Immunochromatographic Detection of Myoglobin as a Specific Biomarker of Porcine Muscle Tissues in Meat Products


 and
and 
Abstract
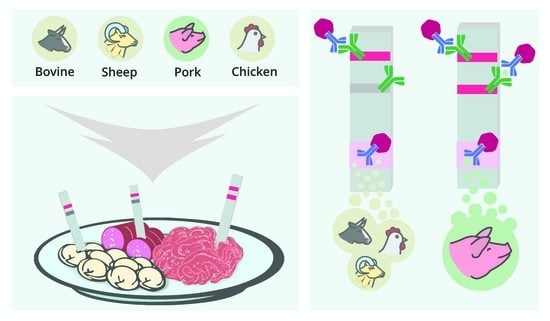
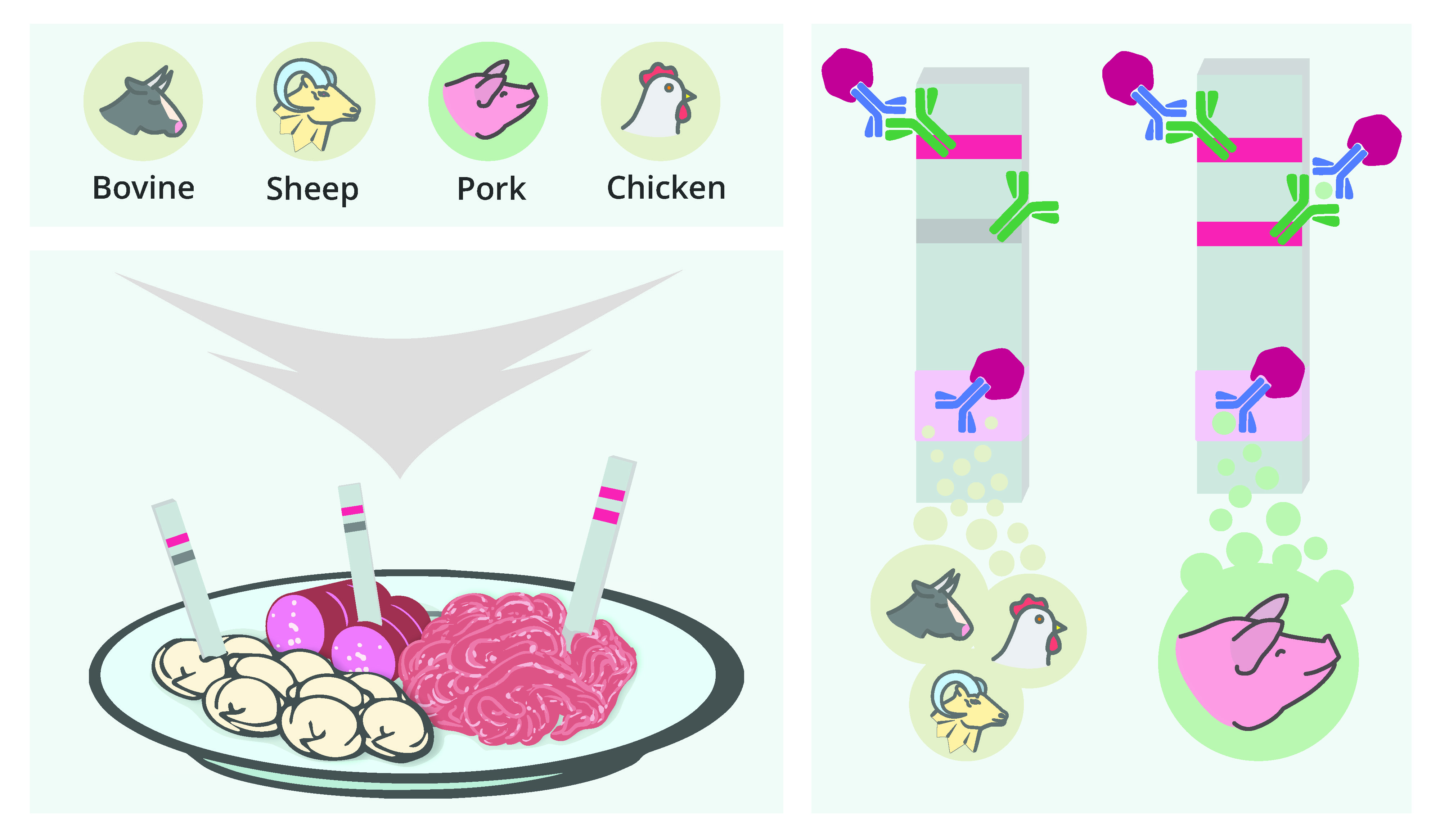
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Determination of the Immune Interactions’ Constants
2.3. Biotinylation of Antibodies
2.4. Sandwich Enzyme Llinked Immunosorbent Assay (ELISA)
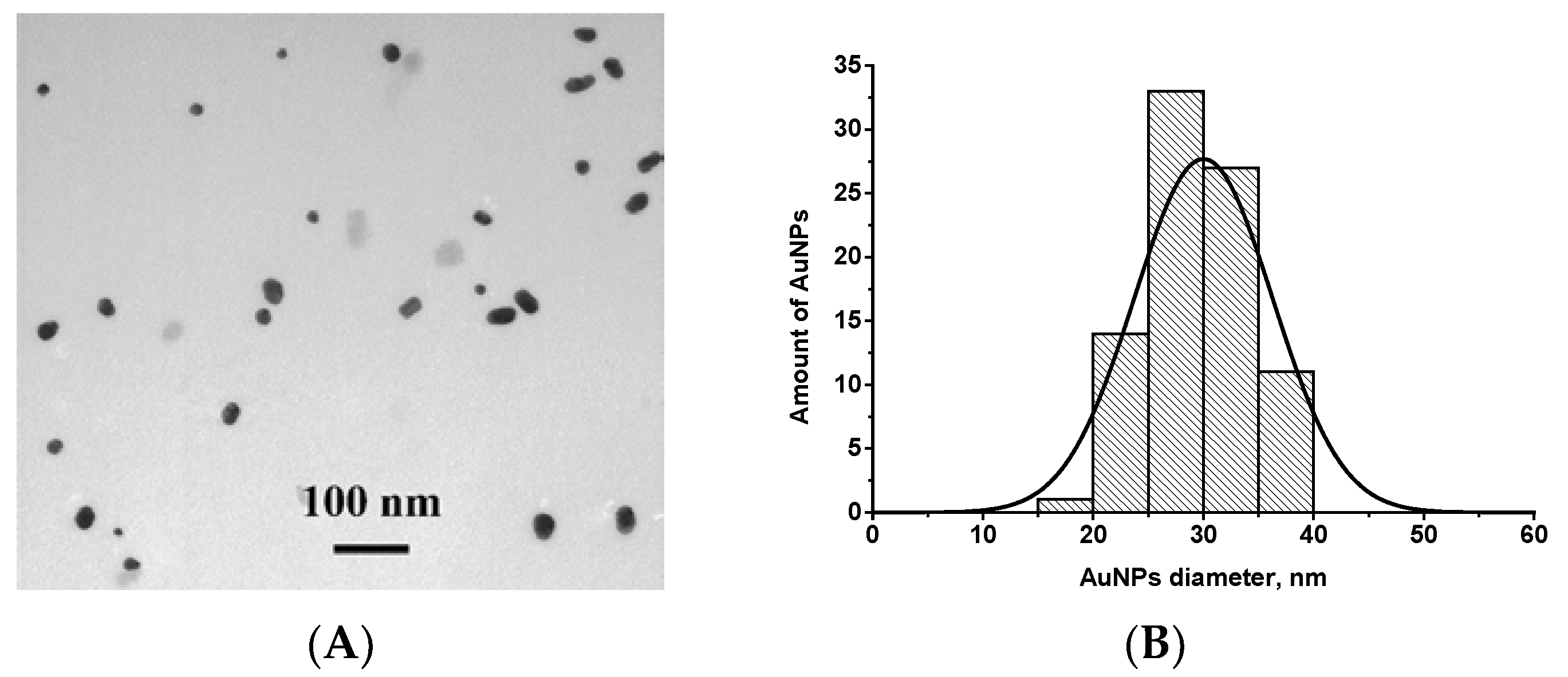
2.5. Synthesis and Characterization of Gold Nanoparticles (AuNPs)
2.6. Immobilization of Antibodies on AuNPs
2.7. Production of Immunochromatographic Tests
2.8. Sample Preparation Before the Lateral Flow Immunoassay (LFIA)
- A total of 100 mg of raw or cooked minced meat is vigorously mixed with 2 mL of PBS, containing 0.1% Triton X-100, and 0.5 M KCl (an extraction buffer), for 10 min. The mixtures are centrifuged for 5 min at 5000× g at room temperature. The obtained supernatants are incubated for 3 min at 100 °C. Then, the samples are centrifuged for 5 min at 7000× g, and supernatants are used for the LFIA [29].
- A total of 250 mg of raw or cooked minced meat is vigorously mixed with 5 mL of the extraction buffer for 30 min, and then sonicates in an ultrasound bath for 30 min. Mixtures are centrifuged for 10 min at 5000× g, and the obtained supernatants are used for the LFIA.
- A total of 150 mg of raw or cooked minced meat is vigorously mixed with 5 mL of PBST for 15 min. The obtained mixtures are used for the LFIA.
- A total of 150 mg of raw or cooked minced meat is vigorously mixed with 5 mL of PBST for 15 min. Mixtures are centrifuged for 5 min at 5000× g, and the obtained supernatants are used for the LFIA.
2.9. LFIA of Myoglobin (MG)
3. Results and Discussion
3.1. Characterization of Monoclonal Antibodies by the ELISA
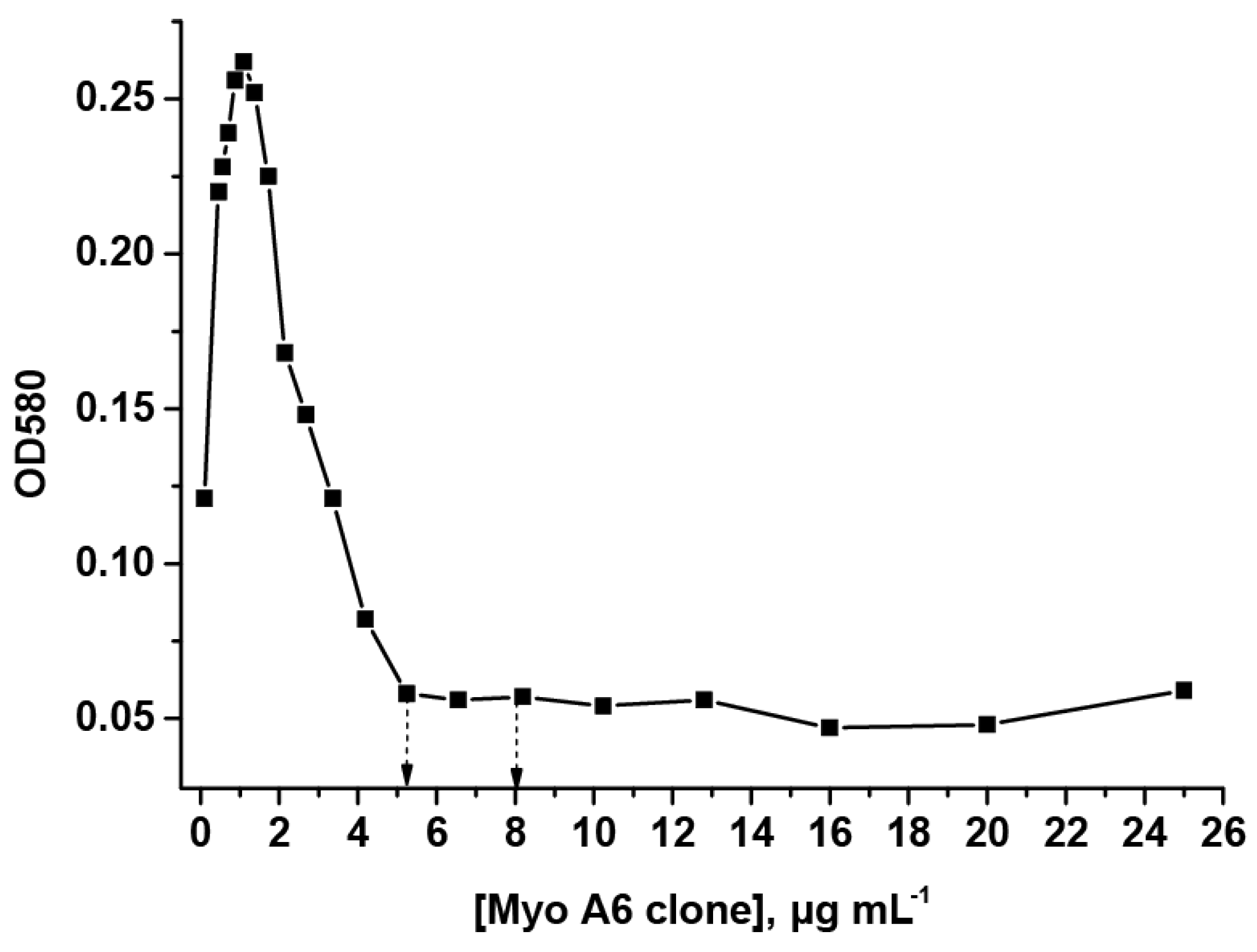
3.2. Obtaining and Characterization of Specific Reagents for the LFIA
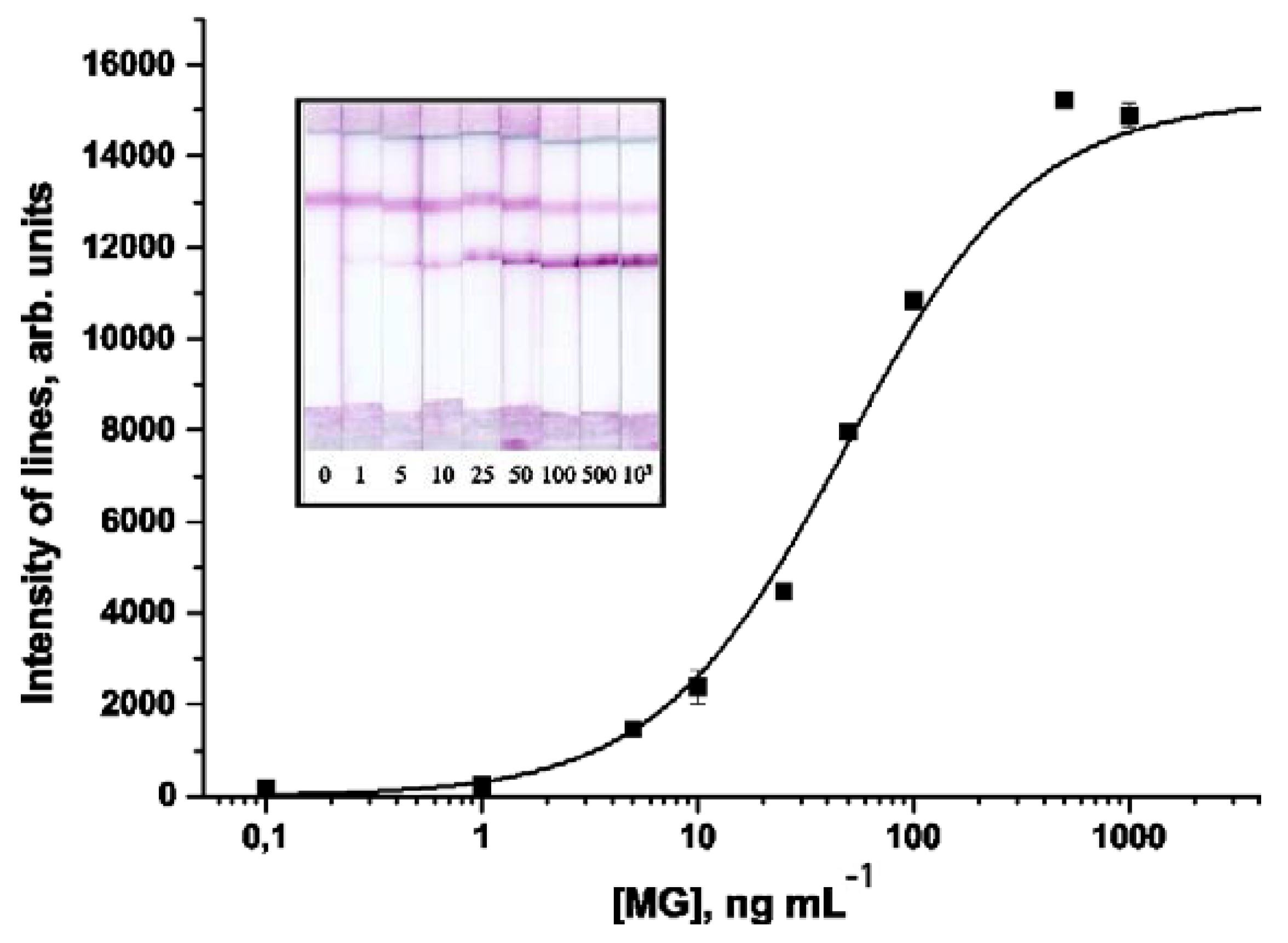
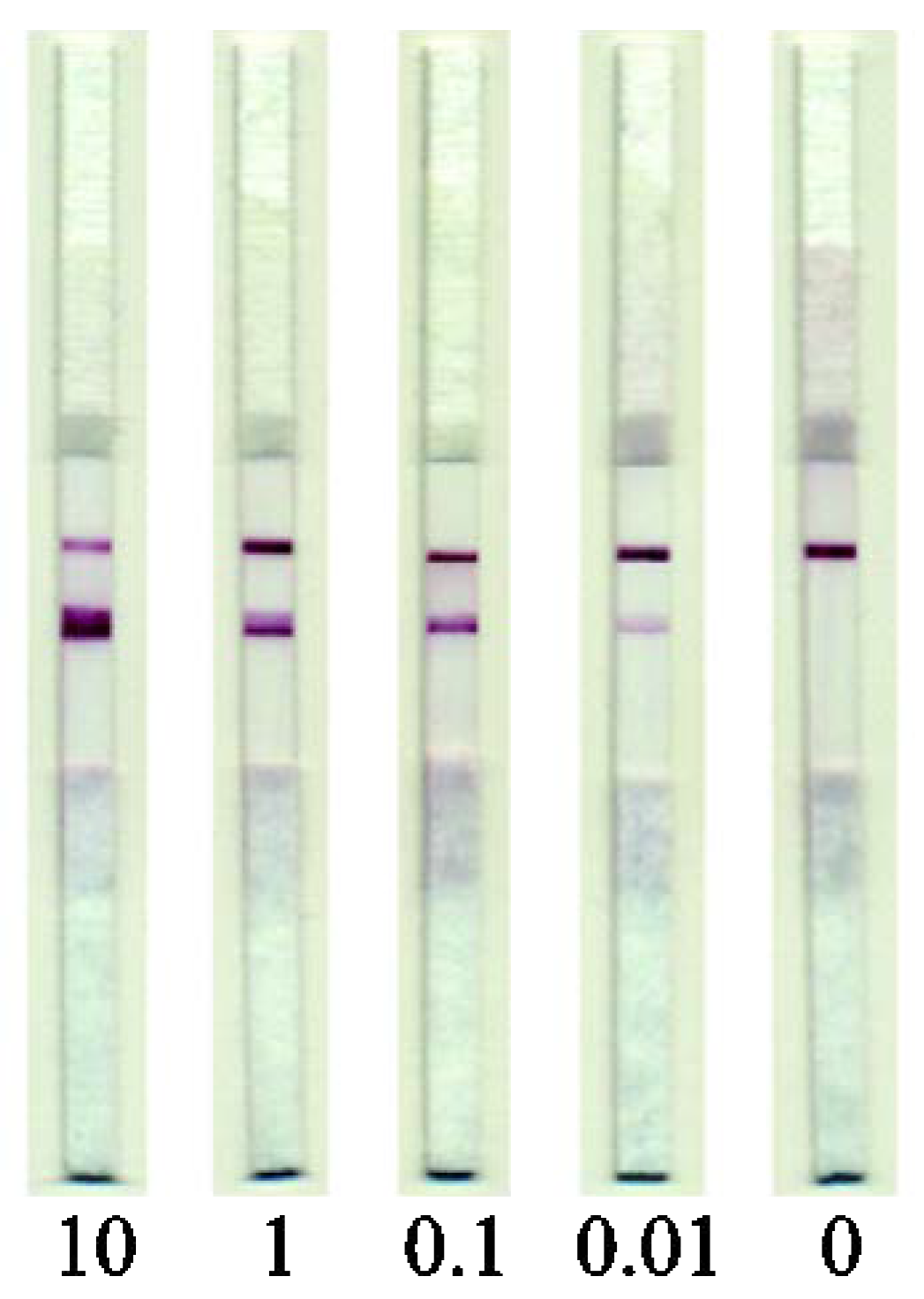
3.3. Development of the LFIA of MG
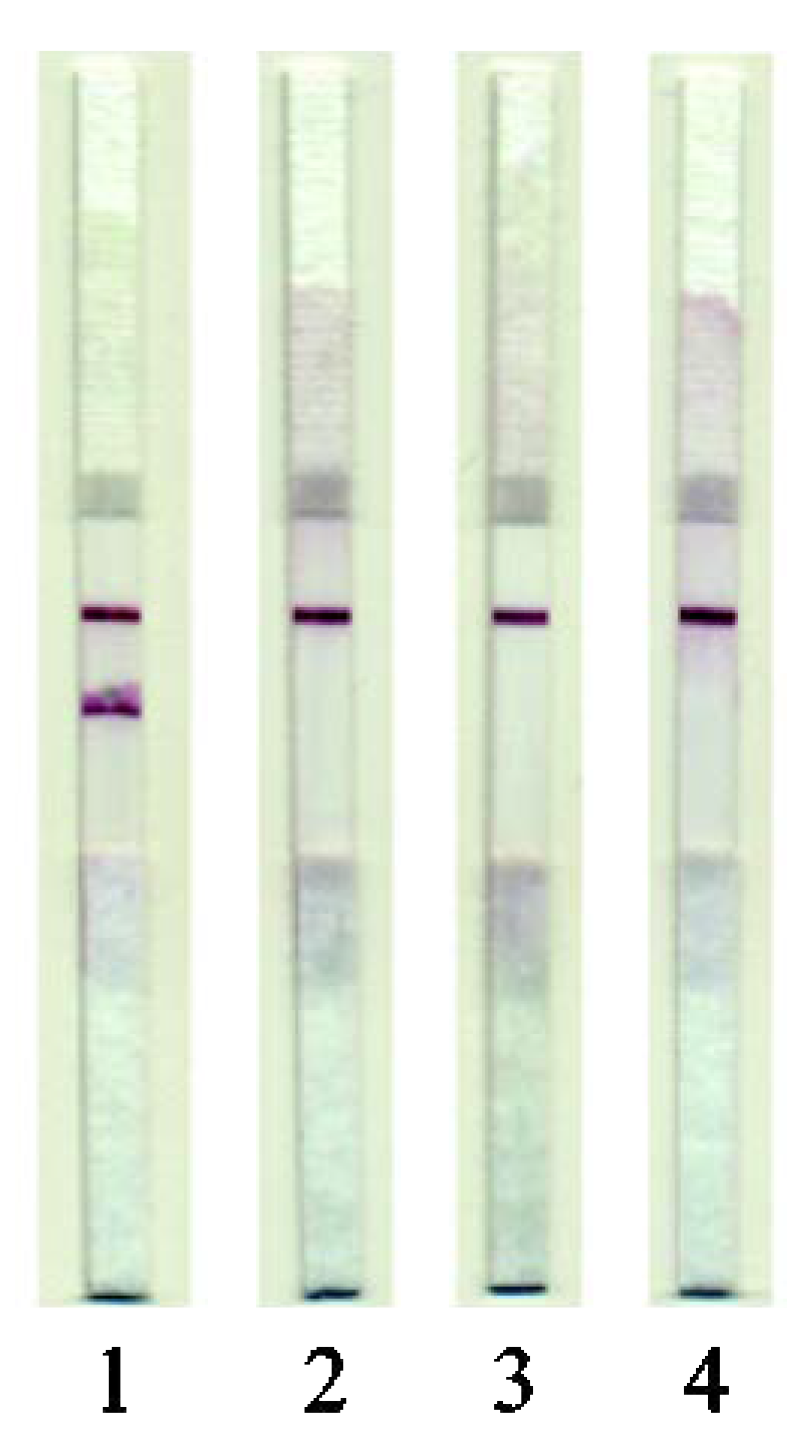
3.4. LFIA of Meat Samples
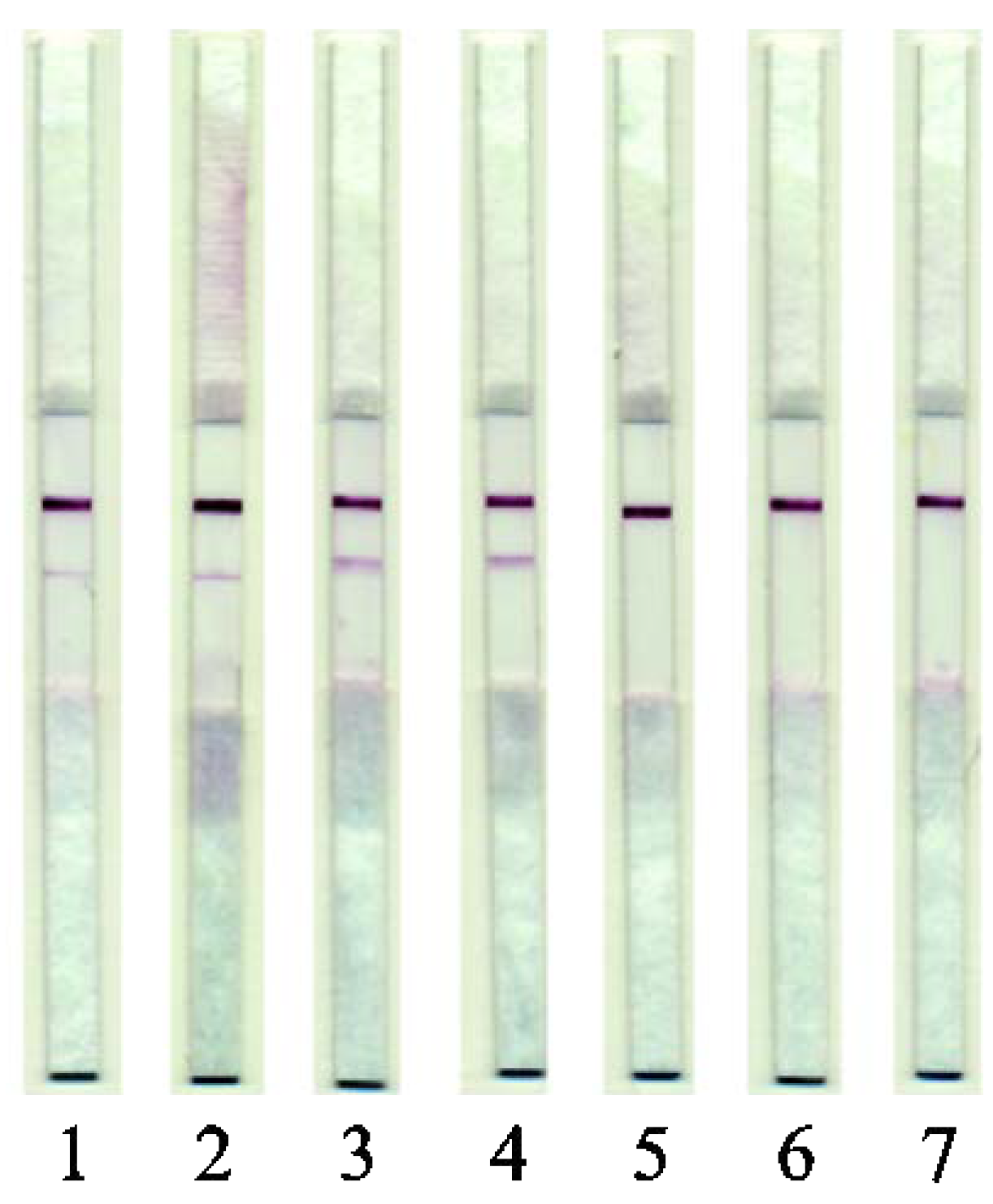
3.5. LFIA of Raw Meat
3.6. LFIA of Cooked Meat
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavin, C.; Cottenet, G.; Cooper, K.M.; Zbinden, P. Meat vulnerabilities to economic food adulteration require new analytical solutions. CHIMIA Int. J. Chem. (Aarau) 2018, 72, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Zia, Q.; Alawami, M.; Mokhtar, N.F.K.; Mohd, R.; Nhari, H.R.; Hanish, I. Current analytical methods for porcine identification in meat and meat products. Food Chem. 2020, 324, 126664. [Google Scholar] [CrossRef] [PubMed]
- Premanandh, J.; Bin Salem, S. Progress and challenges associated with Halal authentication of consumer packaged goods. J. Sci. Food Agric. 2017, 97, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Ballin, N.Z. Authentication of meat and meat products. Meat Sci. 2010, 86, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Chernukha, I.M.; Vostrikova, N.L.; Khvostov, D.V.; Zvereva, E.A.; Taranova, N.A.; Zherdev, A.V. Methods of identification of muscle tissue in meat products. prerequisites for creating a multi–level control system. Theory Pract. Meat Process. 2019, 4, 32–40. [Google Scholar] [CrossRef]
- Al-Jowder, O.; Casuscelli, F.; Defernez, M.; Kemsley, E.K.; Wilson, R.H.; Colquhoun, I.J. High resolution NMR studies of meat composition and authenticity. In Magnetic Resonance in Food Science: A View to the Future, 1st ed.; Webb, G.A., Belton, P.S., Gil, A.M., Delgadillo, I., Eds.; Royal Society of Chemistry: Cambridge, UK, 2001; pp. 232–238. [Google Scholar]
- Jakes, W.; Gerdova, A.; Defernez, M.; Watson, A.D.; McCallum, C.; Limer, E.; Colquhoun, I.J.; Williamson, D.C.; Kemsley, E.K. Authentication of beef versus horse meat using 60 MHz 1H NMR spectroscopy. Food Chem. 2015, 175, 1–9. [Google Scholar] [CrossRef]
- Mandli, J.; El Fatimi, I.; Seddaoui, N.; Amine, A. Enzyme immunoassay (ELISA/immunosensor) for a sensitive detection of pork adulteration in meat. Food Chem. 2018, 255, 380–389. [Google Scholar] [CrossRef]
- Soares, S.; Amaral, J.S.; Oliveira, M.B.P.P.; Mafra, I. A SYBR Green real-time PCR assay to detect and quantify pork meat in processed poultry meat products. Meat Sci. 2013, 94, 115–120. [Google Scholar] [CrossRef]
- Sentandreu, M.A.; Sentandreu, E. Authenticity of meat products: Tools against fraud. Food Res. Int. 2014, 60, 19–29. [Google Scholar] [CrossRef]
- Magiati, M.; Myridaki, V.M.; Christopoulos, T.K.; Kalogianni, D.P. Lateral flow test for meat authentication with visual detection. Food Chem. 2019, 274, 803–807. [Google Scholar] [CrossRef]
- Yin, R.; Sun, Y.; Wang, K.; Feng, N.; Zhang, H.; Xiao, M. Development of a PCR-based lateral flow strip assay for the simple, rapid, and accurate detection of pork in meat and meat products. Food Chem. 2020, 318, 126541. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, L.; Di Nardo, F.; Cavalera, S.; Giovannoli, C.; Baggiani, C. Multiplex lateral flow immunoassay: An overview of strategies towards high-throughput point-of-need testing. Biosensors 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Dzantiev, B.B.; Byzova, N.A.; Urusov, A.E.; Zherdev, A.V. Immunochromatographic methods in food analysis. TrAC Trends Anal. Chem. 2014, 55, 81–93. [Google Scholar] [CrossRef]
- Chen, F.C.; Hsieh, Y.-H.P. Porcine troponin I: A thermostable species marker protein. Meat Sci. 2002, 61, 55–60. [Google Scholar] [CrossRef]
- Masiri, J.; Benoit, L.; Barrios-Lopez, B.; Thienes, C.; Meshgi, M.; Agapov, A.; Dobritsa, A.; Nadala, C.; Samadpour, M. Development and validation of a rapid test system for detection of pork meat and collagen residues. Meat Sci. 2016, 121, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Depamede, S.N. Development of a rapid immunodiagnostic test for pork components in raw beef and chicken meats: A preliminary study. Media Peternak. 2011, 34, 83–87. [Google Scholar] [CrossRef]
- Kuswandi, B.; Gani, A.A.; Ahmad, M. Immuno strip test for detection of pork adulteration in cooked meatballs. Food Biosci. 2017, 19, 1–6. [Google Scholar] [CrossRef]
- Joseph, P.; Suman, S.P.; Li, S.; Claus, J.R.; Fontaine, M.; Steinke, L. Primary structure of turkey myoglobin. Food Chem. 2011, 129, 175–178. [Google Scholar] [CrossRef]
- Ragucci, S.; Russo, R.; Landi, N.; Valletta, M.; Chambery, A.; Esposito, S.; Raundrup, K.; Di Maro, A. Muskox myoglobin: Purification, characterization and kinetics studies compared with cattle and water buffalo myoglobins. J. Sci. Food Agric. 2019, 99, 6278–6286. [Google Scholar] [CrossRef]
- Kotoura, S.; Murakami-Yamaguchi, Y.; Kizu, K.; Nakamura, M.; Fuchu, H.; Miake, K.; Sugiyama, M.; Narita, H. Establishment of a sandwich ELISA for the determination of beef content in processed foods by using monoclonal antibodies to myoglobin. Food Agric. Immunol. 2012, 23, 289–301. [Google Scholar] [CrossRef]
- Chan, K.W.; Lo, C.; Chu, C.S.; Chin, L.T.; Wang, Y.T.; Yang, W.C. Development of a colloidal gold-based immunochromatographic test strip for detection of cetacean myoglobin. J. Vis. Exp. 2016, 113, e53433. [Google Scholar] [CrossRef]
- Giaretta, N.; Di Giuseppe, A.M.A.; Lippert, M.; Parente, A.; Di Maro, A. Myoglobin as marker in meat adulteration: A UPLC method for determining the presence of pork meat in raw beef burger. Food Chem. 2013, 141, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Di Giuseppe, A.M.A.; Giarretta, N.; Lippert, M.; Severino, V.; Di Maro, A. An improved UPLC method for the detection of undeclared horse meat addition by using myoglobin as molecular marker. Food Chem. 2015, 169, 241–245. [Google Scholar] [CrossRef]
- Biacore, A.B. Biacore X Handbook; Biacore AB: Uppsala, Sweden, 2001; p. 108. [Google Scholar]
- Bayer, E.A.; Wilchek, M. Protein biotinylation. Methods Enzymol. 1990, 184, 138–160. [Google Scholar] [CrossRef]
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Zvereva, E.A.; Hendrickson, O.D.; Zherdev, A.V.; Dzantiev, B.B. Immunochromatographic tests for the detection of microcystin-LR toxin in water and fish samples. Anal. Methods 2020, 12, 392–400. [Google Scholar] [CrossRef]
- Zvereva, E.A.; Kovalev, L.I.; Ivanov, A.V.; Kovaleva, M.A.; Zherdev, A.V.; Shishkin, S.S.; Lisitsyn, A.B.; Chernukha, I.M.; Dzantiev, B.B. Enzyme immunoassay and proteomic characterization of troponin I as a marker of mammalian muscle compounds in raw meat and some meat products. Meat Sci. 2015, 105, 46–52. [Google Scholar] [CrossRef]
- Wild, D.G. Immunoassay Handbook: Theory and Applications of Ligand Binding, ELISA and Related Techniques, 4th ed.; Elsevier Science: Amsterdam, The Netherlands, 2013; 1036p. [Google Scholar]
- Lo, C.; Chin, L.T.; Chu, C.S.; Wang, Y.T.; Chan, K.W.; Yang, W.C. Rapid immune colloidal gold strip for cetacean meat restraining illegal trade and consumption: Implications for conservation and public health. PLoS ONE 2013, 8, e60704. [Google Scholar] [CrossRef]
- Safenkova, I.; Zherdev, A.; Dzantiev, B. Factors influencing the detection limit of the lateral-flow sandwich immunoassay: A case study with potato virus X. Anal. Bioanal. Chem. 2012, 403, 1595–1605. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press, Elsevier: Amsterdam, The Netherlands, 2008; 1200p. [Google Scholar]
- Zvereva, E.A.; Zherdev, A.V.; Xu, C.; Dzantiev, B.B. Highly sensitive immunochromatographic assay for qualitative and quantitative control of beta-agonist salbutamol and its structural analogs in foods. Food Control 2018, 86, 50–58. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | ka, M−1 × c−1 | kd, c−1 | KA, M−1 | KD, M | χ2 |
|---|---|---|---|---|---|
| 1B4 | (2.0 ± 0.5) × 105 | (7.1 ± 1.6) × 10−5 | (3.6 ± 1.0) × 109 | (4.7 ± 1.6) × 10−10 | 0.104 |
| 4E2cc | (2.4 ± 0.6) × 105 | (5.1 ± 1.7) × 10−5 | (7.6 ± 3.2) × 109 | (2.9 ± 1.1) × 10−10 | 0.842 |
| 7C3 | (6.5 ± 3.4)·× 105 | (3.2 ± 0.8) × 10−4 | (2.8 ± 1.0) × 109 | (1.8 ± 1.3) × 10−9 | 0.158 |
| MyoA6 | (6.3 ± 1.4) × 104 | (2.8 ± 0.1) × 10−4 | (2.2 ± 0.5) × 108 | (5.9 ± 1.6) × 10−9 | 0.196 |
| MyoB1 | (5.0 ± 1.2) × 104 | (3.6 ± 0.5) × 10−3 | (1.4 ± 0.3) × 107 | (9.6 ± 3.4) × 10−8 | 0.227 |
| MyoB12 | (3.7 ± 0.8) × 104 | (3.6 ± 0.5) × 10−3 | (7.1 ± 1.6) × 106 | (1.8 ± 0.3) × 10−7 | 0.122 |
| MyoD4 | (1.2 ± 0.3) × 105 | (7.0 ± 2.0) × 10−5 | (1.9 ± 0.3) × 109 | (6.2 ± 1.3) × 10−10 | 0.287 |
| MyoE4 | (1.5 ± 0.2) × 105 | (5.1 ± 0.8) × 10−4 | (3.4 ± 0.7) × 108 | (3.9 ± 1.0) × 10−9 | 0.124 |
| MyoF5 | (1.9 ± 0.4) × 105 | (6.2 ± 2.1) × 10−5 | (6.1 ± 3.7) × 109 | (4.1 ± 1.8) × 10−10 | 0.085 |
| MyoF7 | (1.4 ± 0.2) × 105 | (1.2 ± 0.3) × 10−3 | (1.8 ± 0.7) × 108 | (9.6 ± 3.3) × 10−9 | 0.138 |
| MyoH9 | (8.7 ± 3.1) × 104 | (4.6 ± 1.5) × 10−4 | (3.2 ± 1.4) × 108 | (7.3 ± 4.1) × 10−9 | 0.265 |
| MyoH11 | (3.0 ± 0.9) × 105 | (8.3 ± 0.6) × 10−4 | (3.9 ± 1.5) × 108 | (4.8 ± 1.2) × 10−9 | 0.091 |
| Immobilized/Biotinylated (AuNPs-Labeled) Antibody | Detection Limit in the ELISA of MG, ng mL−1 | Detection Limit in the LFIA of MG, ng mL−1 | Color Intensity of the Analytical Zone, % |
|---|---|---|---|
| 7C3/MyoA6 | 9.2 | 5.0 | 100 |
| 7C3/MyoD4 | 9.1 | 24.1 | 75 |
| 7C3/MyoE4 | 12.8 | 35.6 | 35 |
| 7C3/MyoF5 | 10.8 | 15.6 | 78 |
| 7C3/MyoH9 | 13.7 | 10.2 | 79 |
| 7C3/1B4 | 14.6 | 19.4 | 53 |
| 7C3/4E2cc | 2.0 | 21.9 | 31/19 |
| 4E2cc/7C3 | 23.0 | 4.5 | 39/25 |
| Method No. | Sample Preparation Procedure | MG, mg g−1 of Pork |
|---|---|---|
| 1 | Extraction with PBS, containing 0.1% Triton X-100 and 0.5 M KCl, followed by an incubation at 100 °C and centrifugation for 5 min at 7000× g | 0.03 ± 0.01 |
| 2 | Extraction with PBS, containing 0.1% Triton X-100 and 0.5 M KCl, followed by a sonication and centrifugation for 10 min at 5000× g | 0.25 ± 0.02 |
| 3 | Extraction with PBST | 0.13 ± 0.03 |
| 4 | Extraction with PBST followed by a centrifugation for 5 min at 5000× g | 0.40 ± 0.03 |
| Method | Analyte | Detection Limit, % | Extraction Time, min | Time of Analysis, min | Reference |
|---|---|---|---|---|---|
| ELISA | IgG | 0.01/0.1 | >40 | >700/45 | [8] |
| Immunosensor | IgG | 0.01 | >40 | 20 | [8] |
| PCR | DNA | 5 | 235 | 80 | [9] |
| Real-time PCR | DNA | 0.1 | 235 | 28 | [9] |
| UPLC | Proteins profile | 5 | 40 | 30 | [23] |
| LFIA | DNA | 0.02 | >60 | 15 | [11] |
| LFIA | DNA | 0.01 | >90 | 5 | [12] |
| LFIA | PSA* | 0.01 | 25 | 15 | [16] |
| LFIA | IgG | 0.1 | 65 | 15 | [18] |
| LFIA | MG | 0.01 | 20 | 15 | Present work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zvereva, E.A.; Byzova, N.A.; Hendrickson, O.D.; Popravko, D.S.; Belichenko, K.A.; Dzantiev, B.B.; Zherdev, A.V. Immunochromatographic Detection of Myoglobin as a Specific Biomarker of Porcine Muscle Tissues in Meat Products. Appl. Sci. 2020, 10, 7437. https://doi.org/10.3390/app10217437
Zvereva EA, Byzova NA, Hendrickson OD, Popravko DS, Belichenko KA, Dzantiev BB, Zherdev AV. Immunochromatographic Detection of Myoglobin as a Specific Biomarker of Porcine Muscle Tissues in Meat Products. Applied Sciences. 2020; 10(21):7437. https://doi.org/10.3390/app10217437
Chicago/Turabian StyleZvereva, Elena A., Nadezhda A. Byzova, Olga D. Hendrickson, Demid S. Popravko, Ksenia A. Belichenko, Boris B. Dzantiev, and Anatoly V. Zherdev. 2020. "Immunochromatographic Detection of Myoglobin as a Specific Biomarker of Porcine Muscle Tissues in Meat Products" Applied Sciences 10, no. 21: 7437. https://doi.org/10.3390/app10217437
APA StyleZvereva, E. A., Byzova, N. A., Hendrickson, O. D., Popravko, D. S., Belichenko, K. A., Dzantiev, B. B., & Zherdev, A. V. (2020). Immunochromatographic Detection of Myoglobin as a Specific Biomarker of Porcine Muscle Tissues in Meat Products. Applied Sciences, 10(21), 7437. https://doi.org/10.3390/app10217437