Thermodynamics and Magnetism of YCo5 Compound Doped with Fe and Ni: An Ab Initio Study




Abstract
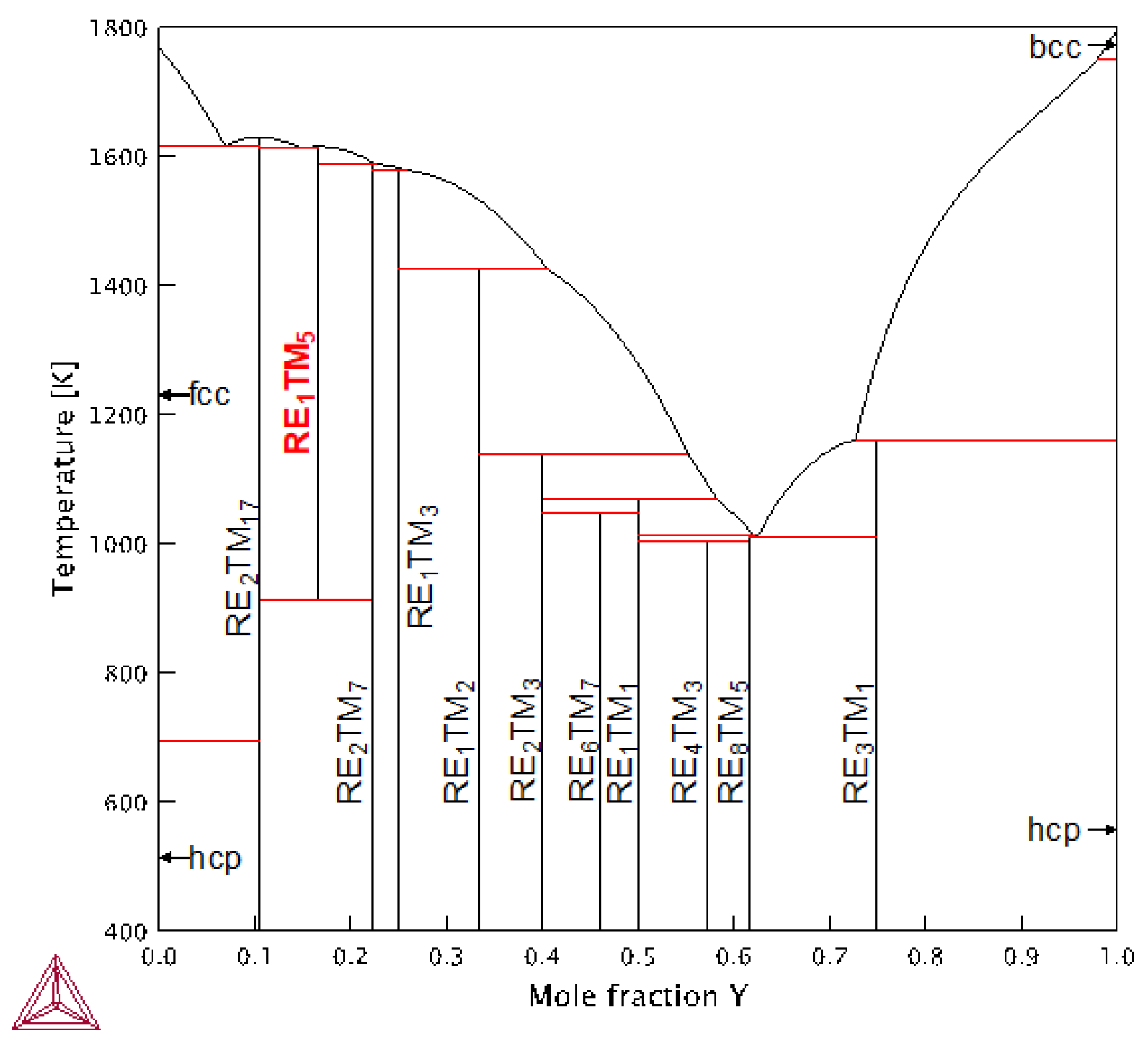
1. Introduction
2. Computational Methods
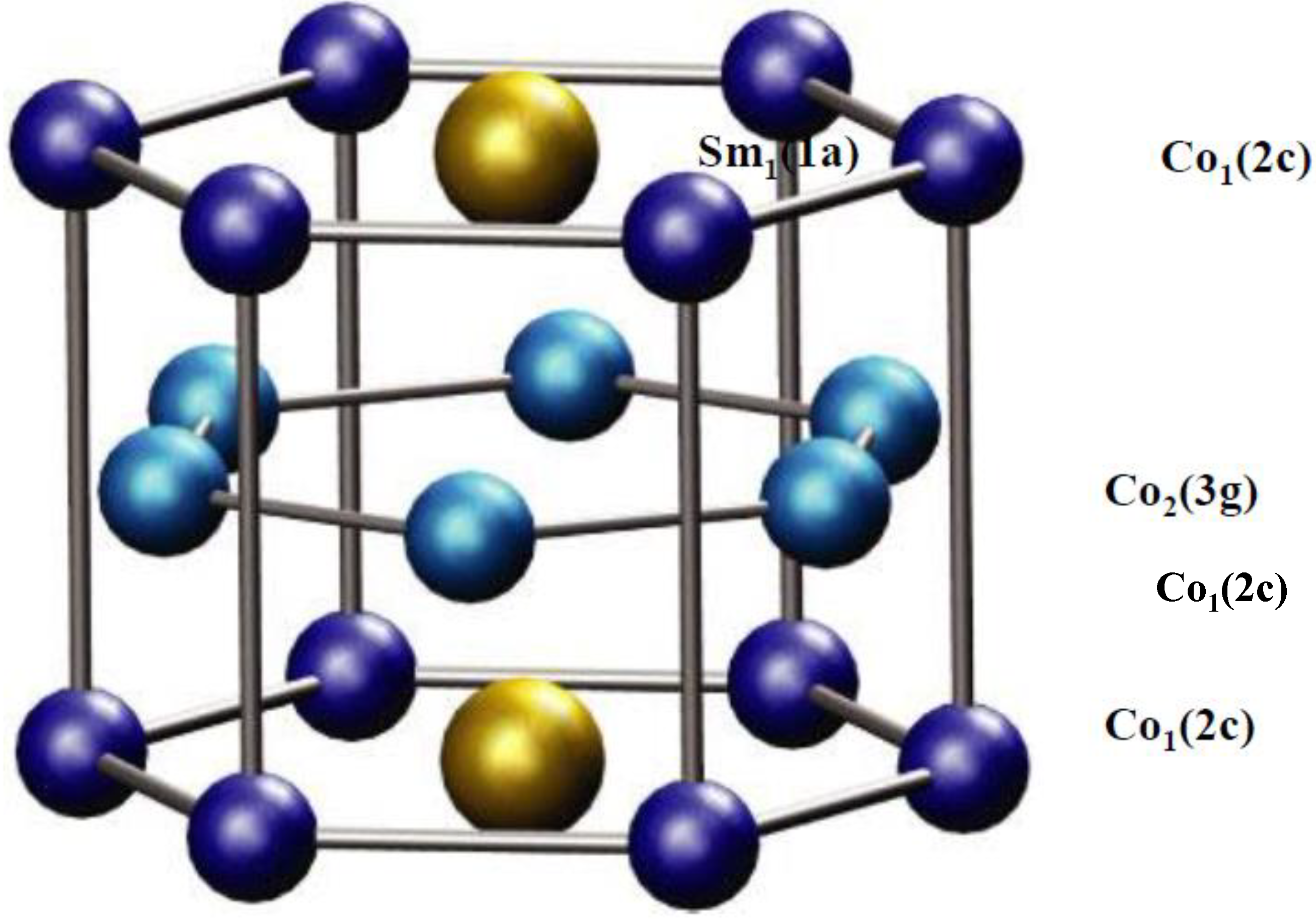
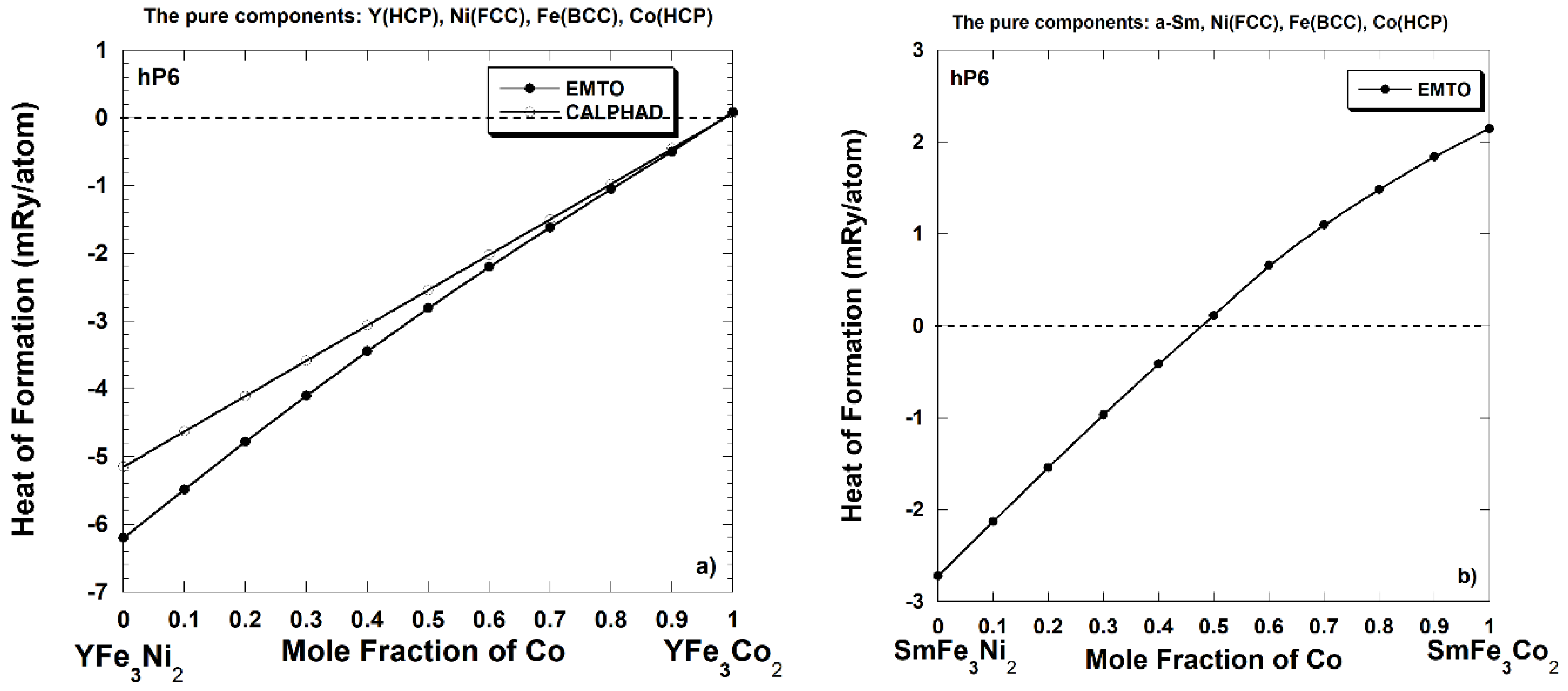
3. Thermodynamics Properties of the Y(Co-Fe-Ni)5 Alloys
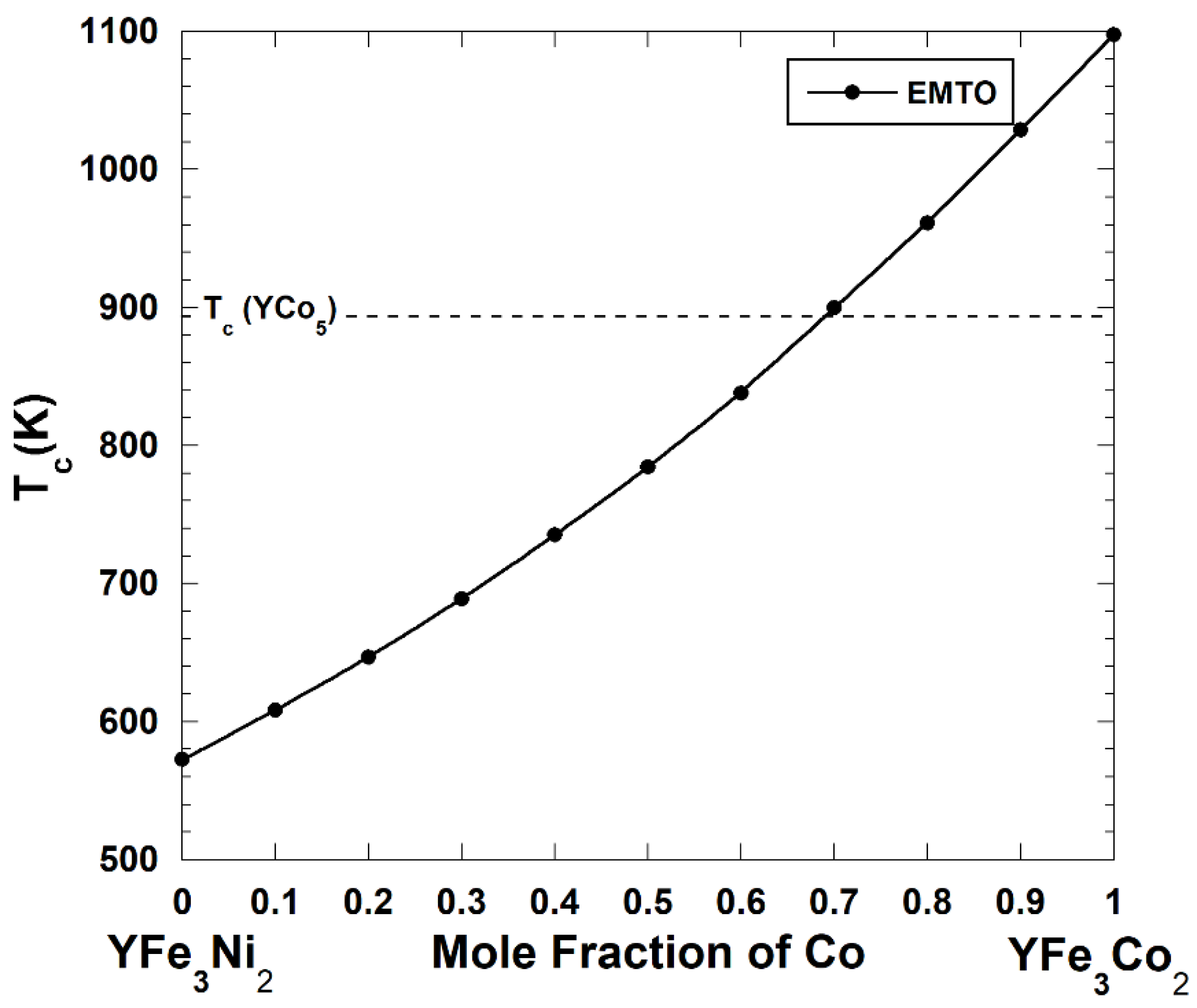
4. Magnetic Properties of the Y(Co-Fe-Ni)5 Alloys
5. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Buschow, K.H.J. New developments in hard magnetic materials. Rep. Prog. Phys. 1991, 54, 1123–1214. [Google Scholar] [CrossRef]
- Buschow, K.H.J.; de Boer, F.R. Physics of Magnetism and Magnetic Materials; Kluwer Academic/Plenum: New York, NY, USA, 2004. [Google Scholar]
- Söderlind, P.; Landa, A.; Locht, I.L.M.; Åberg, D.; Kvashnin, Y.; Pereiro, M.; Däne, M.; Turchi, P.E.A.; Antropov, V.P.; Eriksson, O. Prediction of the new efficient permanent magnet SmCoNiFe3. Phys. Rev. B 2017, 96, 100404-1–100404-5(R). [Google Scholar] [CrossRef]
- Landa, A.; Söderlind, P.; Parker, D.; Åberg, D.; Lordi, V.; Perron, A.; Turchi, P.E.A.; Chouhan, R.K.; Paudyal, D.; Lograsso, T.A. Thermodynamics of SmCo5 compound doped with Fe and Ni: An ab initio study. J. Alloy. Compd. 2018, 765, 659–663. [Google Scholar] [CrossRef]
- Zhang, D.-T.; Cai, N.-X.; Zhu, R.-C.; Liu, W.-Q.; Yue, M. Low-cost Sm0.7Y0.3Co5 sintered magnet produced by traditional powder metallurgical techniques. Rare Met. 2019, 39, 421–428. [Google Scholar] [CrossRef]
- Coey, J.M.D. Permanent magnets: Plugging the gap. Scr. Mater. 2012, 67, 524–529. [Google Scholar] [CrossRef]
- Coey, J.M.D. Hard magnetic materials: A Perspective. IEEE Trans. Magn. 2011, 47, 4671–4681. [Google Scholar] [CrossRef]
- Gutfleisch, O.; Willard, M.A.; Brück, E.; Chen, C.H.; Sankar, S.G.; Liu, J.P. Magnetic materials, and devices for the 21st century: Stronger, lighter, and more energy efficient. Adv. Mater. 2011, 23, 821–842. [Google Scholar] [CrossRef]
- Kramer, M.J.; McCallum, R.W.; Anderson, I.A.; Constantinides, S. Prospects for non-rare earth permanent magnets for traction motors and generators. JOM 2012, 64, 752–763. [Google Scholar] [CrossRef]
- Hoffer, G.; Strnat, K. Magnetocrystalline anisotropy of YCo5 and Y2Co17. IEEE Trans. Magn. 1966, 2, 487–489. [Google Scholar] [CrossRef]
- Strnat, K.; Hoffer, G.; Olson, J.; Ostertag, W. A Family of new cobalt-base permanent magnet materials. J. Appl. Phys. 1967, 38, 1001–1002. [Google Scholar] [CrossRef]
- Strnat, K.J. The recent development of permanent magnet materials containing rare earth metals. IEEE Trans. Magn. 1970, 6, 182–189. [Google Scholar] [CrossRef]
- Buschow, K.H. Composition and stability of CaCu5-type compounds of yttrium with iron and cobalt. J. Less-Common Met. 1973, 31, 359–364. [Google Scholar] [CrossRef]
- Laforest, J.; Shan, J.S. Neutron diffraction study of the Th(Co1-xFex)5 alloys. IEEE Trans. Magn. 1973, 9, 217–220. [Google Scholar] [CrossRef]
- Klein, H.P.; Menth, A.; Perkins, R.S. Magnetocrystalline anisotropy of light rare-earth cobalt compounds. Physica B+C 1975, 80, 153–163. [Google Scholar] [CrossRef]
- Heidemann, A.; Richter, R.; Buschow, K.H.J. Investigation of the hyperfine fields in the compounds LaCo13, LaCo5, YCo5 and ThCo5 by means of inelastic neutron scattering. Eur. Phys. J. B 1975, 22, 367–372. [Google Scholar] [CrossRef]
- Ermolenko, A.S. Magnetocrystalline anisotropy of rare earth intermetallics. IEEE Trans. Magn. 1976, 12, 992–996. [Google Scholar] [CrossRef]
- Déportes, J.; Givord, D.; Lemaire, R.; Nagai, H.; Yang, Y.T. Influence of substitutional pairs of cobalt atoms on the magnetocrystalline anisotropy of cobalt-rich rare-earth compounds. J. Less-Common Met. 1976, 44, 273–279. [Google Scholar] [CrossRef]
- Déportes, J.; Givord, D.; Scweizer, J.; Tasset, F. Different contributions of the two cobalt sites to the magnetocrystalline anisotropy of YCo5 and related compounds. IEEE Trans. Magn. 1976, 12, 1000–1002. [Google Scholar] [CrossRef]
- Buschow, K.H.J. Intermetallic compounds of rare-earth and 3d transition metals. Rep. Prog. Phys. 1977, 40, 1179–1256. [Google Scholar] [CrossRef]
- Malik, S.K.; Arlinghaus, F.A.; Wallace, W.E. Spin-polarized energy-band structure of YCo5, SmCo5, and GdCo5. Phys. Rev. B 1977, 16, 1242–1248. [Google Scholar] [CrossRef]
- Kirchmayar, H.R.; Poldy, C.A. Magnetism in rare earth—3d intermetallics. J. Magn. Magn. Mater. 1978, 8, 1–42. [Google Scholar] [CrossRef]
- Streever, R.L. NMR investigation of Co magnetic anisotropy in RCo5 compounds. Phys. Lett. A 1978, 65, 360–362. [Google Scholar] [CrossRef]
- Alameda, J.M.; Déportes, J.; Givord, D.; Lemaire, R.; Lu, Q. Large magnetization anisotropy in uniaxial YCo5 intermetallic. J. Magn. Magn. Mater. 1980, 15–18, 1257–1258. [Google Scholar] [CrossRef]
- Streever, R.L. Individual Co site contributions to the magnetic anisotropy of RCo5 compounds and related structures. Phys. Rev. B 1979, 19, 2704–2711. [Google Scholar] [CrossRef]
- Wohlfarth, E.P. First and second order transitions in some metallic ferromagnets. J. Appl. Phys. 1979, 50, 7542–7544. [Google Scholar] [CrossRef]
- Wohlfarth, E.P. The Curie temperatures of compounds of the heavy rare earths and yttrium with cobalt. J. Phys. F Met. Phys. 1979, 9, L123–L128. [Google Scholar] [CrossRef]
- Schweitzer, J.; Tasset, F. Polarized neutron study of the RCo5 intermetallic compounds. I. The cobalt magnetization in YCo5. J. Phys. F Met. Phys. 1980, 10, 2799–2817. [Google Scholar] [CrossRef]
- Alameda, J.M.; Givord, D.; Lemaire, R.; Lu, Q. Co energy and magnetization anisotropies in RCo5 intermetallics between 4.2 K and 300 K. J. Appl. Phys. 1981, 52, 2079–2081. [Google Scholar] [CrossRef]
- Inomata, K. Individual Co site contributions to the magnetic anisotropy and NMR investigation of Y2(Co1−xMx)17 (M=Cu,Al). Phys. Rev. B 1981, 23, 2076–2081. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Wu, C.H.; Chang, Y.C. Structure and magnetic properties of Y(Co1−xMx)5 compounds. J. Less Common Met. 1982, 84, 201–213. [Google Scholar] [CrossRef]
- Pasturel, A.; Colinet, C.; Allibert, C.; Hicter, P.; Percheron-Guegan, A.; Achard, J.C. A Theoretical and Experimental Study of the Enthalpies of Formation of LaNi5-Type Compounds. Phys. Status Solidi B 1984, 125, 101–106. [Google Scholar] [CrossRef]
- Meyer-Liautaud, F.; Derkaout, S.; Allibert, C.H.; Castanet, R. Structural and thermodynamic data on the pseudobinary phases R(Co1−xCux)5 WITH R ≡ Sm, Y, Ce. J. Less Common Met. 1987, 127, 231–242. [Google Scholar] [CrossRef]
- Franse, J.J.M.; Thuy, N.P.; Hong, N.M. Individual site magnetic anisotropy of the iron and cobalt ions in rare earth-iron and rare earth-cobalt intermetallic compounds. J. Magn. Magn. Mater. 1988, 72, 361–366. [Google Scholar] [CrossRef]
- Strnat, K.J.; Strnat, R.M.W. Rare earth-cobalt permanent magnets. J. Magn. Magn. Mater. 1991, 100, 38–56. [Google Scholar] [CrossRef]
- Zhao, T.-S.; Jin, H.-M.; Gua, G.-H.; Han, X.-F.; Chen, H. Magnetic properties of R ions in RCo5 compounds (R=Pr, Nd, Sm, Gd, Tb, Dy, Ho, and Er). Phys. Rev. B 1991, 43, 8593–8598. [Google Scholar] [CrossRef]
- Nordström, L.; Brooks, M.S.S.; Johansson, B. Calculation of orbital magnetism and magnetocrystalline anisotropy energy in YCo5. J. Phys. Condens. Matter 1992, 4, 3261–3272. [Google Scholar] [CrossRef]
- Coehoorn, R.; Daalderop, G.H.O. Magnetocrystalline anisotropy in new magnetic materials. J. Magn. Magn. Mater. 1992, 104–107, 1081–1085. [Google Scholar] [CrossRef]
- Daalderop, G.H.O.; Kelly, P.J.; Schuurmans, M.F.H. Magnetocrystalline anisotropy of RECo5 compounds. J. Magn. Magn. Mater. 1992, 104–107, 737–738. [Google Scholar] [CrossRef]
- Trygg, J.; Nordström, L.; Johansson, B. First-principles calculations of the mgnetocrystalline anisotropy energy for the pseudobinary compound. In Physics of Transition Metals; Oppeneer, P.M., Kübler, J., Eds.; Publisher Word Scientific: Singapore, 1993; pp. 745–748. [Google Scholar]
- Radwanski, R.J.; Franse, J.J.M. Magnetic properties of the 3d sublattice in RnTm intermetallics. Int. J. Mod. Phys. 1993, 7, 782–785. [Google Scholar] [CrossRef]
- Paoluzi, A.; Pareti, L.; Solzi, M.; Albertini, F. Study of the iron contribution to the 3d-sublattice anisotropy in some uniaxial YCoFe structures derived from the CaCu5 unit cell. J. Magn. Magn. Mater. 1994, 132, 185–190. [Google Scholar] [CrossRef]
- Colinet, C. The thermodynamic properties of rare earth metallic systems. J. Alloy. Compd. 1995, 225, 409–422. [Google Scholar] [CrossRef]
- Crisan, V.; Popescu, V.; Vernes, A.; Andreica, D.; Burda, I.; Cristea, S. On the electronic properties of YCo5−xNix. J. Alloy. Compd. 1995, 223, 147–150. [Google Scholar] [CrossRef]
- Daalderop, G.H.O.; Kelly, P.J.; Schuurmans, M.F.H. Magnetocrystalline anisotropy of YCo5 and related RECo5 compounds. Phys. Rev. B 1996, 53, 14415–14433. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Asano, S. First-principles calculation of the 3d magnetocrystalline anisotropy energy of YCo5. J. Appl. Phys. 1996, 79, 5952–5954. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Asano, S. First-principles calculations of the 3d magnetocrystalline anisotropy energy of YCo5, Y2Co7, YCo3, and Y2Co17. J. Magn. Magn. Mater. 1997, 168, 161–168. [Google Scholar] [CrossRef]
- Kitagawa, I.; Terao, K.; Aoki, M.; Yamada, H. Electronic structure, and magnetism of YCo5, YNi5, and YCo3Ni2. J. Phys. Condens. Matter 1997, 9, 231–239. [Google Scholar] [CrossRef]
- Zhang, G.W.; Feng, Y.P.; Ong, C.K. Electronic and magnetic properties of intermetallic compound YCo5. J. Magn. Magn. Mater. 1998, 184, 215–221. [Google Scholar] [CrossRef]
- Gratz, E.; Lindbaum, A.; Markosyan, A.S.; Milnera, M. Magnetoresistance in Y(Ni1−xCox)5 around the critical concentration for the onset of ferromagnetism. J. Magn. Magn. Mater. 1998, 184, 372–374. [Google Scholar] [CrossRef]
- Yamada, H.; Terao, K.; Ishikawa, F.; Yamaguchi, Y.; Mitamura, H.; Goto, T. Itinerant-electron metamagnetism of Y(Co,Ni)5. J. Phys. Condens. Matter 1999, 11, 483–492. [Google Scholar] [CrossRef]
- Maruyama, F.; Nagai, H.; Amako, Y.; Yoshie, H.; Adachi, K. Magnetic properties of the hypothetical compound YFe5. Physica B 1999, 266, 356–360. [Google Scholar] [CrossRef]
- Téllez-Blanko, J.C.; Grössinger, R.; Sato Turtelli, R.; Estévez-Rams, E. Magnetic and Structural Properties of YCo5xCux. IEEE Trans. Magn. 2000, 36, 3333–3335. [Google Scholar] [CrossRef]
- Al-Omari, I.A.; Skomski, R.; Thomas, R.A.; Lieslie-Pelesky, D.; Sellmyer, D.J. High-temperature magnetic properties of mechanically alloyed SmCo5 and YCo5 magnets. IEEE Trans. Magn. 2001, 37, 2534–2536. [Google Scholar] [CrossRef]
- Steinbeck, L.; Richter, M.; Eschrig, H. Magnetocrystalline anisotropy of RCo5 intermetallics: Itinerant-electron contribution. J. Magn. Magn. Mater. 2001, 226–230, 1011–1013. [Google Scholar] [CrossRef]
- Steinbeck, L.; Richter, M.; Eschrig, H. Itinerant-electron magnetocrystalline anisotropy energy of YCo5 and related compounds. Phys. Rev. B 2001, 63, 184431-1–184431-14. [Google Scholar] [CrossRef]
- Kashyap, A.; Skomski, R.; Sabiryanov, R.R.F.; Jaswal, S.S.; Sellmyer, D.J. Exchange interactions and Curie temperature of Y-Co compounds. IEEE Trans. Magn. 2003, 39, 2908–2910. [Google Scholar] [CrossRef]
- Larson, P.; Mazin, I.I. Calculation of magnetic anisotropy energy in YCo5. J. Magn. Magn. Mater. 2003, 264, 7–13. [Google Scholar] [CrossRef]
- Larson, P.; Mazin, I.I. Magnetic properties of SmCo5 and YCo5. J. Appl. Phys. 2003, 93, 6888–6890. [Google Scholar] [CrossRef]
- Larson, P.; Mazin, I.I.; Papaconstantopoulos, D.A. Effects of doping on the magnetic anisotropy energy in SmCo5−xFex and YCo5−xFex. Phys. Rev. B 2004, 69, 134408-1–134408-7. [Google Scholar] [CrossRef]
- Larson, P.; Mazin, I.I. Effect of impurities on magnetic properties of Y(Co5−xCux) and Y2(Co7−xNix). J. Magn. Magn. Mater. 2004, 269, 176–183. [Google Scholar] [CrossRef]
- Rosner, N.; Koudela, D.; Schwarz, U.; Handstein, A.; Hafland, M.; Opahle, I.; Koepernik, K.; Kuz’min, M.D.; Müller, K.-H.; Mydosh, J.A.; et al. Magneto-elastic lattice collapse in YCo5. Nat. Phys. 2006, 2, 469–472. [Google Scholar] [CrossRef]
- Wu, Q.; Chen, Z.; Xu, G.; Lu, X.; Zhong, K.; Huang, Z. Effects of doping on magnetic properties of YCo5-xFex and YCo5-xAgx—First Principles Calculation. J. Rare Earths 2006, 24 (Suppl. 1), 293–297. [Google Scholar] [CrossRef]
- Koudela, D.; Schwarz, U.; Rosner, H.; Burkhardt, U.; Handstein, A.; Hanfland, M.; Kuz’min, M.D.; Opahle, I.; Koepernik, K.; Müller, K.-H.; et al. Magnetic and elastic properties of YCo5 and LaCo5 under pressure. Phys. Rev. B 2008, 77, 024411-1–024411-7. [Google Scholar] [CrossRef]
- Castillo, M.C.G.; Aquino, J.A.M. Magnetic and electronic properties of the compound Y(Co,Fe)5 calculated by the augmented spherical wave method. Adv. Mater. Res. 2009, 68, 145–151. [Google Scholar]
- Colin, C.V.; Isnard, O.; Guillot, M. High magnetic field study of the anisotropy and neutron diffraction investigation of the crystal and magnetic structure of YCo4. 5Ge0.5. J. Alloy. Compd. 2010, 505, 11–16. [Google Scholar] [CrossRef]
- Liu, X.B.; Altounian, Z.; Yue, M. Effect of Fe partial substitution for Co on the magnetic properties of Y(Co,Fe)5 from first-principles. J. Appl. Phys. 2010, 107, 09A718-1–09A718-3. [Google Scholar] [CrossRef]
- Liu, X.B.; Altounian, Z. Magnetic moments and exchange interaction in Sm(Co, Fe)5 from first-principles. Comput. Mater. Sci. 2011, 50, 841–846. [Google Scholar] [CrossRef]
- Ohashi, K. Present and future of Sm2Co17 magnets. J. Jpn. Inst. Met. 2012, 76, 96–106. [Google Scholar] [CrossRef]
- Zhu, J.-X.; Janoscheck, M.; Rosenberg, R.; Ronning, F.; Thomson, J.D.; Torrez, M.; Bauer, E.D.; Bastia, C.D. LDA+DMFT approach to magnetocrystalline anisotropy of strong magnets. Phys. Rev. X 2014, 4, 021027-1–021027-7. [Google Scholar] [CrossRef]
- Matsumoto, M.; Banerjee, R.; Staunton, J.B. Improvement of magnetic hardness at finite temperatures: Ab initio disordered local-moment approach for YCo5. Phys. Rev. B 2014, 90, 054421-1–054421-11. [Google Scholar] [CrossRef]
- Tozman, P.; Vinkatesan, M.; Zickle, G.A.; Fidler, J.; Coey, J.M.D. Enhanced energy product in Y-Co-Fe magnets intermediate between Nd-Fe-B and ferrite. Appl. Phys. Lett. 2015, 107, 032405-1–032405-4. [Google Scholar] [CrossRef]
- Tozman, P.; Vinkatesan, M.; Coey, J.M.D. Optimization of the magnetic properties of nanostructured Y-Co-Fe alloys for permanent magnets. AIP Adv. 2016, 6, 056016-1–056016-5. [Google Scholar] [CrossRef]
- Patrick, C.E.; Kumar, S.; Balakrishnan, G.; Edwards, R.S.; Lees, M.R.; Mendive-Tapia, E.; Petit, L.; Staunton, J.B. Rare-earth/transition-metal magnetic interactions in pristine and (Ni,Fe)-doped YCo5 and GdCo5. Phys. Rev. Mater. 2017, 1, 024411-1–024411-13. [Google Scholar] [CrossRef]
- Patrick, C.E.; Staunton, J.B. Rare-earth/transition-metal magnets at finite temperature: Self-interaction-corrected relativistic density functional theory in the disordered local moment picture. Phys. Rev. B 2018, 97, 224415-1–224415-15. [Google Scholar] [CrossRef]
- Patrick, C.E.; Kumar, S.; Balakrishnan, G.; Edwards, R.S.; Lees, M.R.; Petit, L.; Staunton, J.B. Calculating the Magnetic Anisotropy of Rare-Earth–Transition-Metal Ferrimagnets. Phys. Rev. Lett. 2018, 120, 097202. [Google Scholar] [CrossRef] [PubMed]
- Burzo, E.; Vlaic, P.; Kozlenko, D.P.; Golosova, N.O.; Kichanov, S.E.; Savenko, B.N.; Östlin, A.; Chionsel, L. Structure and magnetic properties of YCo5 compound at high pressures. JMST 2020, 42, 106–112. [Google Scholar] [CrossRef]
- Sakurai, M.; Wu, S.; Zhao, X.; Nguyen, M.C.; Wang, C.-Z.; Ho, K.-M.; Chelikowsky, J.R. Magnetocrystalline anisotropy in YCo5 and ZrCo5 compounds from first-principles real-space pseudopotentials calculations. Phys. Rev. Mater. 2018, 2, 084410-1–084410-5. [Google Scholar] [CrossRef]
- Nguyen, M.C.; Yao, Y.; Wang, C.-Z.; Ho, K.-M.; Andropov, V.P. Magnetocrystalline anisotropy in cobalt based magnets: A choice of correlation parameters and the relativistic effects. J. Phys. Condens. Matter 2018, 30, 195801-1–195801-8. [Google Scholar] [CrossRef]
- Soderžnik, M.; Korent, M.; Žagar Soderžnik, K.; Dubois, J.-M.; Tozman, P.; Venkatesan, M.; Coey, J.M.D.; Kobe, S. Hot-compaction of YCo4.8Fe0.2 nanocrystals for metal-bonded magnets. J. Magn. Magn. Mater. 2018, 460, 401–408. [Google Scholar] [CrossRef]
- Patrick, C.E.; Matsumoto, M.; Staunton, J.B. First-principles calculations of the magnetocrystalline anisotropy of the prototype 2:17 cell boundary phase. J. Magn. Magn. Mater. 2019, 477, 147–155. [Google Scholar] [CrossRef]
- Patrick, C.E.; Staunton, J.B. Temperature-dependent magnetocrystalline anisotropy of rare earth/transition metal permanent magnets from first principles: The light RCo5 (R=Y, La-Gd) intermetallics. Phys. Rev. Mater. 2019, 3, 101401-1–101401-7. [Google Scholar] [CrossRef]
- Asali, A.; Fidler, J.; Suess, D. Influence of changes in electronic structure on magnetocrystalline anisotropy of YCo5 and related compounds. J. Magn. Magn. Mater. 2019, 485, 61–68. [Google Scholar] [CrossRef]
- Ucar, H.; Choudhary, R.; Paudyal, D. Substitutional and interstitial doping in LaCo5 system for the development of hard magnetic properties: A first principles study. J. Magn. Magn. Mater. 2020, 496, 165902-1–165902-8. [Google Scholar] [CrossRef]
- Subramanian, P.R.; Smith, J.F. Thermodynamics of formation of Y-Co alloys. Metall. Trans. A 1985, 16, 1195–1201. [Google Scholar] [CrossRef]
- Wu, C.H.; Chuang, Y.C. The Co-Y (Cobalt-Yttrium) system. J. Phase Equilibria Diffus. 1991, 12, 587–592. [Google Scholar] [CrossRef]
- Shevchenko, M.A.; Ivanov, M.I.; Berezutski, V.V.; Kudin, V.G.; Sudavtsova, V.S. Thermodynamic properties of alloys of the Co-Sc and Co-Y systems. Russ. J. Phys. Chem. A 2015, 89, 931–940. [Google Scholar] [CrossRef]
- Du, Z.; Zhang, W.; Zhao, L. Thermodynamic Assessment of Co-Y System. Rare Metals 1996, 15, 185–190. [Google Scholar]
- Du, Z.; Lü, D. Thermodynamic modelling of the Co–Y system. J. Alloy. Compd. 2004, 373, 171–178. [Google Scholar] [CrossRef]
- Golumbfskie, W.; Liu, Z.-K. CALPHAD/first-principles re-modeling of the Co–Y binary system. J. Alloy. Compd. 2006, 407, 193–200. [Google Scholar] [CrossRef]
- Gupta, K.G. The Co-Ni-Y (Cobalt-Nickel-Yttrium) system. J. Phase Equilibria Diffus. 2010, 31, 389–394. [Google Scholar] [CrossRef]
- Du, Z.; Lü, D. Thermodynamic modeling of the Co–Ni–Y system. Intermetallics 2005, 13, 586–595. [Google Scholar] [CrossRef]
- Golumbfskie, W.J.; Prins, S.N.; Eden, T.J.; Liu, Z.-K. Predictions of the Al-rich region of the Al–Co–Ni–Y system based upon first-principles and experimental data. Calphad 2009, 33, 124–135. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Wu, C.H.; Chang, Y.C. Study of the 1050 °C isothermal section of the ternary system Y-Co-Fe. J. Less-Common Met. 1986, 118, 7–20. [Google Scholar] [CrossRef]
- Kharchenko, O.I.; Bodak, O.I.; Gladyshevskii, E.I. Interaction of Yttrium with metals of the iron family. Russ. Metall. 1977, 1, 170–176. [Google Scholar]
- Söderlind, P.; Turchi, P.E.A.; Landa, A.; Lordi, V. Ground-state properties of rare-earth metals: An evaluation of density-functional theory. J. Phys. Condens. Matter 2014, 26, 416001-1–416001-8. [Google Scholar] [CrossRef]
- Landa, A.; Söderlind, P.; Parker, D.; Åberg, D.; Lordi, V.; Perron, A.; Turchi, P.E.A.; Chouhan, R.K.; Paudyal, D.; Lograsso, T.A. Thermodynamics of the SmCo5 compound doped with Fe and Ni: An ab initio study. J. Alloys Compd. 2018, 765, 659–663. [Google Scholar] [CrossRef]
- Vitos, L. Total-energy method based on the exact muffin-tin orbital theory. Phys. Rev. B 2001, 64, 014107-1–014107-11. [Google Scholar] [CrossRef]
- Vitos, L. Computational Quantum Mechanics for Materials Engineers: The EMTO Method and Application; Springer: London, UK, 2007. [Google Scholar]
- Kollar, J.; Vitos, L.; Skriver, H.L. From ASA towards the Full Potential. In Lecture Notes in Physics; Dreyssé, H., Ed.; Springer: Berlin, Germany, 2000; pp. 85–113. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Chadi, D.J.; Cohen, M.L. Special Points in the Brillouin Zone. Phys. Rev. B 1973, 8, 5747–5753. [Google Scholar] [CrossRef]
- Pourovskii, L.V.; Ruban, A.V.; Vitos, L.; Ebert, H.; Johansson, B.; Abrikosov, I.A. Fully relativistic spin-polarized exact muffin-tin-orbital method. Phys. Rev. B 2005, 71. [Google Scholar] [CrossRef]
- Faulkner, J.S. The modern theory of alloys. Prog. Mater. Sci. 1982, 27, 1–187. [Google Scholar] [CrossRef]
- Vitos, L.; Abrikosov, I.A.; Johansson, B. Anisotropic lattice distortions in random alloys from first-principles theory. Phys. Rev. Lett. 2001, 87. [Google Scholar] [CrossRef] [PubMed]
- Ruban, A.V.; Skriver, H.L. Screened Coulomb interaction in metalic alloys. I. Univesal screening in the atomic sphere approximations. Phys. Rev. B 2002, 66, 66. [Google Scholar] [CrossRef]
- Ruban, A.V.; Simak, S.I.; Korzhavyi, P.A.; Skriver, H.L. Screened Coulomb interaction in metalic alloys. II. Screening beyond the single-site and atomic-sphere approximations. Phys. Rev. B 2002, 66, 024202-1–024202-12. [Google Scholar] [CrossRef]
- Ruban, A.V.; Simak, S.I.; Shallcross, S.; Skriver, H.L. Local Lattice relaxations in random metallic alloys: Effective tetrahedron model and supercell approach. Phys. Rev. B 2003, 67, 214302-1–214302-12. [Google Scholar] [CrossRef]
- Staunton, J.; Gyorffy, B.; Pindor, A.; Stocks, G.M.; Winter, H. The “disordered local moment” picture of itinerant magnetism at finite temperatures. J. Magn. Magn. Mater. 1984, 45, 15–22. [Google Scholar] [CrossRef]
- Gyorffy, B.L.; Pindor, A.; Staunton, J.; Stocks, G.M.; Winter, H. A first-principles theory of ferromagnetic phase transitions in metals. J. Phys. F Met. Phys. 1985, 15, 1337–1386. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B 1975, 12, 3060–3083. [Google Scholar] [CrossRef]
- Söderlind, P. Delocalization and phase transitions in Pr: Theory. Phys. Rev. B 2002, 65, 115105-1–115105-5. [Google Scholar] [CrossRef]
- Wills, J.M.; Alouani, M.; Andersson, P.; Delin, A.; Eriksson, O.; Grechnyev, O. Full-Potential Electronic Structure Method; Springer Series in Solid-State Science; Springer: Berlin/Heidelberg, Germany, 2010; Volume 167. [Google Scholar]
- von Barth, U.; Hedin, L. A local exchange-correlation potential for the spin polarized case. i. J. Phys. C 1972, 5, 1629–1642. [Google Scholar] [CrossRef]
- Kaufman, L.; Bernstein, H. Computer Calculation of Phase Diagrams with Special Reference to Refractory Metals; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Saunders, N.; Miodownik, A. CALPHAD Calculation of Phase Diagrams: A Comprehensive Guide; Elsevier Science: New York, NY, USA, 1998. [Google Scholar]
- Lukas, H.; Fries, S.; Sundman, B. Computational Thermodynamics: The CALPHAD Method; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Sundman, B.; Jansson, B.; Andersson, J.-O. The Thermo-Calc databank system. Calphad 1985, 9, 153–190. [Google Scholar] [CrossRef]
- Andersson, J.-O.; Helander, T.; Höglund, L.; Shi, P.; Sundman, B. Thermo-Calc & DICTRA, computational tools for materials science. Calphad 2002, 26, 273–312. [Google Scholar] [CrossRef]
- Du, Z.; Zhang, W.; Zhuang, Y. Thermodynamic Assessment of the Fe-Y System. Rare Metals 1997, 16, 52–58. [Google Scholar]
- Lü, D.; Guo, C.; Li, C.; Du, Z. Thermodynamic description of Fe−Y and Fe−Ni−Y systems. Phys. Procedia 2013, 50, 383–387. [Google Scholar] [CrossRef]
- Konar, B.; Kim, J.; Jung, I.-H. Critical systematic evaluation, and thermodynamic optimization of the Fe-RE system: RE = Gd, Tb, Dy, Ho, Er, Tm, Lu, and Y. J. Phase Equilibria Diffus. 2017, 38, 509–542. [Google Scholar] [CrossRef]
- Kardellass, S.; Servant, C.; Selhaoui, N.; Iddaoudi, A.; Ait Amar, M.; Bouirden, L. Thermodynamic assessments of the Fe-Y and Ni-Sc systems. MATEC Web Conf. 2013, 3, 01008-1–01008-6. [Google Scholar] [CrossRef]
- Kardellass, S.; Servant, C.; Selhaoui, N.; Iddaoudi, A.; Ait Amar, M.; Bouirden, L. A thermodynamic assessment of the iron–yttrium system. J. Alloy. Compd. 2014, 583, 598–606. [Google Scholar] [CrossRef]
- Saenko, I.; Fabrichnaya, O.; Udovsky, A. new thermodynamic assessment of the Fe-Y system. J. Phase Equilibria Diffus. 2017, 38, 684–699. [Google Scholar] [CrossRef]
- Du, Z.; Zhang, W. Thermodynamic assessment of the Ni-Y system. J. Alloy. Compd. 1996, 245, 164–167. [Google Scholar] [CrossRef]
- Mattern, N.; Zinkevich, M.; Löser, W.; Behr, G.; Acker, J. Experimental and Thermodynamic Assessment of the Nb-Ni-Y System. J. Phase Equilibria Diffus. 2008, 29, 141–155. [Google Scholar] [CrossRef]
- Huang, J.; Yang, B.; Chen, H.; Wang, H. Thermodynamic optimization of the Ni-Al-Y ternary system. J. Phase Equilibria Diffus. 2015, 36, 357–365. [Google Scholar] [CrossRef]
- Mezbahul-Islam, M.; Medraj, M. A critical thermodynamic assessment of the Mg–Ni, Ni–Y binary and Mg–Ni–Y ternary systems. Calphad 2009, 33, 478–486. [Google Scholar] [CrossRef]
- Subramanian, P.R.; Smith, J.F. Thermodynamics of formation of Y-Ni alloys. Metall. Trans. B 1985, 16, 577–584. [Google Scholar] [CrossRef]
- Okamoto, H. Supplemental Literature Review of Binary Phase Diagrams: Cd-Se, Cu-Hg, Cu-Ho, Eu-Mg, H-Sr, Hf-Si, La-Mn, Mn-Nd, Nb-Y, Ni-Y, Pb-Se, and Sc-Sr. J. Phase Equilibria Diffus. 2013, 34, 430–436. [Google Scholar] [CrossRef]
- Sato, K.; Katayama-Yoshida, H.; Dederichs, P. Dilute magnetic semiconductors. Newsletter 2005, 70, 93–110. [Google Scholar]
- Ma, D.; Grabowski, B.; Körmann, F.; Neugebauer, J.; Raabe, D. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: Importance of entropy contributions beyond the configurational one. Acta Mater. 2015, 100, 90–97. [Google Scholar] [CrossRef]
- Coey, M.D. Magnetism and Magnetic Materials; Cambridge University Press: Cambridge, UK, 2010; p. 471. [Google Scholar]
- Ekholm, M.; Gambino, D.; Jönsson, H.J.M.; Tasnádi, F.; Alling, B.; Abrikosov, I.A. Assessing the SCAN functional for itinerant electron ferromagnets. Phys. Rev. B 2018, 98, 094413-1–094413-7. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | SREMTO | FREMTO | FPLMTO | Expt. [29,55] |
|---|---|---|---|---|
| Unit cell volume (Å3) | 85.07 | 85.24 | 82.65 | 82.50 |
| c/a axial ratio | 0.80 | |||
| Bulk modulus (GPa) | 134.63 | 128.65 | 164 | |
| Bulk modulus pressure derivative | 3.70 | 3.81 | 2.62 |
| Phase | Model | Reference |
|---|---|---|
| Y2TM17 | Sublattice model: (Co,Fe,Ni)17(Y)2 | |
| This work | ||
| [126] | ||
| [121] | ||
| YTM5 | Sublattice model: (Co,Fe,Ni)5(Y)1 | |
| This work | ||
| This work | ||
| [121] |
| Component: | Y1(1a) | Co1(2c) | Co2(3g) |
|---|---|---|---|
| m(s) (μB) | +0.31 | −1.55 | −1.47 |
| m(o)(μB) | −0.01 | −0.14 | −0.11 |
| Material | Unit Cell Volume (Å3) | c/a Ratio | K1 (meV/cell) | K1 (MJ/m3) | ||||
|---|---|---|---|---|---|---|---|---|
| GGA | LDA | GGA | LDA | GGA | LDA | GGA | LDA | |
| YFe5 | 84.84 | 77.46 | 0.79 | 0.79 | 0.51 | 1.17 | 0.96 | 2.42 |
| YCo5 | 82.65 | 76.38 | 0.80 | 0.80 | 9.89 | 6.44 | 19.17 | 13.51 |
| YNi5 | 81.96 | 74.94 | 0.81 | 0.81 | 1.40 | 0.48 | 2.74 | 1.03 |
| YFe3Co2 | 86.40 | 77.28 | 0.79 | 0.79 | 2.01 | 6.71 | 3.73 | 13.91 |
| YFe3CoNi | 86.22 | 76.38 | 0.81 | 0.82 | 4.51 | 5.04 | 8.38 | 10.57 |
| YFe3Ni2 | 86.34 | 76.64 | 0.84 | 0.83 | 1.93 | 3.69 | 3.58 | 7.69 |
| Material | Ms (MA/m) | Tc (K) | K1 (MJ/m3) | |BH|max (kJ/m3) |
|---|---|---|---|---|
| Nd2Fe14B | 1.28 | 588 | 4.9 | 512 |
| SmCo5 | 0.86 | 1020 | 17.2 | 231 |
| YCo5 | 0.85 | 987 | 6.5 | 224 |
| SmFe3CoNi | 1.08 | 1103 | 9.2 | 361 |
| YFe3CoNi | 1.00 | 785 | 10.6 | 309 |
| YFe3(Ni0.3Co0.7)2 | 1.06 | 900 | 351 | |
| YFe3Co2 | 1.14 | 1098 | 365 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landa, A.; Söderlind, P.; Moore, E.E.; Perron, A. Thermodynamics and Magnetism of YCo5 Compound Doped with Fe and Ni: An Ab Initio Study. Appl. Sci. 2020, 10, 6037. https://doi.org/10.3390/app10176037
Landa A, Söderlind P, Moore EE, Perron A. Thermodynamics and Magnetism of YCo5 Compound Doped with Fe and Ni: An Ab Initio Study. Applied Sciences. 2020; 10(17):6037. https://doi.org/10.3390/app10176037
Chicago/Turabian StyleLanda, Alexander, Per Söderlind, Emily E. Moore, and Aurelien Perron. 2020. "Thermodynamics and Magnetism of YCo5 Compound Doped with Fe and Ni: An Ab Initio Study" Applied Sciences 10, no. 17: 6037. https://doi.org/10.3390/app10176037
APA StyleLanda, A., Söderlind, P., Moore, E. E., & Perron, A. (2020). Thermodynamics and Magnetism of YCo5 Compound Doped with Fe and Ni: An Ab Initio Study. Applied Sciences, 10(17), 6037. https://doi.org/10.3390/app10176037