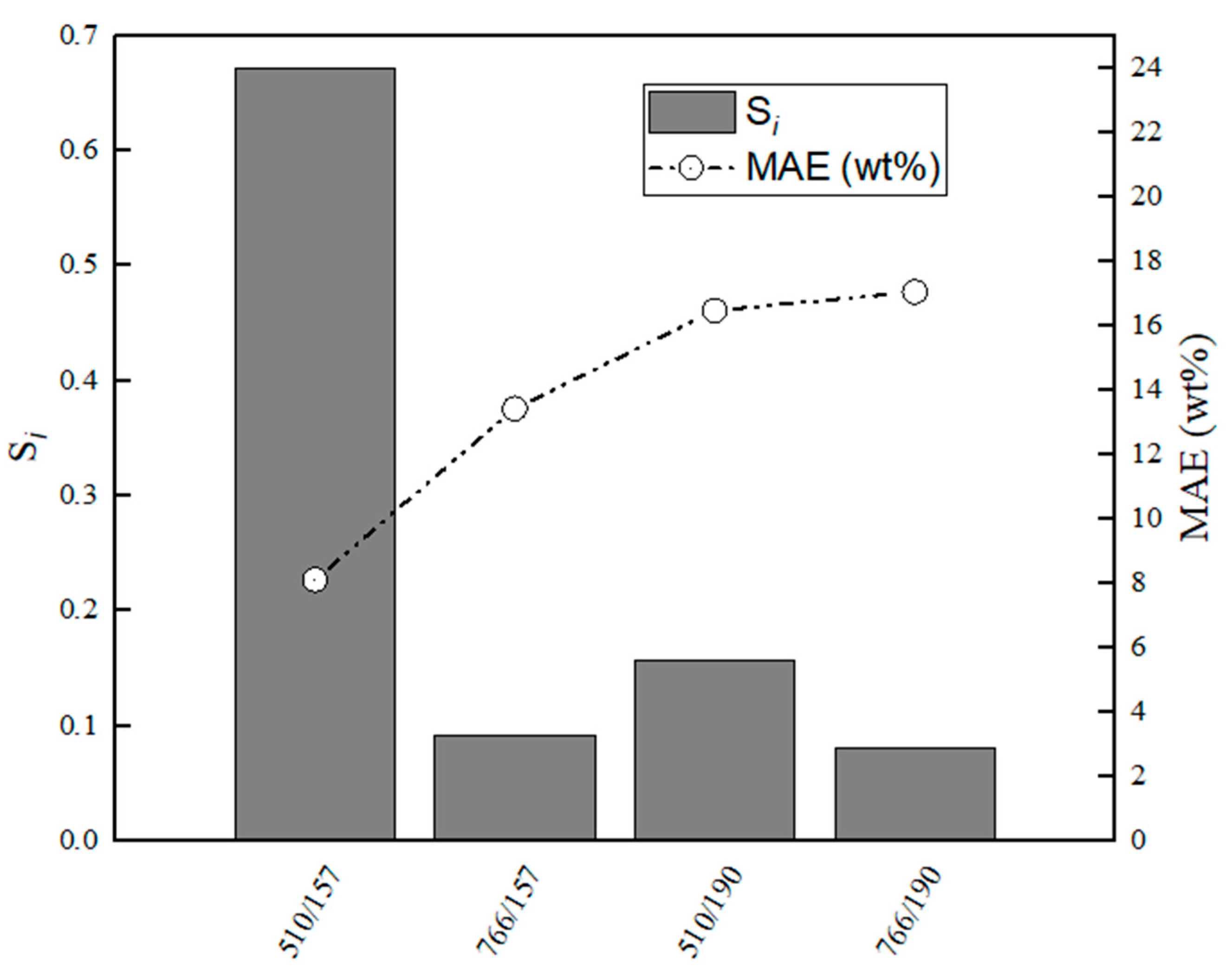
Figure 15.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for C3A–CaS.
Figure 15.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for C3A–CaS.
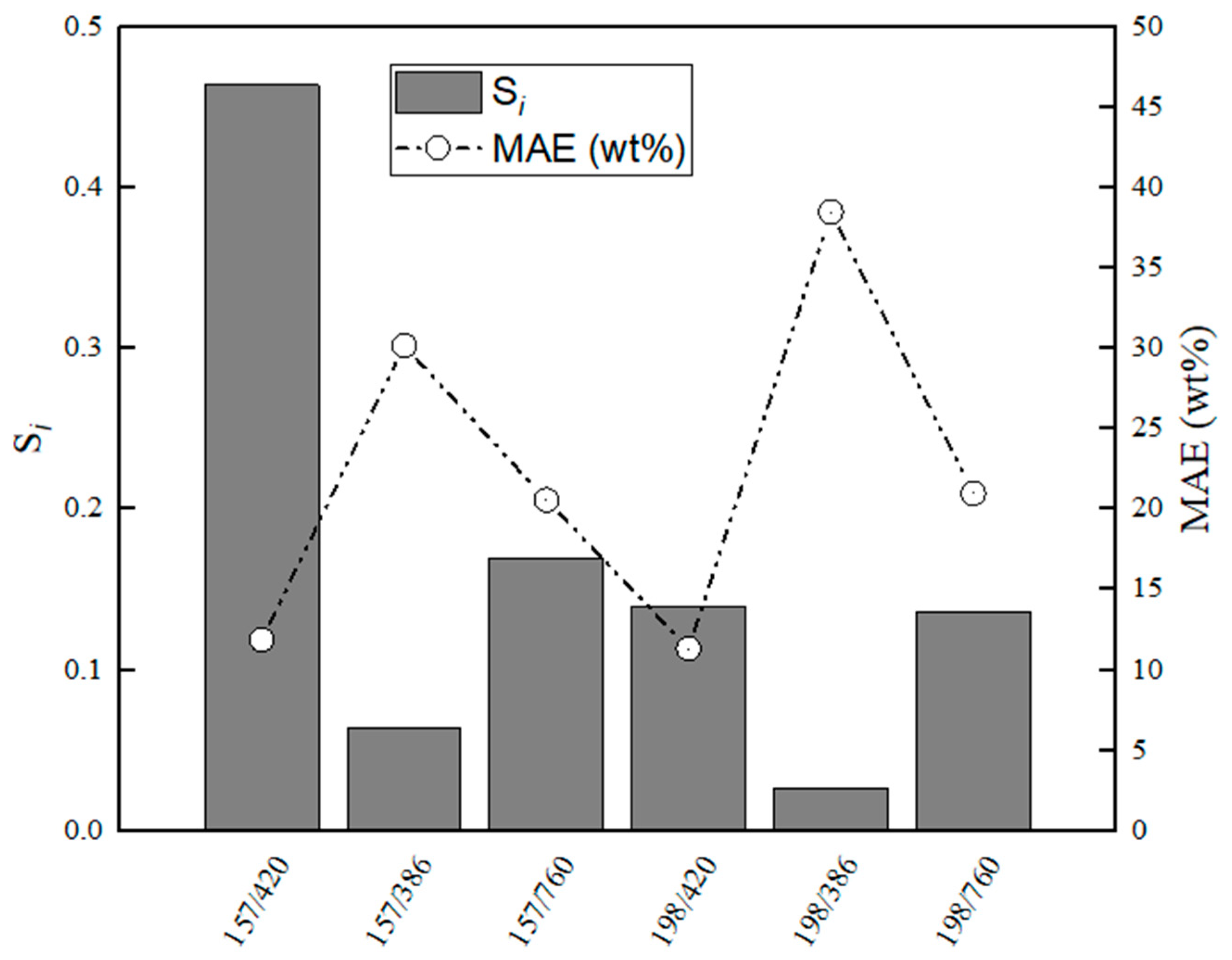
Figure 16.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for CaS–Al2O3.
Figure 16.
Evaluation of relative stabilities (Si) of the calibration variable candidates and mean absolute error values obtained for training and validation sets for CaS–Al2O3.
Figure 17.
Raman spectroscopy estimation for the phase content (wt. %) for MA as a function of the phase content of MA in C3A–CaS sample.
Figure 17.
Raman spectroscopy estimation for the phase content (wt. %) for MA as a function of the phase content of MA in C3A–CaS sample.
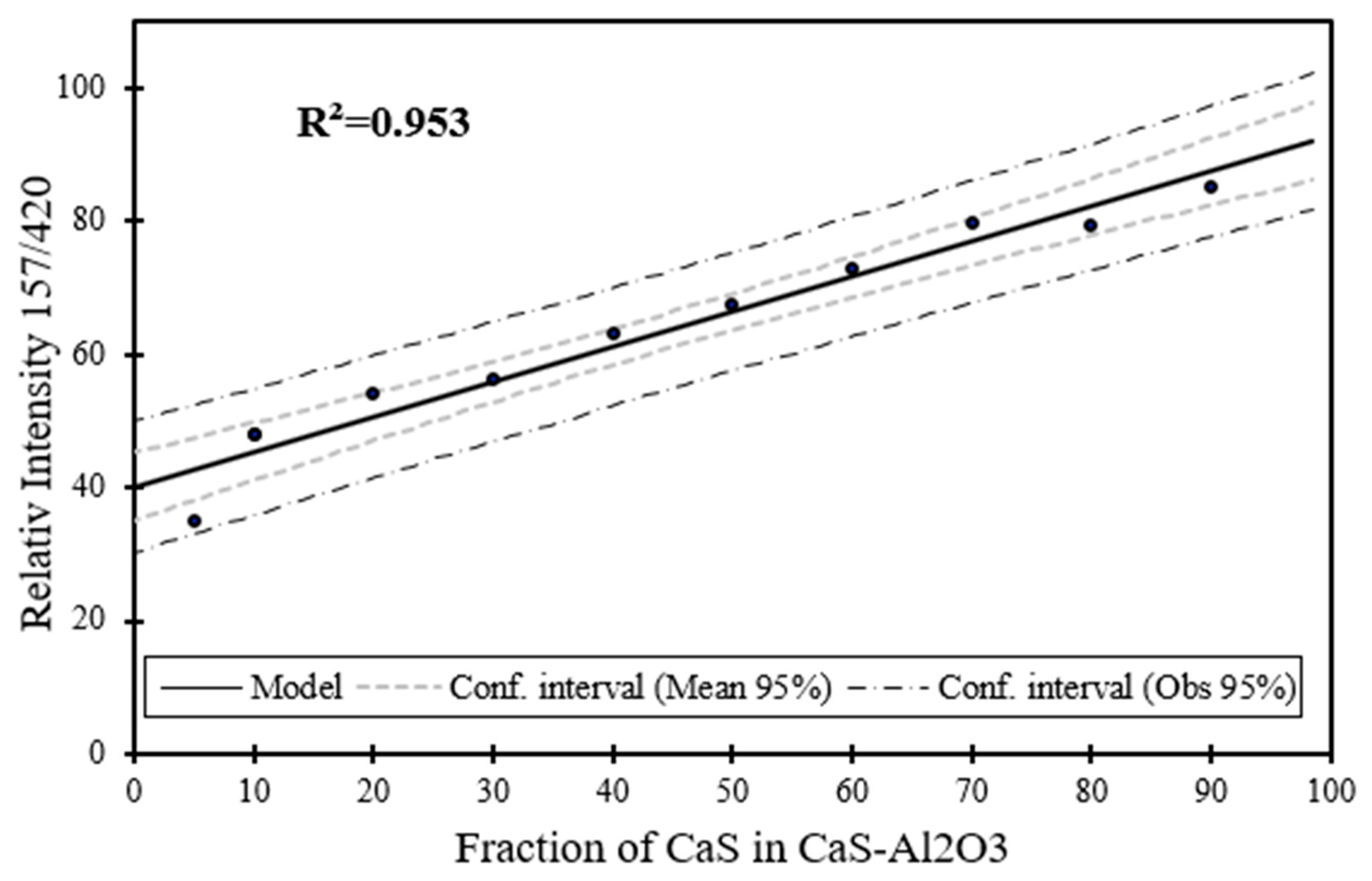
Figure 18.
Raman spectroscopy estimation for the phase content (wt. %) for MA as a function of the phase content of MA in CaS–Al2O3 sample.
Figure 18.
Raman spectroscopy estimation for the phase content (wt. %) for MA as a function of the phase content of MA in CaS–Al2O3 sample.
Table 1.
Initial sample composition, XRD and XRF analyses for binary calcium sulphide-aluminate spinel phases of MgO∙Al2O3 (MA)–CaS.
Table 1.
Initial sample composition, XRD and XRF analyses for binary calcium sulphide-aluminate spinel phases of MgO∙Al2O3 (MA)–CaS.
| Initial Phase Composition (wt%) | XRD Phase Composition (wt%) | XRF Calculated Phase Composition (wt%) and Elemental Data |
|---|
| MgO∙Al2O3 | CaS | MgO∙Al2O3 | CaS | MgO∙Al2O3 | CaS | Ca | S | Al | Mg |
| 90 | 10 | 91 | 9 | 87 | 13 | 7.27 | 7.03 | 33.27 | 12.68 |
| 80 | 20 | 84 | 16 | 76 | 24 | 13.29 | 12.63 | 27.77 | 11.22 |
| 30 | 70 | 34 | 66 | 31 | 69 | 43.78 | 35.02 | 7.97 | 3.70 |
| 20 | 80 | 25 | 75 | 21 | 79 | 38.48 | 30.86 | 11.63 | 5.17 |
| 10 | 90 | 12 | 88 | 7 | 93 | 51.94 | 41.55 | 2.31 | 1.30 |
Table 2.
Initial sample composition, XRD and XRF analyses for binary aluminum oxide-calcium sulphide phases of Al2O3–CaS.
Table 2.
Initial sample composition, XRD and XRF analyses for binary aluminum oxide-calcium sulphide phases of Al2O3–CaS.
| Initial Phase Composition (wt%) | XRD Phase Composition (wt%) | XRF Calculated Phase Composition (wt%) and Elemental Data |
|---|
| Al2O3 | CaS | Al2O3 | CaS | Al2O3 | CaS | Ca | S | Al |
| 10 | 90 | 8 | 92 | 15 | 85 | 46.81 | 37.44 | 8.16 |
| 20 | 80 | 18 | 82 | 24 | 76 | 42.08 | 33.66 | 12.66 |
| 30 | 70 | 28 | 72 | 34 | 66 | 36.26 | 29.01 | 18.23 |
| 60 | 40 | 58 | 42 | 57 | 43 | 23.55 | 18.83 | 30.41 |
| 70 | 30 | 72 | 28 | 69 | 31 | 17.28 | 13.82 | 36.38 |
| 90 | 10 | 91 | 9 | 89 | 11 | 6.28 | 5.02 | 46.95 |
Table 3.
Reference and measured Raman shift (cm−1) for starting phases of CA, CaS, C12A7, C3A, Al2O3, and MgO∙Al2O3 (where s is strong, m is medium, and w is weak).
Table 3.
Reference and measured Raman shift (cm−1) for starting phases of CA, CaS, C12A7, C3A, Al2O3, and MgO∙Al2O3 (where s is strong, m is medium, and w is weak).
| Phase | Measured Peaks [Raman Shift, cm−1] | Reference [Raman Shift, cm−1] | Reference |
|---|
| CaS | 156–160 s, 190–200 m, 478 w | 160 s, 185 m, 215 (±15), 485 (±10) w | [12] |
| C12A7 | 517 s, 781 m, 314 m | 312–333 m, 516–517 m, 772 m, 779 m | [13,15] |
| C3A | 766 s, 512 m, | 756–760 s, 506–508 m | [13,15] |
| Al2O3 | 420 s, 386 m, 760 m | 413–420 s, 375–382 m, 748–751 m | [11] |
| MgO∙Al2O3 | 412 s, 674 m, 773 m | 409–412 s, 767–772 m, 666–674 m | [14] |
Table 4.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for CaS–CA.
Table 4.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for CaS–CA.
| | | Training Data | Validation | |
|---|
| Phases | Relative Intensity | Mean (R2) | Mean (MAE) | Mean (R2) | Mean (MAE) | Sum MAE | Relative Stability |
|---|
| CaS-CA | 157/524 | 0.97 | 3.49 | 0.98 | 3.71 | 7.20 | 0.48 |
| CaS-CA | 157/790 | 0.92 | 6.08 | 0.93 | 6.12 | 12.20 | 0.15 |
| CaS-CA | 200/524 | 0.94 | 5.30 | 0.94 | 5.50 | 10.80 | 0.17 |
| CaS-CA | 200/790 | 0.83 | 9.27 | 0.84 | 9.49 | 18.76 | 0.07 |
| CaS-CA | 478/524 | 0.75 | 11.72 | 0.76 | 12.12 | 23.84 | 0.05 |
| CaS-CA | 478/790 | 0.84 | 9.22 | 0.84 | 9.36 | 18.58 | 0.07 |
Table 5.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for MA–CaS.
Table 5.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for MA–CaS.
| | | Training Data | Validation | | |
|---|
| Phases | Relative Intensity | Mean (R2) | Mean (MAE) | Mean (R2) | Mean (MAE) | Sum MAE | Relative Stability |
|---|
| MA-CaS | 412//157 | 0.96 | 4.35 | 0.96 | 4.52 | 8.87 | 0.56 |
| MA-CaS | 776/157 | 0.72 | 11.00 | 0.72 | 11.92 | 22.92 | 0.06 |
| MA-CaS | 412//200 | 0.87 | 7.53 | 0.87 | 7.84 | 15.37 | 0.14 |
| MA-CaS | 412//478 | 0.76 | 11.01 | 0.77 | 11.47 | 22.48 | 0.13 |
| MA-CaS | 776//478 | 0.75 | 10.65 | 0.74 | 11.29 | 21.94 | 0.07 |
| MA-CaS | 776//200 | 0.63 | 12.68 | 0.62 | 13.57 | 26.26 | 0.04 |
Table 6.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for C12A7–CaS.
Table 6.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for C12A7–CaS.
| | | Training Data | Validation | | |
|---|
| Phases | Relative Intensity | Mean (R2) | Mean (MAE) | Mean (R2) | Mean (MAE) | Sum MAE | Relative Stability |
|---|
| C12A7-CaS | 517/157 | 0.98 | 2.76 | 0.98 | 2.96 | 5.72 | 0.98 |
| C12A7-CaS | 517/200 | 0.84 | 8.03 | 0.84 | 8.72 | 16.75 | 4.44 |
| C12A7-CaS | 781/157 | 0.90 | 6.06 | 0.90 | 6.43 | 12.49 | 1.54 |
| C12A7-CaS | 781/200 | 0.75 | 10.49 | 0.72 | 11.31 | 21.80 | 3.78 |
| C12A7-CaS | 774/157 | 0.76 | 10.30 | 0.80 | 11.10 | 21.40 | 4.78 |
| C12A7-CaS | 774/200 | 0.77 | 9.55 | 0.77 | 11.16 | 20.71 | 5.73 |
Table 7.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for C3A-CaS.
Table 7.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for C3A-CaS.
| | | Training Data | Validation | | |
|---|
| Phases | Relative Intensity | Mean (R2) | Mean (MAE) | Mean (R2) | Mean (MAE) | Sum MAE | Relative Stability |
|---|
| C3A-CaS | 510/157 | 0.96 | 4.03 | 0.97 | 4.07 | 8.10 | 0.67 |
| C3A-CaS | 766/157 | 0.88 | 6.50 | 0.88 | 6.92 | 13.42 | 0.09 |
| C3A-CaS | 510/190 | 0.87 | 7.72 | 0.86 | 8.74 | 16.46 | 0.16 |
| C3A-CaS | 766/190 | 0.85 | 8.07 | 0.82 | 8.97 | 17.04 | 0.08 |
Table 8.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for CaS- Al2O3.
Table 8.
Evaluation of the coefficient of determination (R2) and mean absolute error (MAE) for the prediction and validation between the phase content and relative intensity of the Raman peaks and phase content for CaS- Al2O3.
| | | Training Data | Validation | | |
|---|
| Phases | Relative Intensity | Mean (R2) | Mean (MAE) | Mean (R2) | Mean (MAE) | Sum MAE | Relative Stability |
|---|
| CaS- Al2O3 | 157/420 | 0.94 | 5.65 | 0.95 | 6.23 | 11.88 | 0.46 |
| CaS- Al2O3 | 157/386 | 0.58 | 14.68 | 0.59 | 15.49 | 30.17 | 0.06 |
| CaS-Al2O3 | 157/760 | 0.82 | 9.72 | 0.83 | 10.86 | 20.58 | 0.17 |
| CaS-Al2O3 | 198/420 | 0.91 | 5.49 | 0.90 | 5.84 | 11.33 | 0.14 |
| CaS-Al2O3 | 198/386 | 0.38 | 18.50 | 0.40 | 19.99 | 38.49 | 0.03 |
| CaS-Al2O3 | 198/760 | 0.80 | 9.88 | 0.82 | 11.10 | 20.98 | 0.14 |

 ,
, 



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}