A Framework for Enhancing Big Data Integration in Biological Domain Using Distributed Processing




Abstract
1. Introduction
2. Background
2.1. Ontology
2.2. Gene Ontology
- A gene is a region of DNA that encodes instructions for the cell to make a large molecule or potentially multiple different macromolecules.
- A macromolecule is a gene product that is generated according to the gene instructions; it can be a protein or a non-coding RNA.
- A gene product can work as a molecular machine, such as by performing a chemical action that is called an activity.
- A macromolecular complex is a set of gene products from different genes combined to represent a larger molecular machine.
3. Literature Review
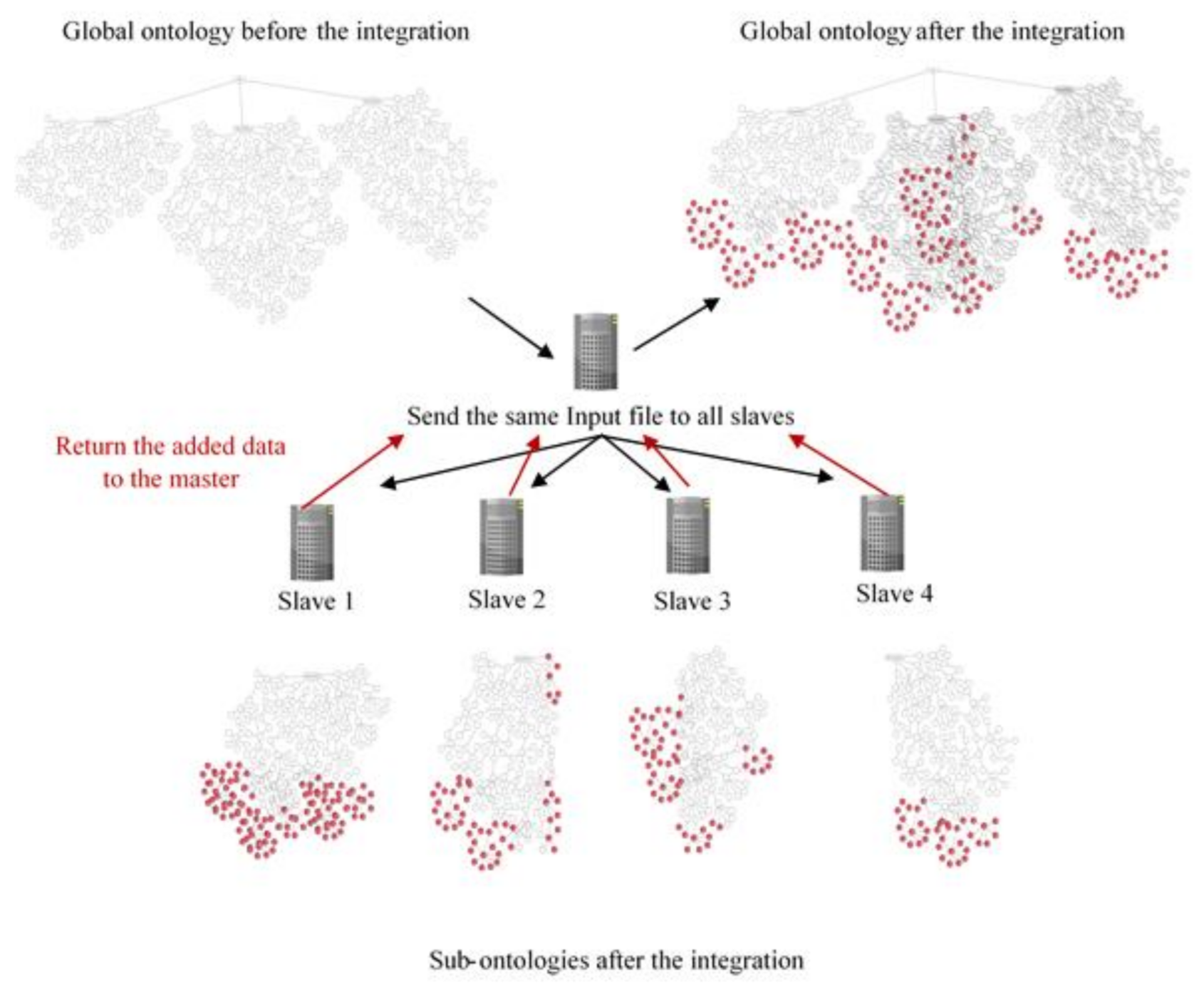
4. Methodology
- Add: each slave loads its sub-ontology then adds any related data from the input file. Data added to the sub-ontology is also added to the global one.
- Compare: the global ontology resulting from the distributed integration is compared to the global ontology resulting from doing the integration locally.
4.1. Environmental Setup
- Equipment:
- Dell PowerEdge T620 server with SATA (7.2K) hard drive is and with a VMware Workstation Pro 14 software to create a set of six virtual machines (VM); each machine runs on Ubuntu 16.04 LTS, Intel® Xeon® processor E5-2600 product family × 4 processors. One VM works as a master with 14 GB of RAM and the rest work as slaves with 8 GB.
- Samba file and print service, which is an open-source implementation of the Server Message Block/Common Internet File System (SMB/CIFS) protocols that provides the sharing of files and printers between master and slaves.
- Protégé [12] is an open-source ontology editor and knowledge management system. We will use it to validate or test the logical consistency of all ontologies.
- Programming language:
- JAVA programming language version 1.8.
- Libraries:
- Semantic measure library and toolkit (SML) [56] to read and process the GO.
- JCIFS library [57] to access and manage shared data on a Samba Server installed on the master node using JAVA.
- Jena is a Java-based programming toolkit.
- Pellet and HermiT are used to check the ontology consistency and identifying subsumption relationships between classes. Pellet reasoner is an open-source based on OWL2 reasoner using Java programming language. It is used with Jena and OWL API libraries. HermiT limitations are based on OWL language.
- Algorithms:
- GO Split Algorithm to generate N GO Splits, where N ranged from 1 to 5, because in our settings we can have 2, 3, or 5 slaves.
- Input:Due to the hardware limitation (hard drive size) in our system, we cannot integrate all the input data; therefore, a sample of input data was selected. Samples are generated based on collecting a line from an input sample file if its gene ID is in the NCBI genes list that has a relation with any gene in GO. To reduce the sample size, one line for each gene ID is taken because some gene IDs are repeated in many lines. Input sample files are:
- gene_info [58]: text file of information that has about 2,013,945 NCBI genes. A sample of 56,603 genes was selected.
- gene2go [59]: text file that reports about 2,070,137 relations between genes from GO and genes from NCBI. A sample of 55,859 relations was selected.
- gene_neighbors [60]: text file that represents neighboring genes for all genes located on a given genomic sequence. A sample of 56,647 relations was selected.
- gene2ensembl [61]: text file of 1,907,407 matches between NCBI genes and Ensembl annotations based on the comparison of RNA and protein features. A sample of 56,647 relations was selected.
- gene2pubmed [62]: text file report that has about 11,165,891 relations to link genes from NCBI to PubMed ID. A sample of 56,044 relations was selected.
- gene2sts [63]: text file report that has about 1,173,647 relations to link genes from NCBI to UniSTS ID. A sample of 56,647 relations was selected.
- gene2accession [64]: text file of 18,142,094 accessions related to GeneID of the genes mentioned in the NCBI gene information file. It contains sequences from the international sequence collaboration, Swiss-Prot, and RefSeq. A sample of 56,498 accessions was selected.
- gene2vega [65]: text file is composed of 84,828 matches between NCBI genes and Vega annotations. A sample of 29,496 matches was selected.
- gene2unigene [66]: text file report that has about 589,221 relations to link genes from NCBI to the UniGene cluster. A sample of 55,891 relations was selected.
Evaluation Measures
- Logical Consistency measure: an ontology is marked as passing if ontology passes the logical consistency test and is marked as failing otherwise. Logical consistency tests are Jena, Pellet, and HermiT tests. The Jena test is done by loading ontology/sub-ontology in a Java program using the Jena library. If it is loaded correctly without any errors, this means the ontology/sub-ontology does not violate any logical consistency. Pellet and HermiT tests are done by loading ontology/sub-ontology in the Protégé program and applying Pellet/HermiT reasoners. If there are no errors, this means the ontology/sub-ontology does not violate Pellet/HermiT logical consistency.
- Equivalence measure: an ontology resulting from the distributed integration is marked as equivalent if it is the same as the ontology resulted from local integration. Otherwise, it is marked as not equivalent. They are equivalent if they have the same ontology size before the integration, number of added items, number of genes after the integration, number of edges, vertices, and roots.
4.2. Test Cases
- Case 1: testing the logical consistency of the resulted ontologies (sub-ontologies and global ones) iteratively after adding each input data source. Logical consistency is checked using Jena Ontology API, Pellet, and HermiT reasoners.
- Case 2: comparing the global ontology resulting from the distributed integration to the global ontology resulting from doing the integration locally. Comparison is based on ontology size before the integration, number of added items, number of added roots, total number of genes after integration, number of edges, vertices, and roots.
5. Results and Discussion
5.1. Test Cases 1 and 2: Big Data Integration Framework
5.1.1. Local Data Integration
5.1.2. Distributed Data Integration
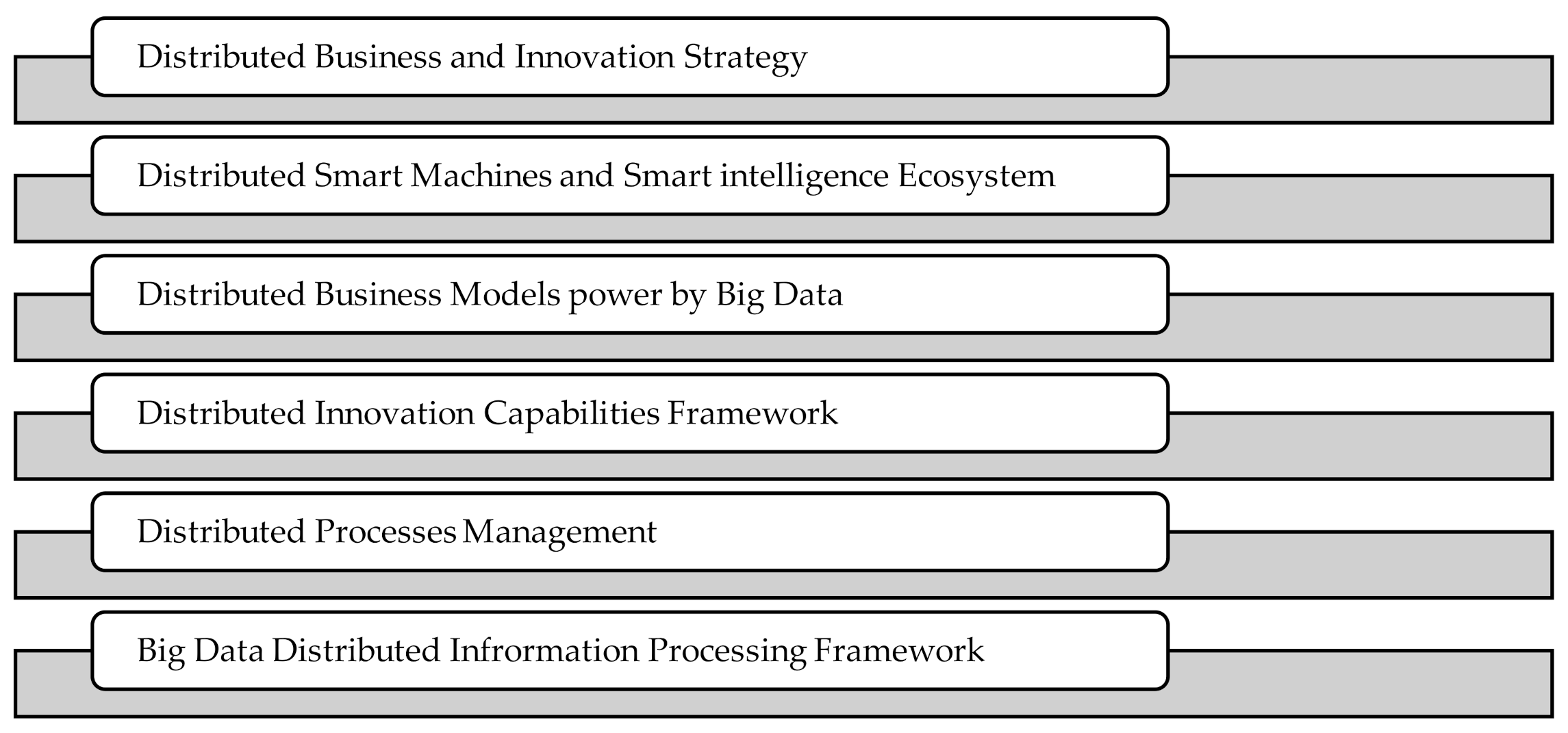
6. Implications of Our Work
- Distributed Business and Innovation Strategy
- Distributed Smart Machines and Smart intelligence Ecosystem
- Distributed Business Models power by Big Data
- Distributed Innovation Capabilities Framework
- Distributed Processes Management
- Big Data Distributed Information Processing Framework
7. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Acronyms
| BDI | Big Data Integration |
| BP | Biological Process |
| CC | Cellular Components |
| DW | Data Warehousing |
| EC | Evidence Code |
| GAF | Gene Association File |
| GO | Gene Ontology |
| GPAD | Gene Product Association Data |
| HPC | High Performance Computing |
| IEA | Inferred from Electronic Annotation |
| MF | Molecular Function |
| OBO | Open Biomedical Ontology |
| ROLAP | Relational Online Analytical Processing |
| RDAG | Rooted Directed Acyclic Graph |
| SML | Semantic Measure Library and Toolkit |
| SSM | Semantic Similarity Measures |
| SMB/CIFS | Server Message Block/Common Internet File System |
| VDI | Virtual Data Integration |
| VM | Virtual Machines |
| OWL | Web Ontology Language |
References
- Sakr, S. Introduction. In Big Data 2.0 Processing Systems: A. Survey; Sakr, S., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–13. [Google Scholar]
- Laney, D. 3-D Data Management: Controlling Data Volume, Velocity, and Variety, META Group Res. Note 6. J. Data Anal. Inf. Process. 2001, 6, 4. [Google Scholar]
- Fan, W.; Bifet, A. Mining big data. ACM SIGKDD Explor. Newsl. 2013, 14, 1–5. [Google Scholar] [CrossRef]
- Abawajy, J. Comprehensive analysis of big data variety landscape. Int. J. Parallel Emergent Distrib. Syst. 2014, 30, 5–14. [Google Scholar] [CrossRef]
- Cordoba, A. Understanding the Predictive Analytics Lifecycle; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Doan, A.; Halevy, A.; Ives, Z. Principles of Data Integration; Elsevier BV: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Arputhamary, B.; Arockiam, L. A Review on Big Data Integration. Int. J. Comput. Appl. 2014, 21–26. Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.742.2276&rep=rep1&type=pdf (accessed on 1 October 2020).
- Dong, X.L.; Srivastava, D. Big data integration. In Proceedings of the 2013 IEEE 29th International Conference on Data Engineering (ICDE), Brisbane, QLD, Australia, 8–11 April 2013; pp. 1245–1248. [Google Scholar]
- Reeve, A. Managing Data in Motion; Elsevier BV: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Hastings, J. Primer on Ontologies. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2016; Volume 1446, pp. 3–13. [Google Scholar]
- Tirmizi, S.H.; Aitken, S.; Moreira, D.A.; Mungall, C.J.; Sequeda, J.F.; Shah, N.H.; Miranker, D.P. Mapping between the OBO and OWL ontology languages. J. Biomed. Semant. 2011, 2, S3. [Google Scholar] [CrossRef]
- Protégé. Available online: https://protege.stanford.edu/products.php (accessed on 20 November 2019).
- Glimm, B.; Horrocks, I.; Motik, B.; Stoilos, G.; Wang, Z. HermiT: An OWL 2 Reasoner. J. Autom. Reason. 2014, 53, 245–269. [Google Scholar] [CrossRef]
- Sirin, E.; Parsia, B.; Grau, B.C.; Kalyanpur, A.; Katz, Y. Pellet: A practical OWL-DL reasoner. J. Web Semant. 2007, 5, 51–53. [Google Scholar] [CrossRef]
- Gaudet, P.; Škunca, N.; Hu, J.C.; Dessimoz, C. Primer on the Gene Ontology. Viruses Hum. Cancer 2016, 1446, 25–37. [Google Scholar] [CrossRef]
- Thomas, P.D. The Gene Ontology and the Meaning of Biological Function. Methods Mol. Biol. 2016, 1446, 15–24. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Ehsani, R.; Drabløs, F. TopoICSim: A new semantic similarity measure based on gene ontology. BMC Bioinform. 2016, 17, 296. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [CrossRef]
- Blake, A.J. MGD: The Mouse Genome Database. Nucleic Acids Res. 2003, 31, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998, 26, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.; Antoshechkin, I.; Bieri, T.; Blasiar, D.; Chan, J.; Chen, W.J.; De La Cruz, N.; Davis, P.; Duesbury, M.; Fang, R.; et al. WormBase: A comprehensive resource for nematode research. Nucleic Acids Res. 2009, 38, D463–D467. [Google Scholar] [CrossRef] [PubMed]
- McQuilton, P.; Pierre, S.E.S.; Thurmond, J. The FlyBase Consortium FlyBase 101—The basics of navigating FlyBase. Nucleic Acids Res. 2011, 40, D706–D714. [Google Scholar] [CrossRef]
- Chisholm, R.L. DictyBase, the model organism database for Dictyostelium discoideum. Nucleic Acids Res. 2006, 34, D423–D427. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2011, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Studer, R.A.; Robinson-Rechavi, M.W.; Dessimoz, C. Resolving the Ortholog Conjecture: Orthologs Tend to Be Weakly, but Significantly, More Similar in Function than Paralogs. PLoS Comput. Biol. 2012, 8, e1002514. [Google Scholar] [CrossRef]
- Guzzi, P.H.; Mina, M.; Guerra, C.; Cannataro, M. Semantic similarity analysis of protein data: Assessment with biological features and issues. Briefings Bioinform. 2011, 13, 569–585. [Google Scholar] [CrossRef]
- Chang, K.; He, B.; Zhang, Z. Toward Large Scale Integration: Building a MetaQuerier over Databases on the Web. In Proceedings of the CIDR 2005 Second Biennial Conference on Innovative Data Systems Research, Asilomar, CA, USA, 4–7 January 2005; pp. 44–55. [Google Scholar]
- Madhavan, J.; Jeffery, S.R.; Cohen, S.; Dong, X.L.; Ko, D.; Yu, C.; Halevy, A. Web-scale data integration: You can only afford to pay as you go. In Proceedings of the Conference on Innovative Data Systems Research (CIDR), Asilomar, CA, USA, 7–10 January 2007; pp. 342–350. [Google Scholar]
- Madhavan, J.; Ko, D.; Kot, Ł.; Ganapathy, V.; Rasmussen, A.; Halevy, A. Google’s Deep Web crawl. Proc. VLDB Endow. 2008, 1, 1241–1252. [Google Scholar] [CrossRef]
- Cafarella, M.J.; Halevy, A.; Wang, D.Z.; Wu, E.; Zhang, Y. Webtables: Exploring the power of tables on the web. Proc. VLDB Endow. 2008, 1, 538–549. [Google Scholar] [CrossRef]
- Pimplikar, R.; Sarawagi, S. Answering table queries on the web using column keywords. Proc. VLDB Endow. 2012, 5, 908–919. [Google Scholar] [CrossRef]
- Gupta, R.; Sarawagi, S. Answering table augmentation queries from unstructured lists on the web. Proc. VLDB Endow. 2009, 2, 289–300. [Google Scholar] [CrossRef]
- Elmeleegy, H.; Madhavan, J.; Halevy, A. Harvesting relational tables from lists on the web. Proc. VLDB Endow. 2009, 2, 1078–1089. [Google Scholar] [CrossRef]
- Sen, S. Integrating XML Data into Multiple Rolap Data Warehouse Schemas. Int. J. Softw. Eng. Appl. 2012, 3, 197–206. [Google Scholar] [CrossRef]
- Schultz, A.; Matteini, A.; Isele, R.; Mendes, P.N.; Bizer, C.; Becker, C. LDIF-a framework for large-scale Linked Data integration. In Proceedings of the 21st International World Wide Web Conference (WWW 2012), Lyon, France, 16–20 April 2012. [Google Scholar]
- Grolinger, K.; Capretz, M.A.; Mezghani, E.; Exposito, E. Knowledge as a Service Framework for Disaster Data Management. In Proceedings of the 2013 Workshops on Enabling Technologies: Infrastructure for Collaborative Enterprises, Hammamet, Tunisia, 17–20 June 2013; pp. 313–318. [Google Scholar]
- Naseer, A.; Laera, L.; Matsutsuka, T. Enterprise BigGraph. In Proceedings of the 2013 46th Hawaii International Conference on System Sciences, Wailea, HI, USA, 7–10 January 2013; pp. 1005–1014. [Google Scholar]
- Bansal, S.K. Towards a Semantic Extract-Transform-Load (ETL) Framework for Big Data Integration. In Procession of the 2014 IEEE International Congress on Big Data, Anchorage, AK, USA, 27 June–2 July 2014; pp. 522–529. [Google Scholar]
- Aggoune, A.; Bouramoul, A.; Kholladi, M.-K. Big data integration: A semantic mediation architecture using summary. In Proceedings of the 2016 2nd International Conference on Advanced Technologies for Signal and Image Processing (ATSIP), Monastir, Tunisia, 21–24 March 2016; pp. 21–25. [Google Scholar]
- Williams, J.W.; Cuddihy, P.; McHugh, J.; Aggour, K.S.; Menon, A.; Gustafson, S.M.; Healy, T. Semantics for Big Data access & integration: Improving industrial equipment design through increased data usability. In Proceedings of the 2015 IEEE International Conference on Big Data (Big Data), Santa Clara, CA, USA, 29 October–1 November 2015; pp. 1103–1112. [Google Scholar]
- Mezghani, E.; Exposito, E.; Drira, K.; Da Silveira, M.; Pruski, C. A Semantic Big Data Platform for Integrating Heterogeneous Wearable Data in Healthcare. J. Med Syst. 2015, 39, 185. [Google Scholar] [CrossRef]
- Ostrowski, D.; Rychtyckyj, N.; Macneille, P.; Kim, M. Integration of Big Data Using Semantic Web Technologies. In Proceedings of the 2016 IEEE Tenth International Conference on Semantic Computing (ICSC), Laguna Hills, CA, USA, 4–6 February 2016; pp. 382–385. [Google Scholar]
- Sandhya, H.; Roy, M.M. Data Integration of Heterogeneous Data Sources Using QR Decomposition. In Advances in Intelligent Systems and Computing; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2015; Volume 385, pp. 333–344. [Google Scholar]
- Legaz-García, M.D.C.; Miñarro-Giménez, J.A.; Menárguez-Tortosa, M.; Fernández-Breis, J.T. Generation of open biomedical datasets through ontology-driven transformation and integration processes. J. Biomed. Semant. 2016, 7, 32. [Google Scholar] [CrossRef]
- Bortoli, S.; Bouquet, P.; Pompermaier, F.; Molinari, A. Semantic big data for tax assessment. In Proceedings of the International Workshop on Software Engineering in Healthcare Systems—SEHS ’16, Austin, TX, USA, 14–15 May 2016; Association for Computing Machinery (ACM): New York, NY, USA, 2016; pp. 1–6. [Google Scholar]
- Sheokand, V.; Singh, V. Modeling Data Heterogeneity Using Big DataSpace Architecture. In Software Engineering in Intelligent Systems; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2016; Volume 452, pp. 259–268. [Google Scholar]
- Iyappan, A.; Kawalia, S.B.; Raschka, T.; Hofmann-Apitius, M.; Senger, P. NeuroRDF: Semantic integration of highly curated data to prioritize biomarker candidates in Alzheimer’s disease. J. Biomed. Semant. 2016, 7, 45. [Google Scholar] [CrossRef][Green Version]
- Obitko, M.; Jirkovský, V. Big Data Semantics in Industry 4.0. In Lecture Notes in Computer Science; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2015; pp. 217–229. [Google Scholar]
- Bansal, S.K.; Kagemann, S. Integrating Big Data: A Semantic Extract-Transform-Load Framework. IEEE Comput. 2015, 48, 42–50. [Google Scholar] [CrossRef]
- Abbes, H.; Gargouri, F. Big Data Integration: A MongoDB Database and Modular Ontologies based Approach. Procedia Comput. Sci. 2016, 96, 446–455. [Google Scholar] [CrossRef]
- Jirkovsky, V.; Obitko, M.; Mařík, V. Understanding Data Heterogeneity in the Context of Cyber-Physical Systems Integration. IEEE Trans. Ind. Informatics 2016, 13, 660–667. [Google Scholar] [CrossRef]
- Sevilla, J.; Segura, V.; Podhorski, A.; Guruceaga, E.; Mato, J.M.; Martínez-Cruz, L.A.; Corrales, F.; Rubio, A. Correlation between Gene Expression and GO Semantic Similarity. IEEE ACM Trans. Comput. Biol. Bioinform. 2005, 2, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Almasoud, A.M.; Al-Khalifa, H.S.; Al-Salman, A.M.S. Handling Big Data Scalability in Biological Domain Using Parallel and Distributed Processing: A Case of Three Biological Semantic Similarity Measures. BioMed Res. Int. 2019, 2019, 6750296. [Google Scholar] [CrossRef]
- Jena Ontology API—Apache Jena. Available online: https://jena.apache.org/documentation/ontology/ (accessed on 11 November 2019).
- Harispe, S.; Ranwez, S.; Janaqi, S.; Montmain, J. The semantic measures library and toolkit: Fast computation of semantic similarity and relatedness using biomedical ontologies. Bioinformatics 2013, 30, 740–742. [Google Scholar] [CrossRef]
- JCIFS. Available online: https://jcifs.samba.org/ (accessed on 5 May 2018).
- Gene_Info. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene_info.gz (accessed on 11 November 2019).
- Gene2go. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2go.gz (accessed on 11 November 2019).
- Gene_Neighbors. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene_neighbors.gz (accessed on 11 November 2019).
- “gene2ensembl”. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2ensembl.gz (accessed on 11 November 2019).
- Gene2pubmed. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2pubmed.gz (accessed on 11 November 2019).
- Gene2sts. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2sts (accessed on 11 November 2019).
- Gene2accession. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2accession.gz (accessed on 11 November 2019).
- Gene2vega. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2vega.gz (accessed on 11 November 2019).
- Gene2unigene. Available online: Ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2unigene (accessed on 11 November 2019).
- Lytras, M.D.; Raghavan, V.; Damiani, E. Big Data and Data Analytics Research. Int. J. Semantic Web Inf. Syst. 2017, 13, 1–10. [Google Scholar] [CrossRef]
- Visvizi, A.; Lytras, M.D. Rescaling and refocusing smart cities research: From mega cities to smart villages. J. Sci. Technol. Policy Manag. 2018, 9, 134–145. [Google Scholar] [CrossRef]
- Spruit, M.; Lytras, M.D. Applied data science in patient-centric healthcare: Adaptive analytic systems for empowering physicians and patients. Telemat. Inform. 2018, 35, 643–653. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ontology Size before | Sample Size | Added Items | Added Roots | Ontology Size after | Edges | Vertices | Roots | |
|---|---|---|---|---|---|---|---|---|
| Original GO | 36,639 | 36,639 | 0 | 0 | 36,639 | 71,577 | 36,639 | 3 |
| gene2go | 36,639 | 55,859 | 55,964 | 1 | 92,604 | 239,470 | 148,568 | 4 |
| gene_info | 92,604 | 56,603 | 55,964 | 1 | 148,569 | 295,435 | 148,569 | 5 |
| gene_neighbors | 148,569 | 56,647 | 55,964 | 1 | 204,534 | 407,364 | 204,534 | 6 |
| gene2pubmed | 204,534 | 56,044 | 55,964 | 1 | 260,499 | 519,293 | 260,499 | 7 |
| gene2ensembl | 260,499 | 56,647 | 55,964 | 1 | 316,464 | 631,222 | 316,464 | 8 |
| gene2sts | 316,464 | 56,647 | 56,647 | 1 | 373,112 | 744,517 | 403,954 | 9 |
| gene2accession | 373,112 | 18,142,094 | 0 | 0 | 373,112 | 744,517 | 403,954 | 9 |
| gene2unigene | 373,112 | 589221 | 0 | 0 | 373,112 | 744,517 | 403,954 | 9 |
| gene2vega | 373,112 | 84828 | 0 | 0 | 373,112 | 744,517 | 403,954 | 9 |
| Jena Ontology API | HermiT Reasoner | Pellet Reasoner | |
|---|---|---|---|
| Original GO | pass | pass | pass |
| gene2go | pass | pass | pass |
| gene_info | pass | pass | pass |
| gene_neighbors | pass | pass | pass |
| gene2pubmed | pass | pass | pass |
| gene2ensembl | pass | pass | pass |
| gene2sts | pass | pass | pass |
| Ontology Size before | Added Items | Added Roots | Skipped Items | Overlapped Item | Ontology Size after | Edges | Vertices | Roots | |
|---|---|---|---|---|---|---|---|---|---|
| Results of adding gene2go | |||||||||
| Master | 36,639 | 55,964 | 1 | 1 | 12,350 | 92,604 | 239,470 | 148,568 | 4 |
| Slave 1 | 14,281 | 8,279 | 1 | 47,743 | 12,350 | 22,561 | 59,222 | 32,433 | 434 |
| Slave 2 | 12,343 | 16,023 | 1 | 39,999 | 12,350 | 28,367 | 70,995 | 45,036 | 692 |
| Slave 3 | 15,827 | 44,012 | 1 | 12,010 | 12,350 | 59,840 | 156,785 | 104,797 | 123 |
| Results of adding gene info | |||||||||
| Master | 92,604 | 55,964 | 1 | 1 | 12,350 | 148,569 | 295,435 | 148,569 | 5 |
| Slave 1 | 22,561 | 8,279 | 1 | 48,369 | 12,350 | 30,841 | 67,502 | 32,434 | 435 |
| Slave 2 | 28,367 | 16,023 | 1 | 40,625 | 12,350 | 44,391 | 87,019 | 45,037 | 693 |
| Slave 3 | 59,840 | 44,012 | 1 | 12,636 | 12,350 | 103,853 | 200,798 | 104,798 | 124 |
| Results of adding gene_ neighbors | |||||||||
| Master | 148,569 | 55,964 | 1 | 1 | 12,349 | 204,534 | 407,364 | 204,534 | 6 |
| Slave 1 | 30,841 | 8,279 | 1 | 48,369 | 12,349 | 39,121 | 84,061 | 40,714 | 436 |
| Slave 2 | 44,391 | 16,023 | 1 | 40,625 | 12,349 | 60,415 | 119,066 | 61,061 | 694 |
| Slave 3 | 103,853 | 44,012 | 1 | 12,636 | 12,349 | 147,866 | 288,823 | 148,811 | 125 |
| Results of adding gene2pubmed | |||||||||
| Master | 204,534 | 55,964 | 1 | 1 | 12,349 | 260,499 | 519,293 | 260,499 | 7 |
| Slave 1 | 39,121 | 8,279 | 1 | 48,369 | 12,349 | 47,401 | 100,620 | 48,994 | 437 |
| Slave 2 | 60,415 | 16,023 | 1 | 40,625 | 12,349 | 76,439 | 151,113 | 77,085 | 695 |
| Slave 3 | 147,866 | 44,012 | 1 | 12,636 | 12,349 | 191,879 | 376,848 | 192,824 | 126 |
| Results of adding gene2ensembl | |||||||||
| Master | 260,499 | 55,964 | 1 | 1 | 12,349 | 316,464 | 631,222 | 316,464 | 8 |
| Slave 1 | 47,401 | 8,279 | 1 | 48,369 | 12,349 | 55,681 | 117,179 | 57,274 | 438 |
| Slave 2 | 76,439 | 16,023 | 1 | 40,625 | 12,349 | 92,463 | 183,160 | 93,109 | 696 |
| Slave 3 | 191,879 | 44,012 | 1 | 12,636 | 12,349 | 235,892 | 464,873 | 236,837 | 127 |
| Results of adding gene2sts | |||||||||
| Master | 316,464 | 56,647 | 1 | 1 | 11,112 | 373,112 | 744,517 | 403,954 | 9 |
| Slave 1 | 55,681 | 8,220 | 1 | 47,984 | 11,112 | 63,902 | 133,620 | 73,061 | 439 |
| Slave 2 | 92,463 | 15,894 | 1 | 40,310 | 11,112 | 108,358 | 214,949 | 123,144 | 697 |
| Slave 3 | 235,892 | 43,646 | 1 | 12,558 | 11,112 | 279,539 | 552,166 | 311,612 | 127 |
| Jena API | HermiT Reasoner | Pellets Reasoner | Compared to Local Integration | |
|---|---|---|---|---|
| Master | pass | pass | pass | equivalent |
| Slave 1 | pass | pass | pass | NA |
| Slave 2 | pass | pass | pass | NA |
| Slave 3 | pass | pass | pass | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almasoud, A.; Al-Khalifa, H.; Al-salman, A.; Lytras, M. A Framework for Enhancing Big Data Integration in Biological Domain Using Distributed Processing. Appl. Sci. 2020, 10, 7092. https://doi.org/10.3390/app10207092
Almasoud A, Al-Khalifa H, Al-salman A, Lytras M. A Framework for Enhancing Big Data Integration in Biological Domain Using Distributed Processing. Applied Sciences. 2020; 10(20):7092. https://doi.org/10.3390/app10207092
Chicago/Turabian StyleAlmasoud, Ameera, Hend Al-Khalifa, AbdulMalik Al-salman, and Miltiadis Lytras. 2020. "A Framework for Enhancing Big Data Integration in Biological Domain Using Distributed Processing" Applied Sciences 10, no. 20: 7092. https://doi.org/10.3390/app10207092
APA StyleAlmasoud, A., Al-Khalifa, H., Al-salman, A., & Lytras, M. (2020). A Framework for Enhancing Big Data Integration in Biological Domain Using Distributed Processing. Applied Sciences, 10(20), 7092. https://doi.org/10.3390/app10207092