

Metabolic Plasticity and Transcriptomic Reprogramming Orchestrate Hypoxia Adaptation in Yak
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Ethics
2.2. Isolation and Culture of Yak Cardiac Fibroblasts
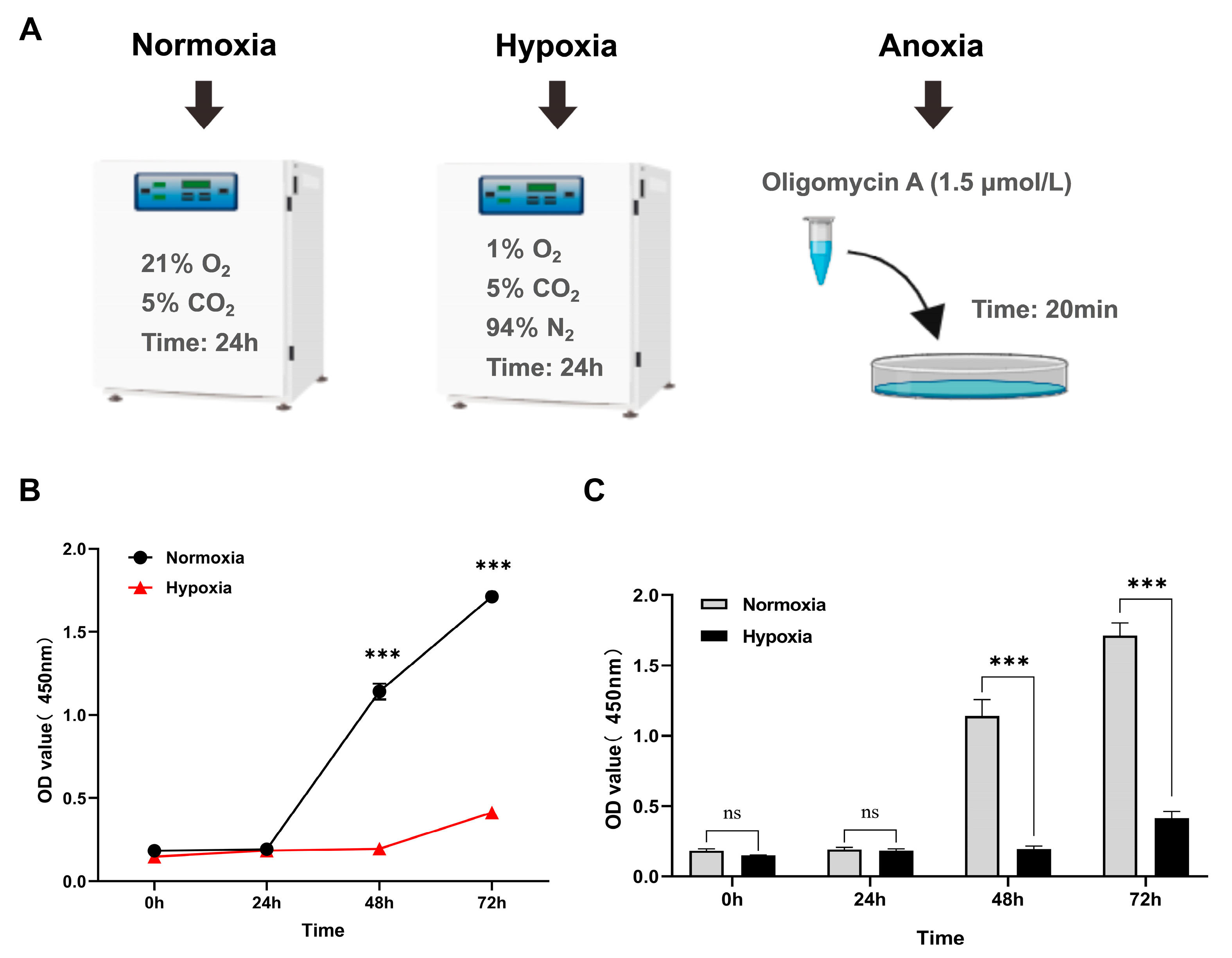
2.3. Treatment of Yak Cardiac Fibroblasts Under Different Oxygen Concentrations
2.4. Measurement of Yak Cardiac Fibroblast Viability Under Different Oxygen Conditions
2.5. Respiration and Glycolysis Analyses
2.6. RNA Extraction and qPCR
2.7. Quantification of mtDNA Copy Number
2.8. RNA Sequencing (RNA-Seq) of Yak Cardiac Fibroblasts
2.9. Transcriptome Sequencing Data Processing and Bioinformatics Analysis
2.10. Protein–Protein Interaction (PPI) Network Analysis
2.11. Data Analysis
3. Results
3.1. Hypoxia Inhibits Cell Proliferation
3.2. Hypoxia Boosts Glycolysis but Limits Aerobic Energy Yield
3.3. Hypoxia Upregulates Mitochondrial Coding Genes Without Changing MtDNA Levels
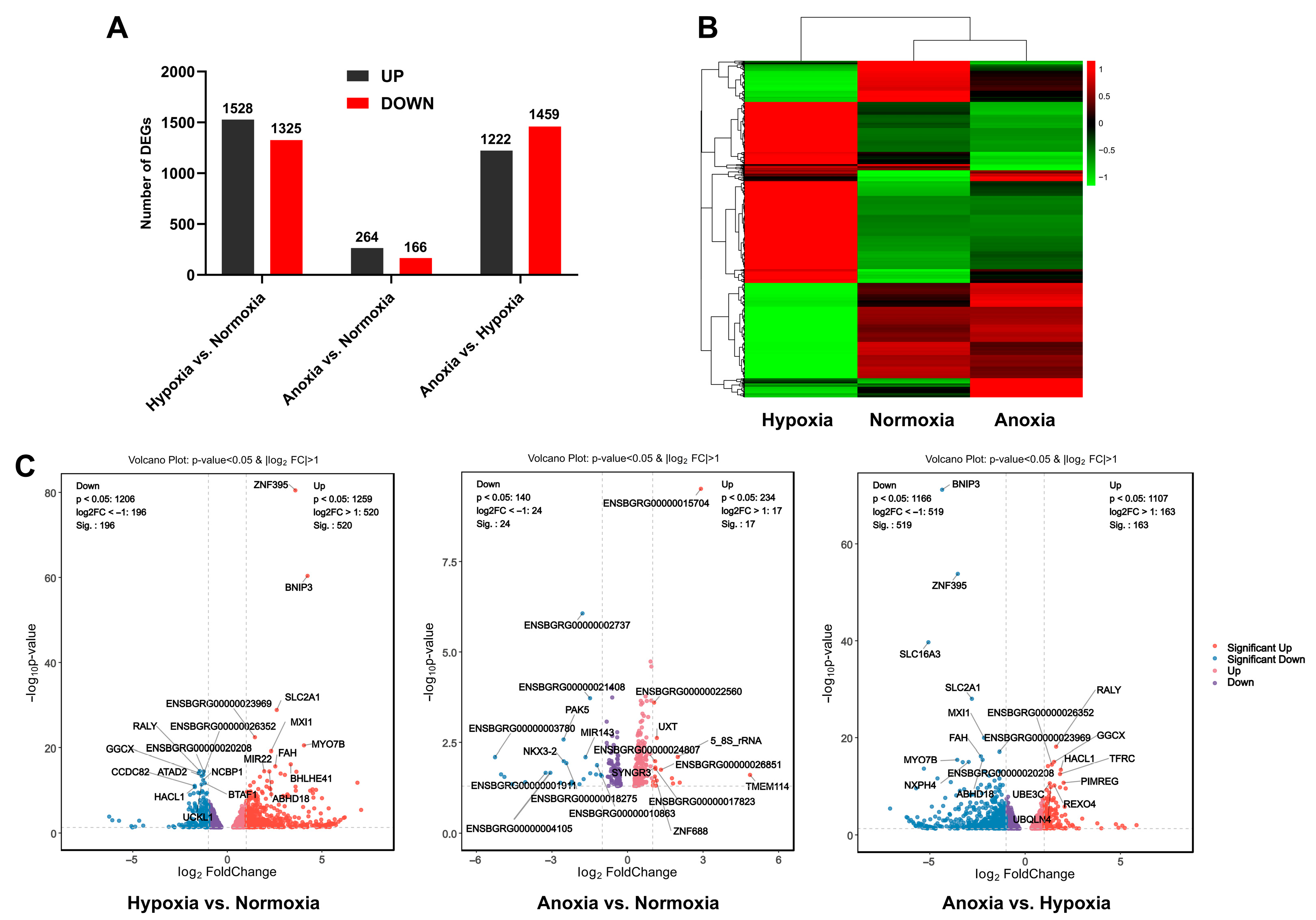
3.4. Transcriptome Sequencing Reveals Gene Expression Differences Under Different Oxygen Partial Pressures
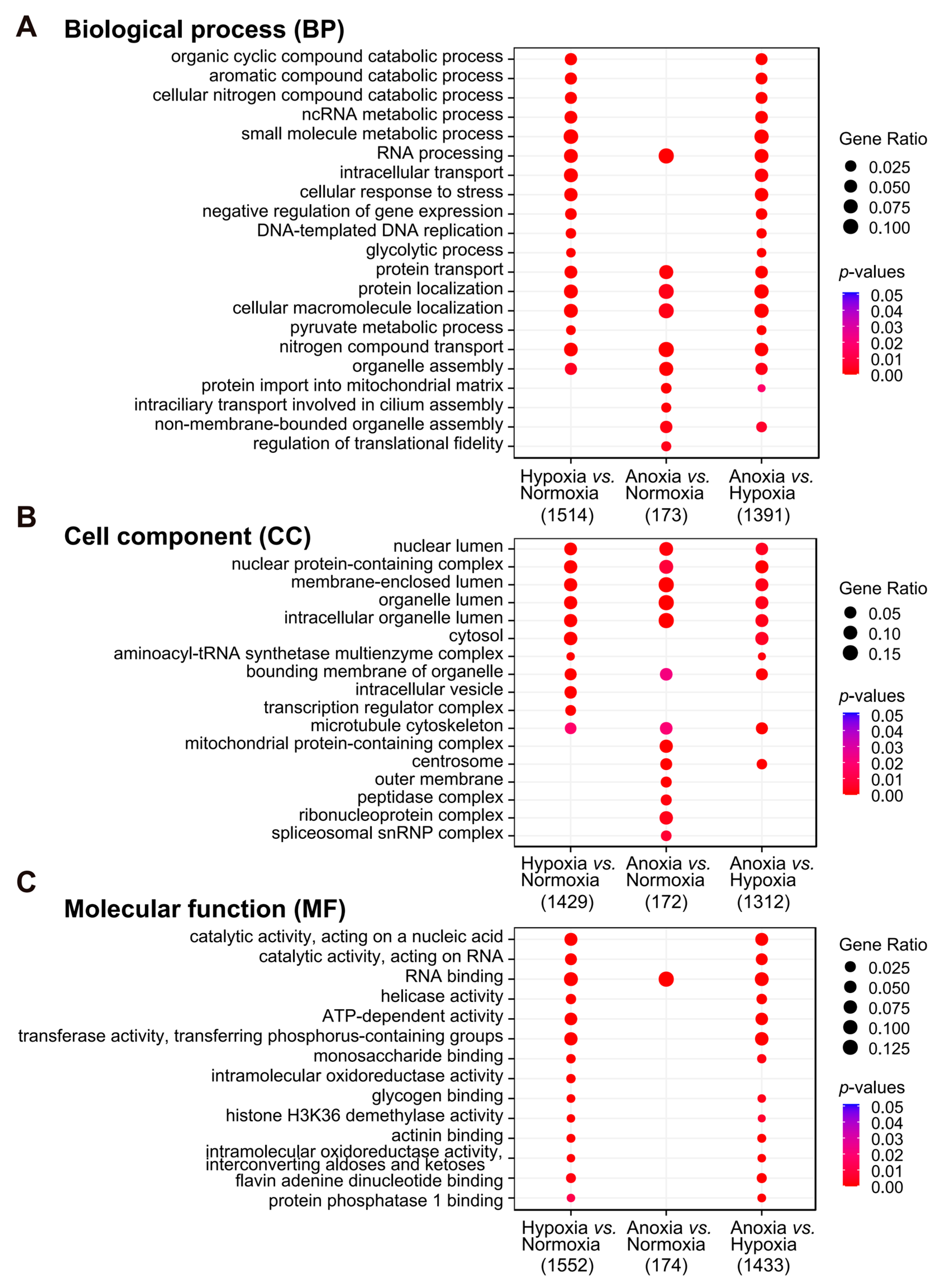
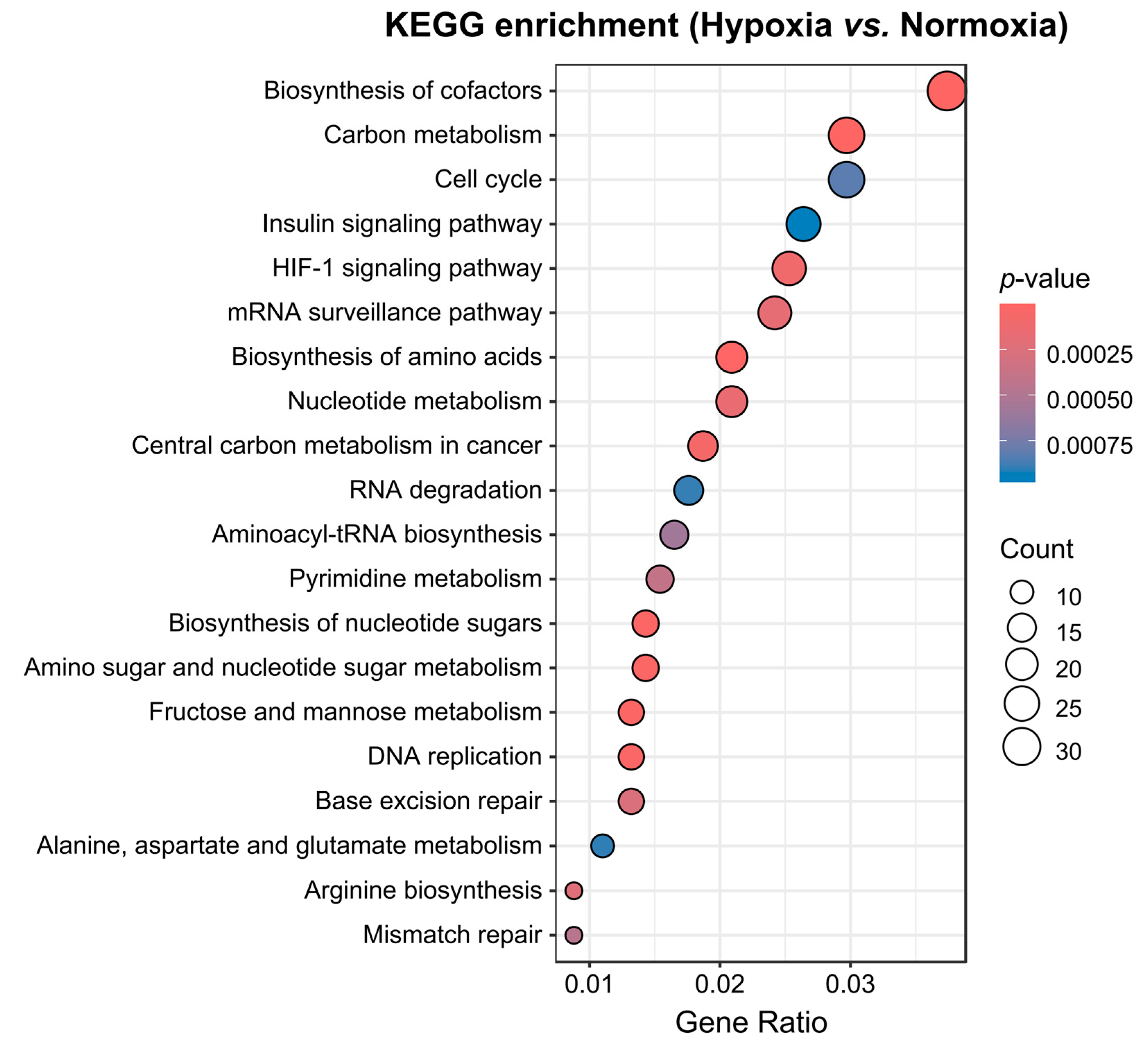
3.5. Functional Analysis of DEGs Revealed That Transcriptional Reprogramming Related to Nucleotide Synthesis, Carbon Metabolism, and Energy Production Plays a Critical Role in Yak Adaptation to Hypoxia
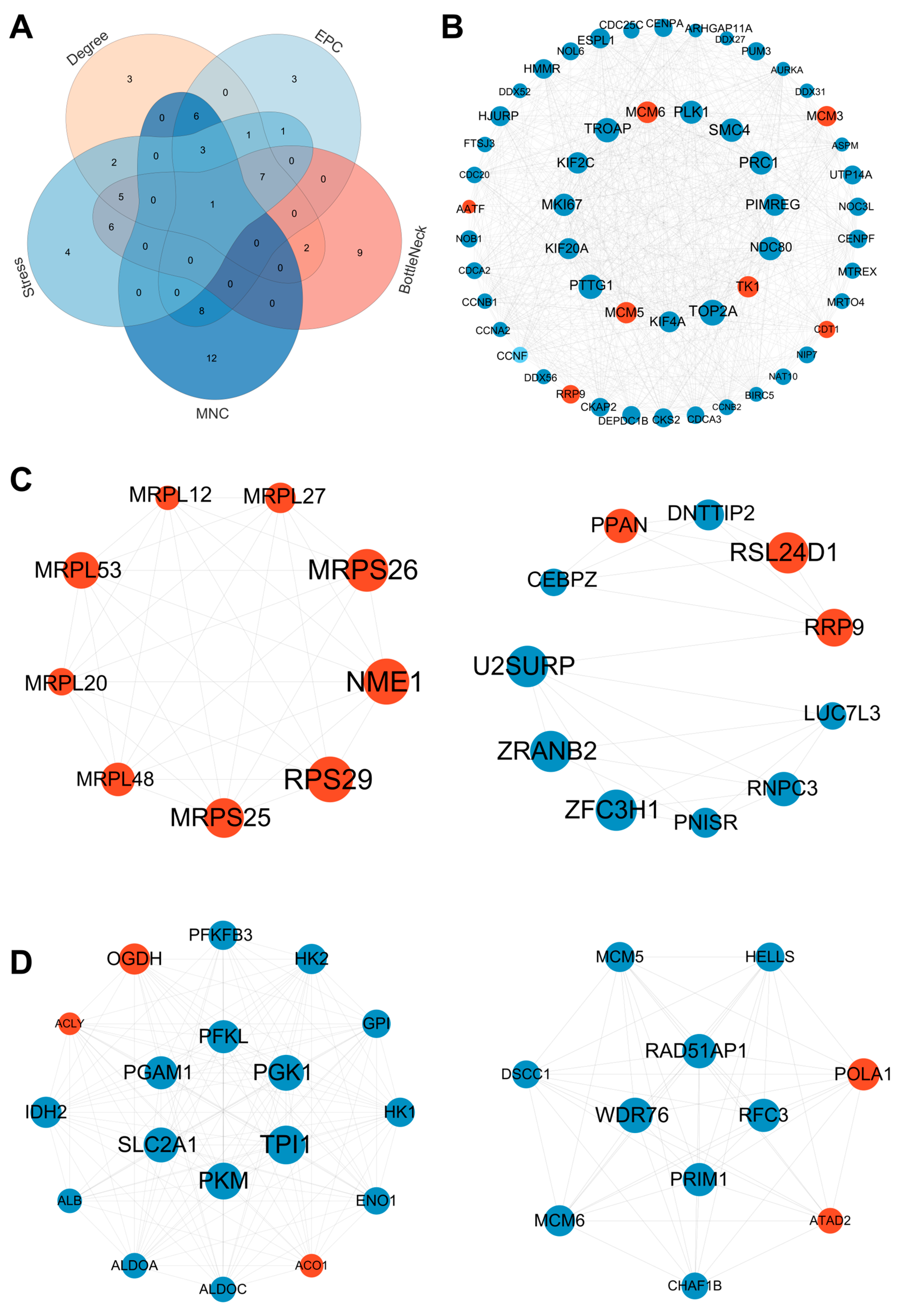
3.6. PPI Network Construction
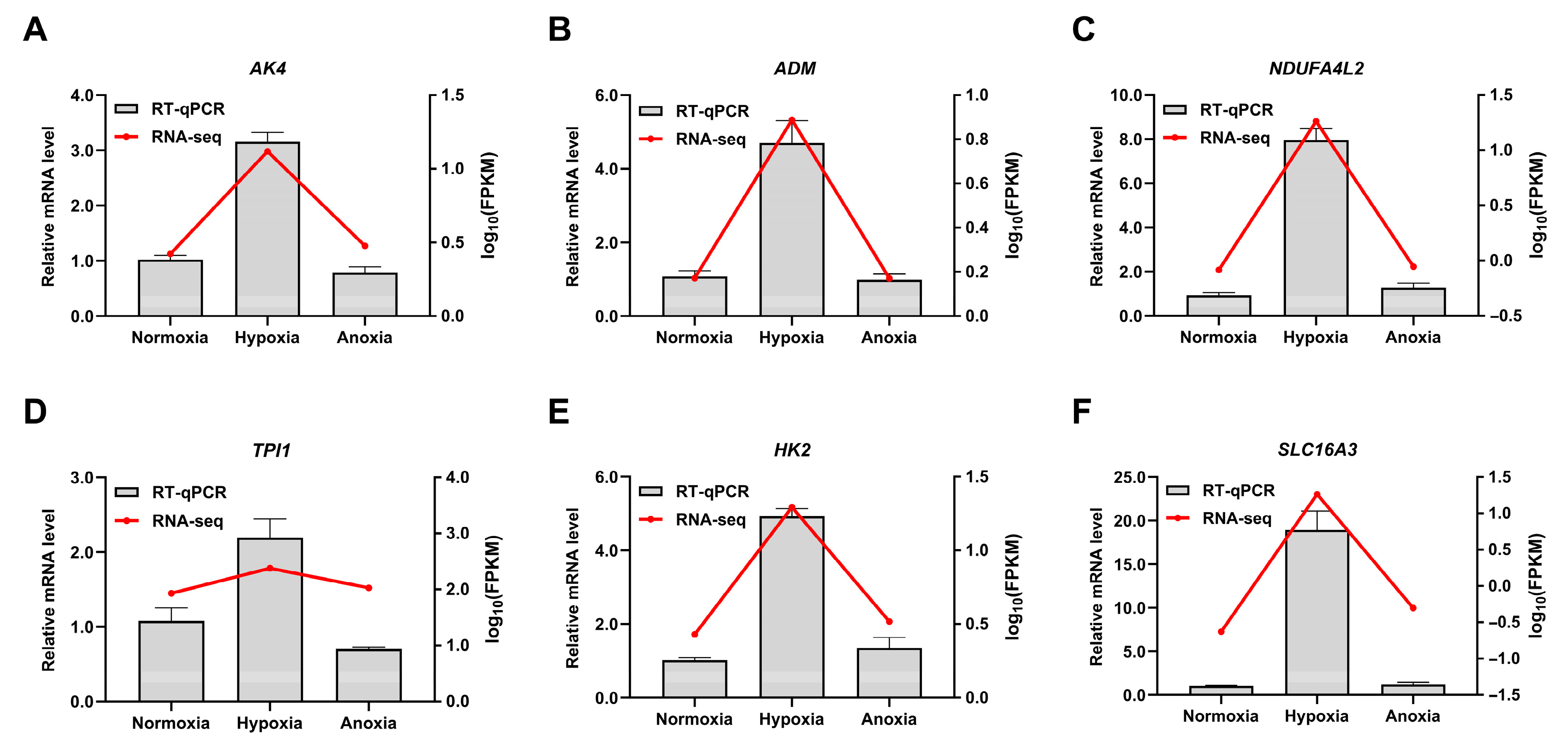
3.7. qPCR Confirms High Consistency and Reproducibility with RNA-Seq Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MtDNA | Mitochondrial DNA |
| ROS | Reactive oxygen species |
| DEGs | Differentially expressed genes |
| PI3K-Akt | Phosphoinositide 3-kinase–Akt |
| HIF-1 | Hypoxia-inducible factor 1 |
| PPAR | Peroxisome proliferator-activated receptor |
| qPCR | Quantitative real-time PCR |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| PBS | Phosphate-buffered saline |
| DMSO | Dimethyl sulfoxide |
| FBS | Fetal bovine serum |
| CCK8 | Cell counting kit-8 |
| NCBI | National Center for Biotechnology Information |
| RNA-seq | RNA sequencing |
| mRNA | Messenger RNA |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PPI | Protein–protein interaction |
| MCODE | Molecular complex detection |
| SEM | Standard error of the mean |
| ECAR | Extracellular acidification rate |
| OCR | Oxygen consumption rate |
| DEG | Differentially expressed gene |
| GO | Gene Ontology |
| OXPHOS | Oxidative phosphorylation |
| ETC | Electron transport chain |
References
- Fuhrmann, D.C.; Brüne, B. Mitochondrial Composition and Function under the Control of Hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Li, N.; Zhao, L.; Geng, X.; Liu, J.; Zhang, X.; Hu, Y.; Qi, J.; Chen, H.; Qiu, J.; Zhang, X.; et al. Stimulation by Exosomes from Hypoxia-Preconditioned Hair Follicle Mesenchymal Stem Cells Facilitates Mitophagy by Inhibiting the PI3K/AKT/mTOR Signaling Pathway to Alleviate Ulcerative Colitis. Theranostics 2024, 14, 4278–4296. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, Y.; Wang, J.; Elzo, M.A.; Yang, X.; Lai, S. Genetic Diversities of MT-ND1 and MT-ND2 Genes Are Associated with High-Altitude Adaptation in Yak. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2017, 29, 485–494. [Google Scholar] [CrossRef]
- Bao, H.G.; Zhao, C.J.; Li, J.Y.; Wu, C. Association of MT-ND5 Gene Variation with Mitochondrial Respiratory Control Ratio and NADH Dehydrogenase Activity in Tibet Chicken Embryos. Anim. Genet. 2007, 38, 514–516. [Google Scholar] [CrossRef]
- Zhou, T.; Shen, X.; Irwin, D.M.; Shen, Y.; Zhang, Y. Mitogenomic Analyses Propose Positive Selection in Mitochondrial Genes for High-Altitude Adaptation in Galliform Birds. Mitochondrion 2014, 18, 70–75. [Google Scholar] [CrossRef]
- Yagi, H.; Boeck, M.; Nian, S.; Neilsen, K.; Wang, C.; Lee, J.; Zeng, Y.; Grumbine, M.; Sweet, I.R.; Kasai, T.; et al. Mitochondrial Control of Hypoxia-Induced Pathological Retinal Angiogenesis. Angiogenesis 2024, 27, 691–699. [Google Scholar] [CrossRef]
- Luo, Y.; Liao, W.; Chen, Y.; Cui, J.; Liu, F.; Jiang, C.; Gao, W.; Gao, Y. Altitude Can Alter the mtDNA Copy Number and nDNA Integrity in Sperm. J. Assist. Reprod. Genet. 2011, 28, 951–956. [Google Scholar] [CrossRef]
- Jiang, J.; Jiang, Y.; Zhang, Y.-G.; Zhang, T.; Li, J.-H.; Huang, D.-L.; Hou, J.; Tian, M.-Y.; Sun, L.; Su, X.-M.; et al. The Effects of Hypoxia on Mitochondrial Function and Metabolism in Gastric Cancer Cells. Transl. Cancer Res. TCR 2021, 10, 817–826. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, M.; Liang, Y.; Li, R.; Zhang, L.; Chen, S.; Yang, K.; Ding, H.; Tan, X.; Zhang, Q.; et al. Study of Transcriptomic Analysis of Yak (Bos grunniens) and Cattle (Bos taurus) Pulmonary Artery Smooth Muscle Cells Under Oxygen Concentration Gradients and Differences in Their Lung Histology and Expression of Pyruvate Dehydrogenase Kinase 1-Related Factors. Animals 2023, 13, 3450. [Google Scholar] [CrossRef]
- Jing, X.; Ding, L.; Zhou, J.; Huang, X.; Degen, A.; Long, R. The Adaptive Strategies of Yaks to Live in the Asian Highlands. Anim. Nutr. 2022, 9, 249–258. [Google Scholar] [CrossRef]
- Long, L.; Zhu, Y.; Li, Z.; Zhang, H.; Liu, L.; Bai, J. Differential Expression of Skeletal Muscle Mitochondrial Proteins in Yak, Dzo, and Cattle: A Proteomics-Based Study. J. Vet. Med. Sci. 2020, 82, 1178–1186. [Google Scholar] [CrossRef]
- Lucero García Rojas, E.Y.; Villanueva, C.; Bond, R.A. Hypoxia Inducible Factors as Central Players in the Pathogenesis and Pathophysiology of Cardiovascular Diseases. Front. Cardiovasc. Med. 2021, 8, 709509. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, J.; Wang, J.; Chai, Z.; Zhang, C.; Xin, J.; Wang, J.; Cai, X.; Wu, Z.; Ji, Q. Whole-Transcriptome Analysis of Yak and Cattle Heart Tissues Reveals Regulatory Pathways Associated with High-Altitude Adaptation. Front. Genet. 2021, 12, 579800. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, Q.; He, Y.; Yang, L.; Zhang, X.; Shi, P.; Yang, L.; Liu, Z.; Zhang, F.; Liu, F.; et al. The Transcriptomic Landscape of Yaks Reveals Molecular Pathways for High Altitude Adaptation. Genome Biol. Evol. 2018, 11, 72–85. [Google Scholar] [CrossRef]
- Jain, I.H.; Calvo, S.E.; Markhard, A.L.; Skinner, O.S.; To, T.-L.; Ast, T.; Mootha, V.K. Genetic Screen for Cell Fitness in High or Low Oxygen Highlights Mitochondrial and Lipid Metabolism. Cell 2020, 181, 716–727.e11. [Google Scholar] [CrossRef]
- Wen, Y.; Li, S.; Zhao, F.; Wang, J.; Liu, X.; Hu, J.; Bao, G.; Luo, Y. Changes in the Mitochondrial Dynamics and Functions Together with the mRNA/miRNA Network in the Heart Tissue Contribute to Hypoxia Adaptation in Tibetan Sheep. Animals 2022, 12, 583. [Google Scholar] [CrossRef]
- Yan, Z.; Yang, J.; Wei, W.-T.; Zhou, M.-L.; Mo, D.-X.; Wan, X.; Ma, R.; Wu, M.-M.; Huang, J.-H.; Liu, Y.-J.; et al. A Time-Resolved Multi-Omics Atlas of Transcriptional Regulation in Response to High-Altitude Hypoxia across Whole-Body Tissues. Nat. Commun. 2024, 15, 3970. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Zhang, M.; Li, X.; Zhang, F.; Zhou, M.; Yang, K.; Chen, W.; Ding, H.; Tan, X.; et al. Study on the Regulatory Mechanism of the PDK1-Mediated TGF-β/Smad Signaling Pathway in Hypoxia-Induced Yak Lungs. Animals 2024, 14, 2422. [Google Scholar] [CrossRef]
- Tian, M.-Y.; Fan, J.-H.; Zhuang, Z.-W.; Dai, F.; Wang, C.-Y.; Hou, H.-T.; Ma, Y.-Z. Effects of Silymarin on P65 NF-κB, P38 MAPK and CYP450 in LPS-Induced Hoof Dermal Inflammatory Cells of Dairy Cows. BMC Vet. Res. 2019, 15, 127. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, H.; Liu, L.; Kong, M.; Yin, T.; Zhao, X. Mitochondrial Haplotypes Influence Metabolic Traits across Bovine Inter- and Intra-Species Cybrids. Sci. Rep. 2017, 7, 4179. [Google Scholar] [CrossRef]
- Tábara, L.C.; Burr, S.P.; Frison, M.; Chowdhury, S.R.; Paupe, V.; Nie, Y.; Johnson, M.; Villar-Azpillaga, J.; Viegas, F.; Segawa, M.; et al. MTFP1 Controls Mitochondrial Fusion to Regulate Inner Membrane Quality Control and Maintain mtDNA Levels. Cell 2024, 187, 3619–3637.e27. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-Induced Energy Stress Regulates mRNA Translation and Cell Growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef]
- Alam, M.R.; Kim, D.-K. Alterations in Telomere Length and Mitochondrial DNA Copy Number in Human Lymphocytes on Short-Term Exposure to Moderate Hypoxia. Toxicol. Rep. 2020, 7, 1443–1447. [Google Scholar] [CrossRef]
- Xu, Y.-Y.; Liu, Y.; Cui, L.; Wu, W.-B.; Quinn, M.J.; Menon, R.; Zhang, H.-J. Hypoxic Effects on the Mitochondrial Content and Functions of the Placenta in Fetal Growth Restriction. Placenta 2021, 114, 100–107. [Google Scholar] [CrossRef]
- Ciano, M.; Fuszard, M.; Heide, H.; Botting, C.H.; Galkin, A. Conformation-Specific Crosslinking of Mitochondrial Complex I. FEBS Lett. 2013, 587, 867–872. [Google Scholar] [CrossRef]
- Wang, J.; Shi, Y.; Elzo, M.A.; Dang, S.; Jia, X.; Lai, S. Genetic Diversity of ATP8 and ATP6 Genes Is Associated with High-Altitude Adaptation in Yak. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2017, 29, 385–393. [Google Scholar] [CrossRef]
- Mahalingam, S.; Cheviron, Z.A.; Storz, J.F.; McClelland, G.B.; Scott, G.R. Chronic Cold Exposure Induces Mitochondrial Plasticity in Deer Mice Native to High Altitudes. J. Physiol. 2020, 598, 5411–5426. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Brand, M.D. The Regulation and Physiology of Mitochondrial Proton Leak. Physiology 2011, 26, 192–205. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Y.; Xia, C.; Gao, P.; Liu, J. Evidence for Involvement of Uncoupling Proteins in Cerebral Mitochondrial Oxidative Phosphorylation Deficiency of Rats Exposed to 5,000 m High Altitude. Neurochem. Res. 2012, 38, 282–289. [Google Scholar] [CrossRef]
- Olesen, K.; Moruzzi, N.; Bulatovic, I.; Folmes, C.; Jeon, R.; Felldin, U.; Terzic, A.; Simonson, O.E.; Le Blanc, K.; Österholm, C. Diversity of Respiratory Parameters and Metabolic Adaptation to Low Oxygen Tension in Mesenchymal Stromal Cells. Metabol. Open 2022, 13, 100167. [Google Scholar] [CrossRef]
- Bendjilali, N.; MacLeon, S.; Kalra, G.; Willis, S.D.; Hossian, A.K.M.N.; Avery, E.; Wojtowicz, O.; Hickman, M.J. Time-Course Analysis of Gene Expression During the Saccharomyces cerevisiae Hypoxic Response. G3 Genes Genomes Genet. 2017, 7, 221–231. [Google Scholar] [CrossRef]
- Arnaiz, E.; Miar, A.; Dias Junior, A.G.; Prasad, N.; Schulze, U.; Waithe, D.; Nathan, J.A.; Rehwinkel, J.; Harris, A.L. Hypoxia Regulates Endogenous Double-Stranded RNA Production via Reduced Mitochondrial DNA Transcription. Front. Oncol. 2021, 11, 779739. [Google Scholar] [CrossRef]
- Peciuliene, I.; Jakubauskiene, E.; Vilys, L.; Zinkeviciute, R.; Kvedaraviciute, K.; Kanopka, A. Short-Term Hypoxia in Cells Induces Expression of Genes Which Are Enhanced in Stressed Cells. Genes 2022, 13, 1596. [Google Scholar] [CrossRef]
- Cheng, L.; Meliala, I.; Kong, Y.; Chen, J.; Proud, C.G.; Björklund, M. PEBP1 Amplifies Mitochondrial Dysfunction Induced Integrated Stress Response. eLife 2025, 13, RP102852. [Google Scholar] [CrossRef]
- Izquierdo, E.; Cuevas, V.D.; Fernández-Arroyo, S.; Riera-Borrull, M.; Orta-Zavalza, E.; Joven, J.; Rial, E.; Corbi, A.L.; Escribese, M.M. Reshaping of Human Macrophage Polarization through Modulation of Glucose Catabolic Pathways. J. Immunol. 2015, 195, 2442–2451. [Google Scholar] [CrossRef]
- Diaz, J.E.; Ahsen, M.E.; Schaffter, T.; Chen, X.; Realubit, R.B.; Karan, C.; Califano, A.; Losic, B.; Stolovitzky, G. The Transcriptomic Response of Cells to a Drug Combination Is More than the Sum of the Responses to the Monotherapies. eLife 2020, 9, e52707. [Google Scholar] [CrossRef]
- Xin, J.-W.; Chai, Z.-X.; Zhang, C.-F.; Zhang, Q.; Zhu, Y.; Cao, H.-W.; Ji, Q.-M.; Zhong, J.-C. Transcriptome Profiles Revealed the Mechanisms Underlying the Adaptation of Yak to High-Altitude Environments. Sci. Rep. 2019, 9, 7558. [Google Scholar] [CrossRef]
- Willson, J.A.; Arienti, S.; Sadiku, P.; Reyes, L.; Coelho, P.; Morrison, T.; Rinaldi, G.; Dockrell, D.H.; Whyte, M.K.B.; Walmsley, S.R. Neutrophil HIF-1α Stabilization Is Augmented by Mitochondrial ROS Produced via the Glycerol 3-Phosphate Shuttle. Blood 2022, 139, 281–286. [Google Scholar] [CrossRef]
- Passarella, S.; Schurr, A.; Portincasa, P. Mitochondrial Transport in Glycolysis and Gluconeogenesis: Achievements and Perspectives. Int. J. Mol. Sci. 2021, 22, 12620. [Google Scholar] [CrossRef]
- Huang, M.; Yang, L.; Peng, X.; Wei, S.; Fan, Q.; Yang, S.; Li, X.; Li, B.; Jin, H.; Wu, B.; et al. Autonomous Glucose Metabolic Reprogramming of Tumour Cells under Hypoxia: Opportunities for Targeted Therapy. J. Exp. Clin. Cancer Res. 2020, 39, 185. [Google Scholar] [CrossRef] [PubMed]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Tao, M.-Q.; Wu, Y.-J.; Xu, L.; Yang, Z.; Li, Y.; Olatunji, O.J.; Wang, X.-W.; Zuo, J. Metabolic Enzyme Triosephosphate Isomerase 1 and Nicotinamide Phosphoribosyltransferase, Two Independent Inflammatory Indicators in Rheumatoid Arthritis: Evidences From Collagen-Induced Arthritis and Clinical Samples. Front. Immunol. 2022, 12, 795626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, J.; Long, Y.; Maimaitijiang, A.; Su, Z.; Li, W.; Li, J. Unraveling the Guardian: P53’s Multifaceted Role in the DNA Damage Response and Tumor Treatment Strategies. Int. J. Mol. Sci. 2024, 25, 12928. [Google Scholar] [CrossRef]
- Voskarides, K.; Giannopoulou, N. The Role of TP53 in Adaptation and Evolution. Cells 2023, 12, 512. [Google Scholar] [CrossRef]
- Deng, H.; Cheng, Y.; Guo, Z.; Zhang, F.; Lu, X.; Feng, L.; Wang, X.; Xu, Z. Overexpression of CyclinA2 Ameliorates Hypoxia-Impaired Proliferation of Cardiomyocytes. Exp. Ther. Med. 2014, 8, 1513–1517. [Google Scholar] [CrossRef]
- Chen, R.; Wang, H.; Zheng, C.; Zhang, X.; Li, L.; Wang, S.; Chen, H.; Duan, J.; Zhou, X.; Peng, H.; et al. Polo-like Kinase 1 Promotes Pulmonary Hypertension. Respir. Res. 2023, 24, 204. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Liao, Y.; Peng, W.; Xiang, H.; Wang, H.; Ma, J.; Chai, Z.; Wu, Z.; Yue, B.; Cai, X.; et al. Metabolic Plasticity and Transcriptomic Reprogramming Orchestrate Hypoxia Adaptation in Yak. Animals 2025, 15, 2084. https://doi.org/10.3390/ani15142084
Huang C, Liao Y, Peng W, Xiang H, Wang H, Ma J, Chai Z, Wu Z, Yue B, Cai X, et al. Metabolic Plasticity and Transcriptomic Reprogramming Orchestrate Hypoxia Adaptation in Yak. Animals. 2025; 15(14):2084. https://doi.org/10.3390/ani15142084
Chicago/Turabian StyleHuang, Ci, Yilie Liao, Wei Peng, Hai Xiang, Hui Wang, Jieqiong Ma, Zhixin Chai, Zhijuan Wu, Binglin Yue, Xin Cai, and et al. 2025. "Metabolic Plasticity and Transcriptomic Reprogramming Orchestrate Hypoxia Adaptation in Yak" Animals 15, no. 14: 2084. https://doi.org/10.3390/ani15142084
APA StyleHuang, C., Liao, Y., Peng, W., Xiang, H., Wang, H., Ma, J., Chai, Z., Wu, Z., Yue, B., Cai, X., Zhong, J., & Wang, J. (2025). Metabolic Plasticity and Transcriptomic Reprogramming Orchestrate Hypoxia Adaptation in Yak. Animals, 15(14), 2084. https://doi.org/10.3390/ani15142084