The Role of Housing Environment and Dietary Protein Source on the Gut Microbiota of Chicken


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
1.1. Role of Environment in Microbiota Structure
1.2. Role of Dietary Protein Source
2. Materials and Methods
2.1. Study Population
2.2. Sample Collection and Storage
2.3. 16S rRNA Gene Amplification and Sequencing
2.4. Microbiota Analyses
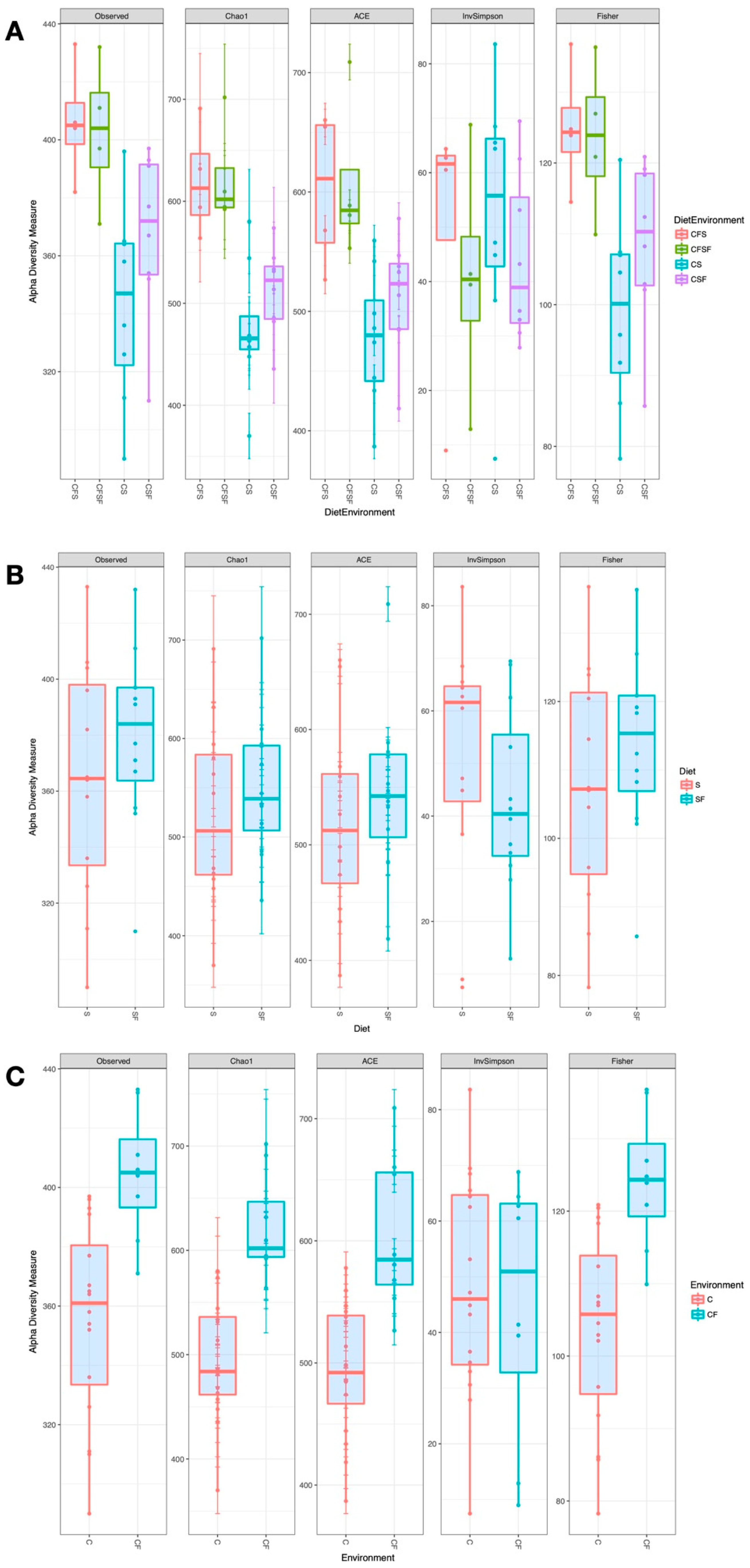
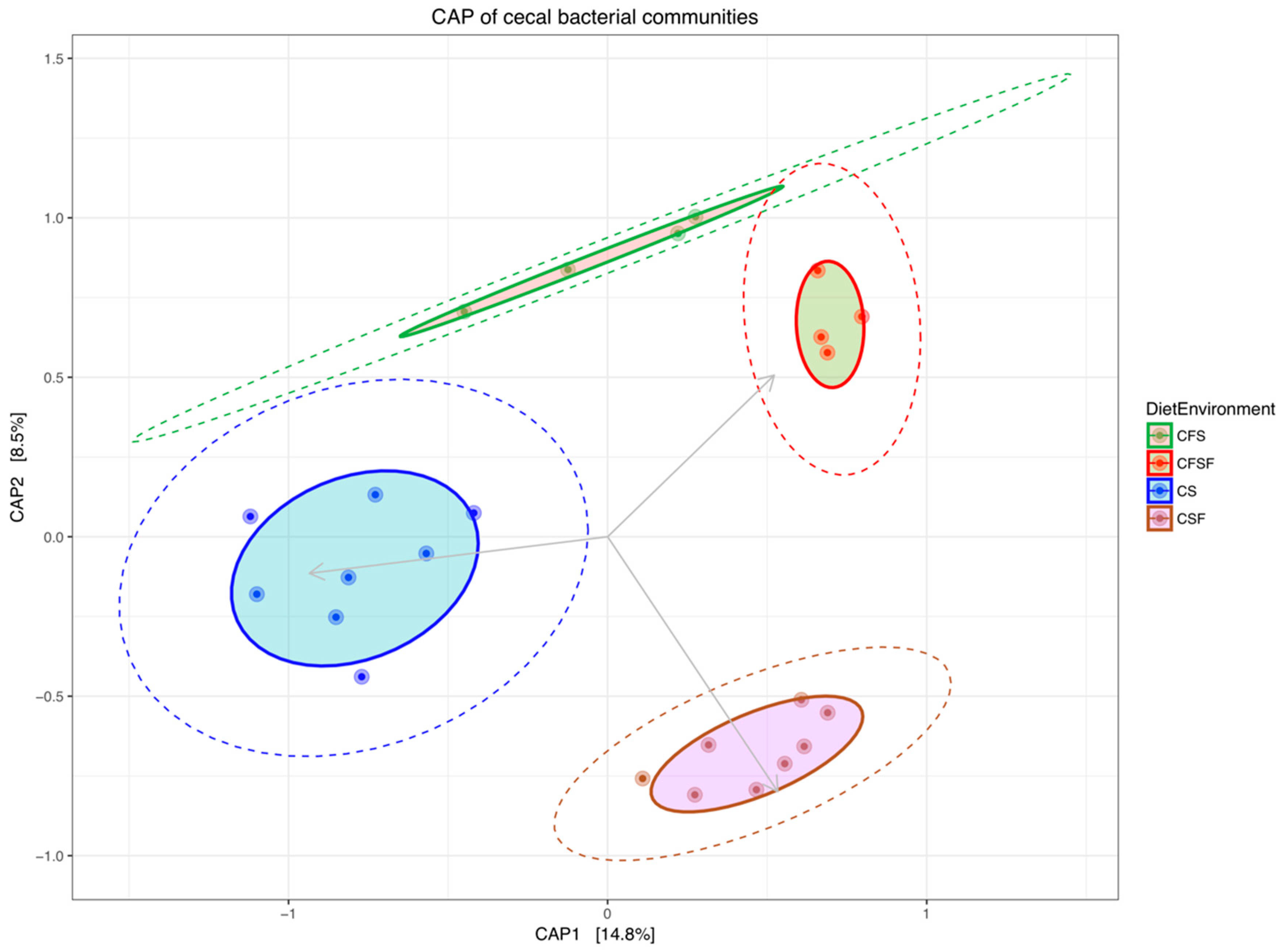
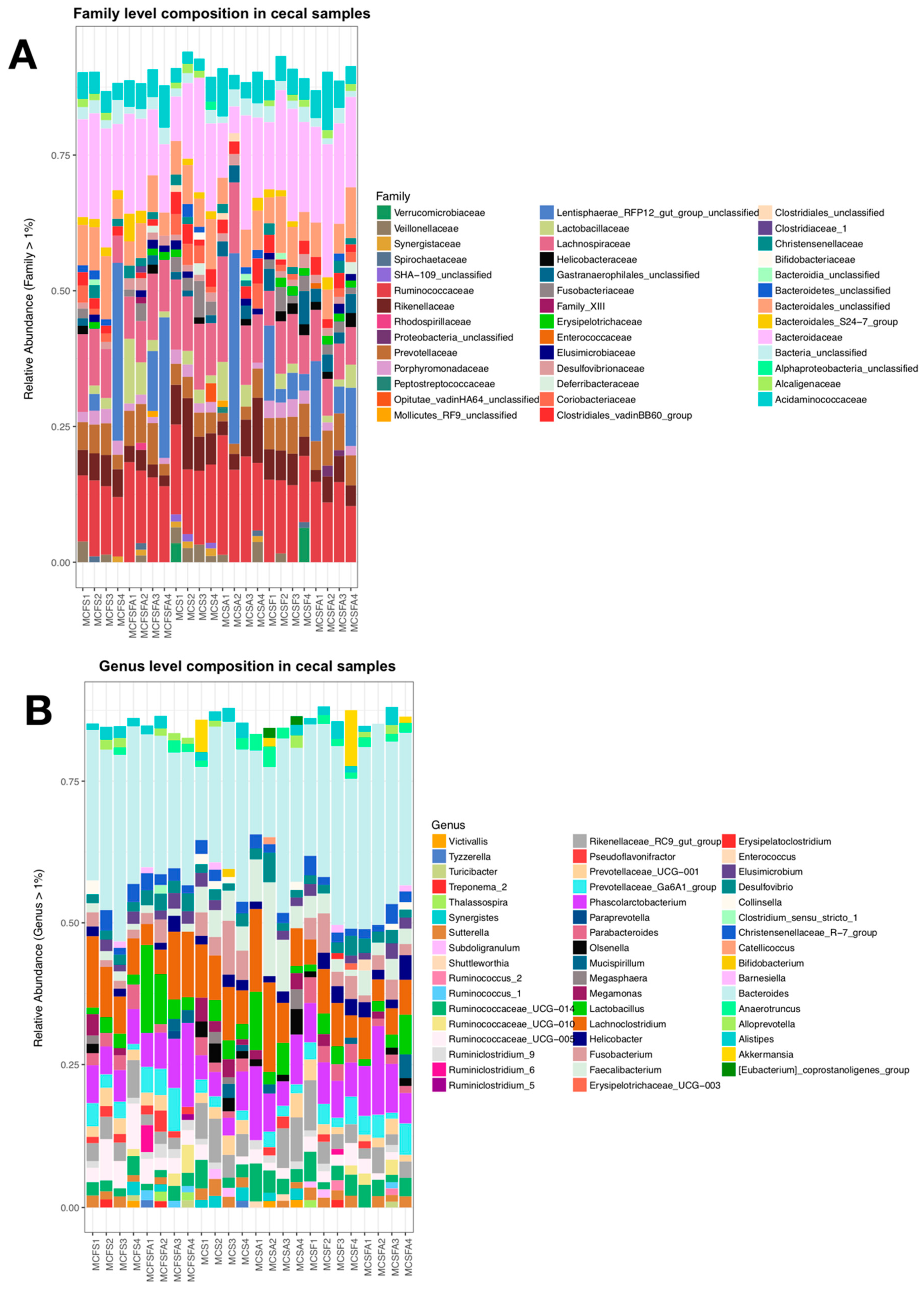
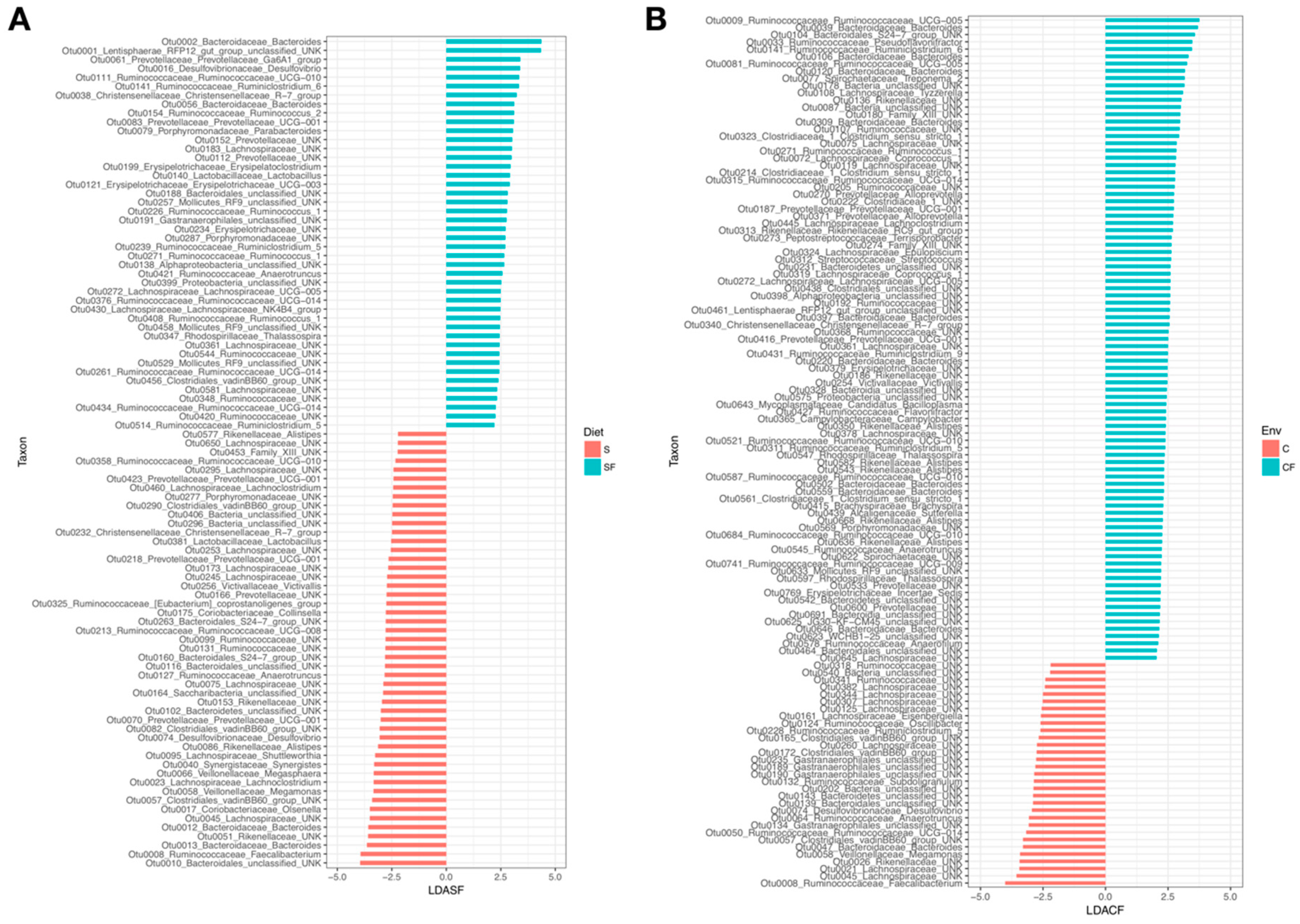
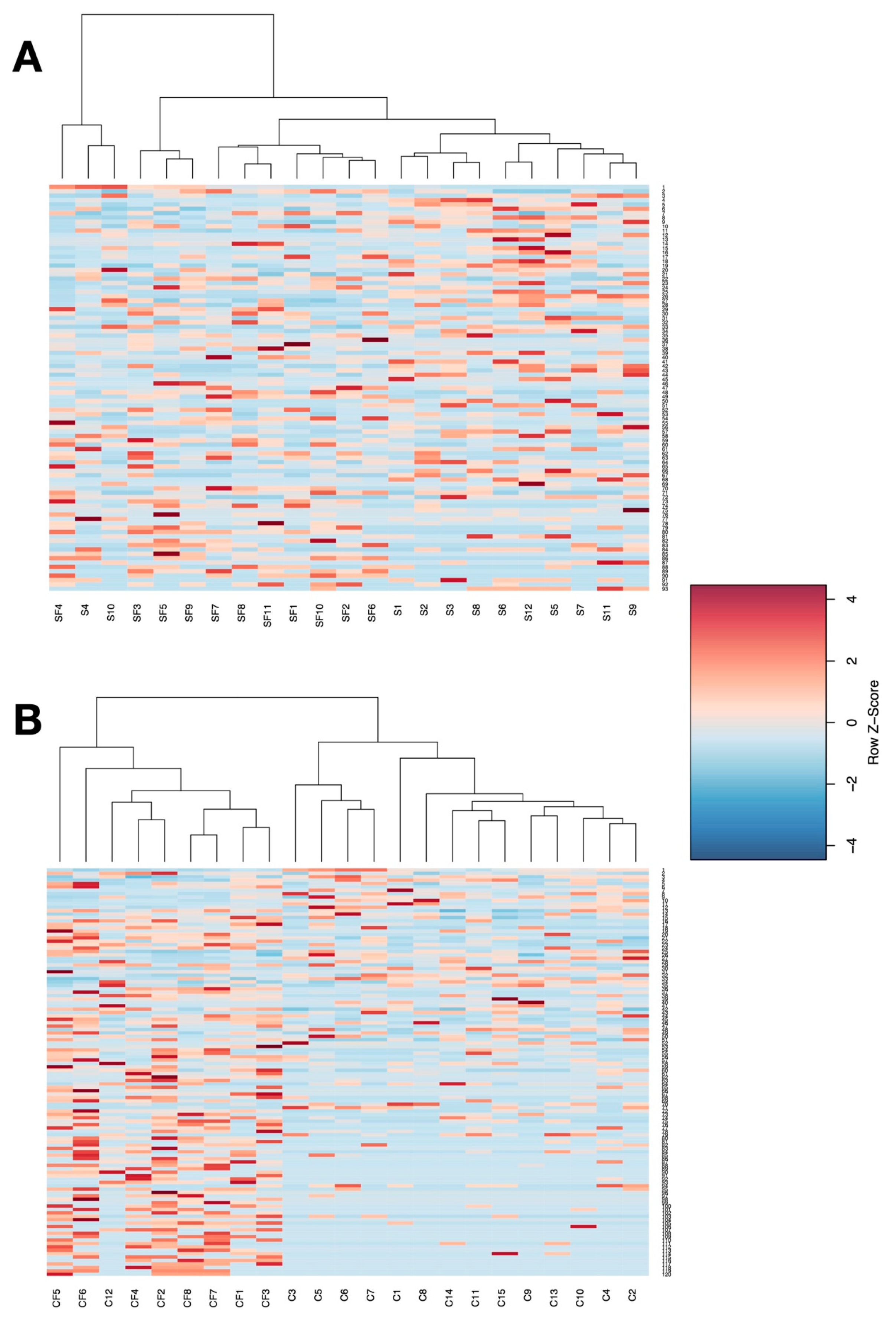
3. Results
4. Discussion
4.1. Lower Richness and Enrichment of Taxa in Caged Environments
4.2. Soy-Based and Cotton-Seed-Based Diets Generate Comparable Microbiota Diversity
4.3. Cage-Free Environments Support Higher Chicken Microbiota Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- OECD; FAO. OECD-FAO Agricultural Outlook 2017–2026; OECD-FAO Agricultural Outlook; OECD Publishing: Paris, France, 2017. [Google Scholar]
- FDA Guidance for Industry > FDA’s Strategy on Antimicrobial Resistance—Questions and Answers. Available online: https://www.fda.gov/animalveterinary/guidancecomplianceenforcement/guidanceforindustry/ucm216939.htm (accessed on 14 July 2018).
- Choi, K.Y.; Lee, T.K.; Sul, W.J. Metagenomic Analysis of Chicken Gut Microbiota for Improving Metabolism and Health of Chickens—A Review. Asian-Australas J. Anim. Sci. 2015, 28, 1217–1225. [Google Scholar] [CrossRef]
- Ding, J.; Dai, R.; Yang, L.; He, C.; Xu, K.; Liu, S.; Zhao, W.; Xiao, L.; Luo, L.; Zhang, Y.; et al. Inheritance and establishment of gut microbiota in chickens. Front. Microbiol. 2017, 8, 1967. [Google Scholar] [CrossRef] [PubMed]
- Mohd Shaufi, M.A.; Sieo, C.C.; Chong, C.W.; Gan, H.M.; Ho, Y.W. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Stanley, D.; Hughes, R.J.; Moore, R.J. Microbiota of the chicken gastrointestinal tract: Influence on health, productivity and disease. Appl. Microbiol. Biotechnol. 2014, 98, 4301–4310. [Google Scholar] [CrossRef] [PubMed]
- Kogut, M.H. The gut microbiota and host innate immunity: Regulators of host metabolism and metabolic diseases in poultry? J. Appl. Poult. Res. 2013, 22, 637–646. [Google Scholar] [CrossRef]
- Kogut, M.H. Issues and consequences of using nutrition to modulate the avian immune response. J. Appl. Poult. Res. 2017, 26, 605–612. [Google Scholar] [CrossRef]
- Cox, L.M.; Yamanishi, S.; Sohn, J.; Alekseyenko, A.V.; Leung, J.M.; Cho, I.; Kim, S.G.; Li, H.; Gao, Z.; Mahana, D.; et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 2014, 158, 705–721. [Google Scholar] [CrossRef]
- Lu, J.; Idris, U.; Harmon, B.; Hofacre, C.; Maurer, J.J.; Lee, M.D. Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl. Environ. Microbiol. 2003, 69, 6816–6824. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Clavijo, V.; Flórez, M.J.V. The gastrointestinal microbiome and its association with the control of pathogens in broiler chicken production: A review. Poult. Sci. 2018, 97, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Oakley, B.B.; Lillehoj, H.S.; Kogut, M.H.; Kim, W.K.; Maurer, J.J.; Pedroso, A.; Lee, M.D.; Collett, S.R.; Johnson, T.J.; Cox, N.A. The chicken gastrointestinal microbiome. FEMS Microbiol. Lett. 2014, 360, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Kogut, M.H. The effect of microbiome modulation on the intestinal health of poultry. Anim. Feed Sci. Technol. 2018, 250, 32–40. [Google Scholar] [CrossRef]
- Pan, D.; Yu, Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes 2014, 5, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Tang, J.W.; Fang, C.L.; Yao, X.H.; Wu, Y.F.; Wang, X.; Feng, J. Molecular analysis of intestinal bacterial microbiota of broiler chickens fed diets containing fermented cottonseed meal. Poult. Sci. 2013, 92, 392–401. [Google Scholar] [CrossRef]
- Kers, J.G.; Velkers, F.C.; Fischer, E.A.J.; Hermes, G.D.A.; Stegeman, J.A.; Smidt, H. Host and environmental factors affecting the intestinal microbiota in chickens. Front. Microbiol. 2018, 9, 235. [Google Scholar] [CrossRef]
- Torok, V.A.; Hughes, R.J.; Mikkelsen, L.L.; Perez-Maldonado, R.; Balding, K.; MacAlpine, R.; Percy, N.J.; Ophel-Keller, K. Identification and characterization of potential performance-related gut microbiotas in broiler chickens across various feeding trials. Appl. Environ. Microbiol. 2011, 77, 5868–5878. [Google Scholar] [CrossRef]
- Torok, V.A.; Hughes, R.J.; Ophel-Keller, K.; Ali, M.; MacAlpine, R. Influence of different litter materials on cecal microbiota colonization in broiler chickens. Poult. Sci. 2009, 88, 2474–2481. [Google Scholar] [CrossRef]
- Elson, H.A. Poultry housing and husbandry. Br. Poult. Sci. 2010, 51 (Suppl. 1), 23–34. [Google Scholar] [CrossRef]
- Questions and Answers—USDA Shell Egg Grading Service | Agricultural Marketing Service. Available online: https://www.ams.usda.gov/publications/qa-shell-eggs (accessed on 24 September 2018).
- Cui, Y.; Wang, Q.; Liu, S.; Sun, R.; Zhou, Y.; Li, Y. Age-Related Variations in Intestinal Microflora of Free-Range and Caged Hens. Front. Microbiol. 2017, 8, 1310. [Google Scholar] [CrossRef]
- D’Alfonso, T.H.; Roush, W.B.; Ventura, J.A. Least Cost Poultry Rations with Nutrient Variability: A Comparison of Linear Programming with a Margin of Safety and Stochastic Programming Models. Poult. Sci. 1992, 71, 255–262. [Google Scholar] [CrossRef]
- Al-Ajeeli, M.N.; Leyva-Jimenez, H.; Abdaljaleel, R.A.; Jameel, Y.; Hashim, M.M.; Archer, G.; Bailey, C.A. Evaluation of the performance of Hy-Line Brown laying hens fed soybean or soybean-free diets using cage or free-range rearing systems. Poult. Sci. 2018, 97, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Choct, M.; Dersjant-Li, Y.; McLeish, J.; Peisker, M. Soy Oligosaccharides and Soluble Non-starch Polysaccharides: A Review of Digestion, Nutritive and Anti-nutritive Effects in Pigs and Poultry. Asian-Australas J. Anim. Sci. 2010, 23, 1386–1398. [Google Scholar] [CrossRef]
- Choct, M. Enzymes for the feed industry: Past, present and future. Worlds. Poult. Sci. J. 2006, 62, 5–16. [Google Scholar] [CrossRef]
- Saitoh, S.; Sato, T.; Harada, H.; Takita, T. Transfer of soy isoflavone into the egg yolk of chickens. Biosci. Biotechnol. Biochem. 2001, 65, 2220–2225. [Google Scholar] [CrossRef] [PubMed]
- Thévenot, A.; Rivera, J.L.; Wilfart, A.; Maillard, F.; Hassouna, M.; Senga-Kiesse, T.; Le Féon, S.; Aubin, J. Mealworm meal for animal feed: Environmental assessment and sensitivity analysis to guide future prospects. J. Clean. Prod. 2018, 170, 1260–1267. [Google Scholar] [CrossRef]
- Khusro, M.; Andrew, N.R. Insects as poultry feed: A scoping study for poultry production systems in Australia. Worlds Poult. Sci. J 2012, 68, 435–446. [Google Scholar] [CrossRef]
- Scott, K.P.; Gratz, S.W.; Sheridan, P.O.; Flint, H.J.; Duncan, S.H. The influence of diet on the gut microbiota. Pharmacol. Res. 2013, 69, 52–60. [Google Scholar] [CrossRef]
- van der Hoeven-Hangoor, E.; van der Vossen, J.M.B.M.; Schuren, F.H.J.; Verstegen, M.W.A.; de Oliveira, J.E.; Montijn, R.C.; Hendriks, W.H. Ileal microbiota composition of broilers fed various commercial diet compositions. Poult. Sci. 2013, 92, 2713–2723. [Google Scholar] [CrossRef]
- Walugembe, M.; Hsieh, J.C.F.; Koszewski, N.J.; Lamont, S.J.; Persia, M.E.; Rothschild, M.F. Effects of dietary fiber on cecal short-chain fatty acid and cecal microbiota of broiler and laying-hen chicks. Poult. Sci. 2015, 94, 2351–2359. [Google Scholar] [CrossRef]
- Danzeisen, J.L.; Kim, H.B.; Isaacson, R.E.; Tu, Z.J.; Johnson, T.J. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS ONE 2011, 6, e27949. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, P.-Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Keller, K.; Brodie, E.L.; Larsen, N.; Piceno, Y.M.; Phan, R.; Andersen, G.L. NAST: A multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006, 34, W394–W399. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Kelly, B.J.; Gross, R.; Bittinger, K.; Sherrill-Mix, S.; Lewis, J.D.; Collman, R.G.; Bushman, F.D.; Li, H. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 2015, 31, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. Permutational multivariate analysis of variance (PERMANOVA). In Wiley Statsref: Statistics Reference Online; Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2014; pp. 1–15. [Google Scholar]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Oakley, B.B.; Morales, C.A.; Line, J.; Berrang, M.E.; Meinersmann, R.J.; Tillman, G.E.; Wise, M.G.; Siragusa, G.R.; Hiett, K.L.; Seal, B.S. The poultry-associated microbiome: Network analysis and farm-to-fork characterizations. PLoS ONE 2013, 8, e57190. [Google Scholar] [CrossRef]
- von Waldburg-Zeil, C.G.; van Staaveren, N.; Harlander-Matauschek, A. Do laying hens eat and forage in excreta from other hens? Animal 2018, 1–7. [Google Scholar] [CrossRef]
- Gadelha, I.C.N.; Fonseca, N.B.S.; Oloris, S.C.S.; Melo, M.M.; Soto-Blanco, B. Gossypol toxicity from cottonseed products. ScientificWorldJournal 2014, 2014, 231635. [Google Scholar] [CrossRef]
- Henry, M.H.; Pesti, G.M.; Brown, T.P. Pathology and histopathology of gossypol toxicity in broiler chicks. Avian Dis. 2001, 45, 598. [Google Scholar] [CrossRef]
- Abou-Donia, M.B.; Lyman, C.M. Metabolic fate of gossypol: The metabolism of gossypol-14C in laying hens. Toxicol. Appl. Pharmacol. 1970, 17, 160–173. [Google Scholar] [CrossRef]
- USDA APHIS USDA Announces Deregulation of GE Low-Gossypol Cotton. Available online: https://www.aphis.usda.gov/aphis/newsroom/stakeholder-info/sa_by_date/sa-2018/sa-10/ge-cotton (accessed on 24 November 2018).
- Sunilkumar, G.; Campbell, L.M.; Puckhaber, L.; Stipanovic, R.D.; Rathore, K.S. Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc. Natl. Acad. Sci. USA 2006, 103, 18054–18059. [Google Scholar] [CrossRef] [PubMed]
- Nordentoft, S.; Mølbak, L.; Bjerrum, L.; De Vylder, J.; Van Immerseel, F.; Pedersen, K. The influence of the cage system and colonisation of Salmonella Enteritidis on the microbial gut flora of laying hens studied by T-RFLP and 454 pyrosequencing. BMC Microbiol. 2011, 11, 187. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef]
- Yegani, M.; Korver, D.R. Factors affecting intestinal health in poultry. Poult. Sci. 2008, 87, 2052–2063. [Google Scholar] [CrossRef]
- Casewell, M.; Friis, C.; Marco, E.; McMullin, P.; Phillips, I. The European ban on growth-promoting antibiotics and emerging consequences for human and animal health. J. Antimicrob. Chemother. 2003, 52, 159–161. [Google Scholar] [CrossRef]
- Bjerrum, L.; Engberg, R.M.; Leser, T.D.; Jensen, B.B.; Finster, K.; Pedersen, K. Microbial community composition of the ileum and cecum of broiler chickens as revealed by molecular and culture-based techniques. Poult. Sci. 2006, 85, 1151–1164. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, H.; Zhang, L.; Su, Y.; Shi, D.; Xiao, H.; Tian, Y. High-throughput sequencing technology to reveal the composition and function of cecal microbiota in Dagu chicken. BMC Microbiol. 2016, 16, 259. [Google Scholar] [CrossRef]
- Ding, J.; Zhao, L.; Wang, L.; Zhao, W.; Zhai, Z.; Leng, L.; Wang, Y.; He, C.; Zhang, Y.; Zhang, H.; et al. Divergent selection-induced obesity alters the composition and functional pathways of chicken gut microbiota. Genet. Sel. Evol. 2016, 48, 93. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, G.; Siegel, P.; He, C.; Wang, H.; Zhao, W.; Zhai, Z.; Tian, F.; Zhao, J.; Zhang, H.; et al. Quantitative genetic background of the host influences gut microbiomes in chickens. Sci. Rep. 2013, 3, 1163. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Bushman, F.D.; FitzGerald, G.A. Time in motion: The molecular clock meets the microbiome. Cell 2014, 159, 469–470. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Korem, T.; Dohnalová, L.; Shapiro, H.; Jaitin, D.A.; David, E.; Winter, D.R.; Gury-BenAri, M.; Tatirovsky, E.; et al. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell 2016, 167, 1495–1510.e12. [Google Scholar] [CrossRef] [PubMed]
- Hieke, A.-S.C.; Hubert, S.M.; Athrey, G. Circadian disruption and divergent microbiota acquisition under extended photoperiod regimens in chicken. PeerJ 2019, 7, e6592. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubert, S.M.; Al-Ajeeli, M.; Bailey, C.A.; Athrey, G. The Role of Housing Environment and Dietary Protein Source on the Gut Microbiota of Chicken. Animals 2019, 9, 1085. https://doi.org/10.3390/ani9121085
Hubert SM, Al-Ajeeli M, Bailey CA, Athrey G. The Role of Housing Environment and Dietary Protein Source on the Gut Microbiota of Chicken. Animals. 2019; 9(12):1085. https://doi.org/10.3390/ani9121085
Chicago/Turabian StyleHubert, Shawna Marie, Morouj Al-Ajeeli, Christopher A. Bailey, and Giridhar Athrey. 2019. "The Role of Housing Environment and Dietary Protein Source on the Gut Microbiota of Chicken" Animals 9, no. 12: 1085. https://doi.org/10.3390/ani9121085
APA StyleHubert, S. M., Al-Ajeeli, M., Bailey, C. A., & Athrey, G. (2019). The Role of Housing Environment and Dietary Protein Source on the Gut Microbiota of Chicken. Animals, 9(12), 1085. https://doi.org/10.3390/ani9121085