
Exploration of Multi-Source Lignocellulose-Degrading Microbial Resources and Bioaugmentation Strategies: Implications for Rumen Efficiency

Simple Summary
Abstract
1. Introduction
2. Lignocellulose Degradation Mechanisms
2.1. Degradation Mechanism of Lignin
2.2. Degradation Mechanism of Cellulose
2.3. Degradation Mechanism of Hemicellulose
3. Sources and Systems of Cellulose-Degrading Microorganisms
3.1. Insect Gut

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Host Source | Cellulose-Degrading Bacteria | Microbe Phylum, Genus | References |
|---|---|---|---|---|
| 2018 | Achatina fulica | B. tequilensis G9 | Firmicutes | [26] |
| 2019 | Bamboo weevils | No specific strain was identified | — | [13] |
| 2021 | Desert Locust | Bacillus safensis MED1, Bacillus sp. CACO | Firmicutes | [29] |
| 2021 | Indralaya | Staphylococcus sp. Microbacterium sp. Bacillus sp. Brevibacterium sp. | Staphylococcus, Microorganisms, Bacillus, Brevibacterium | [27] |
| 2021 | Cotton bollworm | 25 strains (Firmicutes, Proteobacteria) | Firmicutes and Proteobacteria | [28] |
3.2. Ruminant Gastrointestinal Tract
3.2.1. Cellulose-Degrading Microorganisms from Cattle Rumen
3.2.2. Cellulose-Degrading Microorganisms from Sheep Rumen
3.2.3. Cellulose-Degrading Microorganisms from Other Ruminants
| Year | Host Source | Isolated Cellulose-Degrading Bacteria | Microbes Phylum, Genus | References |
|---|---|---|---|---|
| 2017 | Indonesian Aceh bovine rumen | Bacteroides sp. S1 | Probably a new species of Enterobacteriaceae. | [14] |
| 2017 | Cattle rumen | ZH-4 | Proteobacteria | [32] |
| 2022 | Rumen microbial community in yak | Uncultured microbiota | — | [33] |
| 2019 | Sheep rumen | Enterobacter cloacae subsp. KLCD08 | Genus Bacillus | [34] |
| 2024 | Sheep digestive tract | Bacillus tequilensis Isolate2 | Bacillus, Lysinibacillus, Priestia | [35] |
| 2022 | Wujimu Sheep | 22 isolates | Actinomycetales, Firmicutes, Proteobacteria and Ascomycetes | [36] |
| 2022 | Camel rumen | Aspergillus sydowii C6d | Ascomycota | [37] |
| 2024 | The feces of Southeast Asian banteng, Javan banteng, muntjac, and Timor deer. | Five facultative anaerobic strains | — | [38] |
3.3. Forest Soil-Derived Cellulose-Degrading Microorganisms
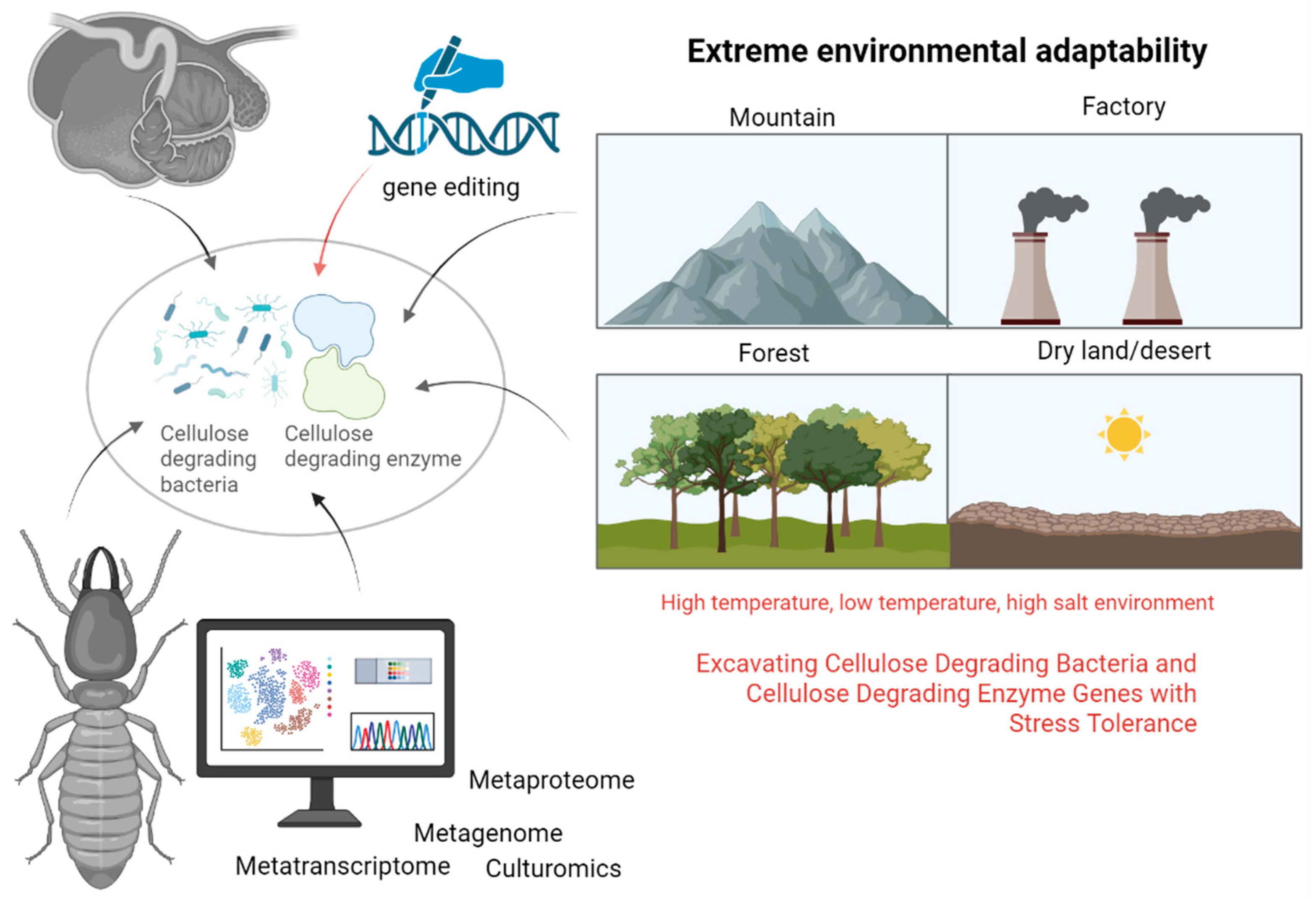
3.4. Other Source Systems
4. Mining and Separation Techniques for Cellulose-Degrading Bacteria
4.1. Screening Strategies for Aerobic and Anaerobic Bacteria
4.2. Microbial Culturomics
5. Classification of Major Cellulose-Degrading Microorganisms
5.1. Bacteria
5.1.1. Fibrobacter succinogenes
5.1.2. Ruminococcus albus
5.1.3. Ruminococcus flavefaciens
5.2. Fungi
5.2.1. White-Rot Fungi
5.2.2. Brown-Rot Fungi
5.2.3. Yeast
6. Mining of Cellulose-Degrading Enzyme Genes
6.1. Metagenomics
6.2. Metatranscriptomics
6.3. Metaproteomics
6.4. Gene Editing Technology
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, A.; Zhang, X.; Liang, Q.; Sun, M. Co-application of straw incorporation and biochar addition stimulated soil N2O and NH3 productions. PLoS ONE 2024, 19, e0289300. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Xu, X.; Wang, C.; Bai, Y.; Fu, C.; Zhang, L.; Fu, R.; Wang, Y. Environmental burdens of the comprehensive utilization of straw: Wheat straw utilization from a life-cycle perspective. J. Clean. Prod. 2020, 259, 120702. [Google Scholar] [CrossRef]
- Chen, L.; Hong, F.; Yang, X.-X.; Han, S.-F. Biotransformation of wheat straw to bacterial cellulose and its mechanism. Bioresour. Technol. 2013, 135, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Zou, H.; Qian, C.; Yu, Y.; Hao, Y.; Li, L.; Wang, Q.; Jiang, Y.; Ma, J. Construction of in situ degradation bacteria of corn straw and analysis of its degradation efficiency. Ann. Microbiol. 2020, 70, 62. [Google Scholar] [CrossRef]
- Trubetskaya, A.; Jensen, P.A.; Jensen, A.D.; Steibel, M.; Spliethoff, H.; Glarborg, P.; Larsen, F.H. Comparison of high temperature chars of wheat straw and rice husk with respect to chemistry, morphology and reactivity. Biomass Bioenergy 2016, 86, 76–87. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Sun, X.; Wang, Y.; Liu, Z. Separation and characterization of biomass components (cellulose, hemicellulose, and lignin) from corn stalk. BioResources 2021, 16, 7205–7219. [Google Scholar] [CrossRef]
- Hsu, T.-C.; Guo, G.-L.; Chen, W.-H.; Hwang, W.-S. Effect of dilute acid pretreatment of rice straw on structural properties and enzymatic hydrolysis. Bioresour. Technol. 2010, 101, 4907–4913. [Google Scholar] [CrossRef]
- Qiu, C.; Liu, N.; Diao, X.; He, L.; Zhou, H.; Zhang, W. Effects of Cellulase and Xylanase on Fermentation Characteristics, Chemical Composition and Bacterial Community of the Mixed Silage of King Grass and Rice Straw. Microorganisms 2024, 12, 561. [Google Scholar] [CrossRef]
- Meng, Z.; Ma, J.; Sun, Z.; Yang, C.; Leng, J.; Zhu, W.; Cheng, Y. Characterization of a novel bifunctional enzyme from buffalo rumen metagenome and its effect on in vitro ruminal fermentation and microbial community composition. Anim. Nutr. 2023, 13, 137–149. [Google Scholar] [CrossRef]
- Fang, M.; Sun, X.; Yao, F.; Lu, L.; Ma, X.; Shao, K.; Kaimoyo, E. A Combination of Transcriptome and Enzyme Activity Analysis Unveils Key Genes and Patterns of Corncob Lignocellulose Degradation by Auricularia heimuer under Cultivation Conditions. J. Fungi 2024, 10, 545. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Ru, H.; He, T.; Sun, N. Study on Microbial Community Succession and Functional Analysis during Biodegradation of Mushroom Residue. BioMed Res. Int. 2021, 2021, 6620574. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Shen, X.; Gang, L.; Xu, H.; Wu, F.; Sheng, L. A novel lignin degradation bacteria-Bacillus amyloliquefaciens SL-7 used to degrade straw lignin efficiently. Bioresour. Technol. 2020, 310, 123445. [Google Scholar] [CrossRef]
- Luo, C.; Li, Y.; Chen, Y.; Fu, C.; Nong, X.; Yang, Y. Degradation of bamboo lignocellulose by bamboo snout beetle Cyrtotrachelus buqueti in vivo and vitro: Efficiency and mechanism. Biotechnol. Biofuels 2019, 12, 75. [Google Scholar] [CrossRef]
- Sari, W.N.; Safika; Darmawi; Fahrimal, Y. Isolation and identification of a cellulolytic Enterobacter from rumen of Aceh cattle. Vet. World 2017, 10, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Chen, H.; Liu, D.; Feng, C.; Hua, Y.; Gu, T.; Guo, X.; Zhou, Y.; Wang, H.; Tong, G.; et al. Soil-derived cellulose-degrading bacteria: Screening, identification, the optimization of fermentation conditions, and their whole genome sequencing. Front. Microbiol. 2024, 15, 1409697. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, B.; Luo, L.; Zhang, F.; Yi, Y.; Shan, Y.; Liu, B.; Zhou, Y.; Wang, X.; Lü, X. A review on recycling techniques for bioethanol production from lignocellulosic biomass. Renew. Sustain. Energy Rev. 2021, 149, 111370. [Google Scholar] [CrossRef]
- Zhang, S.; Dong, Z.; Shi, J.; Yang, C.; Fang, Y.; Chen, G.; Chen, H.; Tian, C. Enzymatic hydrolysis of corn stover lignin by laccase, lignin peroxidase, and manganese peroxidase. Bioresour. Technol. 2022, 361, 127699. [Google Scholar] [CrossRef] [PubMed]
- Falade, A.O.; Nwodo, U.U.; Iweriebor, B.C.; Green, E.; Mabinya, L.V.; Okoh, A.I. Lignin peroxidase functionalities and prospective applications. Microbiol. Open 2017, 6, e00394. [Google Scholar] [CrossRef]
- Atiwesh, G.; Parrish, C.C.; Banoub, J.; Le, T.-A.T. Lignin degradation by microorganisms: A review. Biotechnol. Prog. 2022, 38, e3226. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.-P.; Sain, M. Biocomposites reinforced with natural fibers: 2000–2010. Prog. Polym. Sci. 2012, 37, 1552–1596. [Google Scholar] [CrossRef]
- Wohlert, M.; Benselfelt, T.; Wågberg, L.; Furó, I.; Berglund, L.A.; Wohlert, J. Cellulose and the role of hydrogen bonds: Not in charge of everything. Cellulose 2022, 29, 1–23. [Google Scholar] [CrossRef]
- Weimer, P.J. Degradation of Cellulose and Hemicellulose by Ruminal Microorganisms. Microorganisms 2022, 10, 2345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Wang, R.X.; Zhou, J.S.; Cheng, J.F.; Li, Y.H. An assessment of the genomics, comparative genomics and cellulose degradation potential of Mucilaginibacter polytrichastri strain RG4-7. Bioresour. Technol. 2020, 297, 122389. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Ríos, K.L.; Dejonghe, W.; Vanbroekhoven, K.; Rakotoarivonina, H.; Rémond, C. Enzymatic Production of Xylo-oligosaccharides from Destarched Wheat Bran and the Impact of Their Degree of Polymerization and Substituents on Their Utilization as a Carbon Source by Probiotic Bacteria. J. Agric. Food Chem. 2021, 69, 13217–13226. [Google Scholar] [CrossRef]
- Tao, J.; Song, S.; Qu, C. Recent Progress on Conversion of Lignocellulosic Biomass by MOF-Immobilized Enzyme. Polymers 2024, 16, 1010. [Google Scholar] [CrossRef]
- Dar, M.A.; Pawar, K.D.; Pandit, R.S. Prospecting the gut fluid of giant African land snail, Achatina fulica for cellulose degrading bacteria. Int. Biodeterior. Biodegrad. 2018, 126, 103–111. [Google Scholar] [CrossRef]
- Oktiarni, D.; Hermansyah; Hasanudin; Miksusanti; Nofyan, E.; Kasmiarti, G. Isolation and Identification Cellulolytic Bacteria from Termite Gut Obtained from Indralaya Peatland area. IOP Conf. Ser. Earth Environ. Sci. 2021, 926, 012024. [Google Scholar] [CrossRef]
- Dar, M.A.; Shaikh, A.F.; Pawar, K.D.; Xie, R.; Sun, J.; Kandasamy, S.; Pandit, R.S. Evaluation of cellulose degrading bacteria isolated from the gut-system of cotton bollworm, Helicoverpa armigera and their potential values in biomass conversion. PeerJ 2021, 9, e11254. [Google Scholar] [CrossRef]
- Nelson, K.; Muge, E.; Wamalwa, B. Cellulolytic Bacillus species isolated from the gut of the desert locust Schistocerca gregaria. Sci. Afr. 2021, 11, e00665. [Google Scholar] [CrossRef]
- Silva, É.B.R.d.; Silva, J.A.R.d.; Silva, W.C.d.; Belo, T.S.; Sousa, C.E.L.; Santos, M.R.P.d.; Neves, K.A.L.; Rodrigues, T.C.G.d.C.; Camargo-Júnior, R.N.C.; Lourenço-Júnior, J.d.B. A Review of the Rumen Microbiota and the Different Molecular Techniques Used to Identify Microorganisms Found in the Rumen Fluid of Ruminants. Animals 2024, 14, 1448. [Google Scholar] [CrossRef]
- Morgavi, D.P.; Kelly, W.J.; Janssen, P.H.; Attwood, G.T. Rumen microbial (meta)genomics and its application to ruminant production. Animal 2013, 7, 184–201. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Liu, Z.-Y.; Hao, M.; Zhang, Y.-F.; Qi, Q.-S. An isolated cellulolytic Escherichia coli from bovine rumen produces ethanol and hydrogen from corn straw. Biotechnol. Biofuels 2017, 10, 165. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, L.; Ke, S.; Chen, X.; Kenéz, Á.; Xu, W.; Wang, D.; Zhang, F.; Li, Y.; Cui, Z.; et al. Yak rumen microbiome elevates fiber degradation ability and alters rumen fermentation pattern to increase feed efficiency. Anim. Nutr. 2022, 11, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Guder, D.G.; Krishna, M.S.R. Isolation and Characterization of Potential Cellulose Degrading Bacteria from Sheep Rumen. J. Pure Appl. Microbiol. 2019, 13, 1831–1839. [Google Scholar] [CrossRef]
- Ben Ghalib, K.; Chadli, M.; Daştan, S.D.; Elmtili, N. Isolation and molecular identification of cellulose-degrading bacteria from rumen sheep ‘’Ovis aries’’ and evaluation of their cellulase production. Sci. Afr. 2024, 26, e02439. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, J.; Wang, B.; Yu, Z. Unraveling aerobic cultivable cellulolytic microorganisms within the gastrointestinal tract of sheep (Ovis aries) and their evaluation for cellulose biodegradation. Can. J. Microbiol. 2022, 68, 237–248. [Google Scholar] [CrossRef]
- Tulsani, N.J.; Jakhesara, S.J.; Hinsu, A.T.; Jyotsana, B.; Dafale, N.A.; Patil, N.V.; Purohit, H.J.; Joshi, C.G. Genome analysis and CAZy repertoire of a novel fungus Aspergillus sydowii C6d with lignocellulolytic ability isolated from camel rumen. Electron. J. Biotechnol. 2022, 59, 36–45. [Google Scholar] [CrossRef]
- Pramartaa, I.Q.; Wiryawan, K.G.; Suharti, S. Microbial Protein Synthesis by Cellulolytic Bacterial Isolates from Feces of Indonesian Endemic Herbivores. Indones. J. Appl. Res. (IJAR) 2024, 5, 147–155. [Google Scholar] [CrossRef]
- Mokale Kognou, A.L.; Chio, C.; Khatiwada, J.R.; Shrestha, S.; Chen, X.; Han, S.; Li, H.; Jiang, Z.-H.; Xu, C.C.; Qin, W. Characterization of Cellulose-Degrading Bacteria Isolated from Soil and the Optimization of Their Culture Conditions for Cellulase Production. Appl. Biochem. Biotechnol. 2022, 194, 5060–5082. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lu, Y.; Yan, H.; Wang, X.; Yi, Y.; Shan, Y.; Liu, B.; Zhou, Y.; Lü, X. Screening of cellulolytic bacteria from rotten wood of Qinling (China) for biomass degradation and cloning of cellulases from Bacillus methylotrophicus. BMC Biotechnol. 2020, 20, 2. [Google Scholar] [CrossRef]
- Naresh, S.; Kunasundari, B.; Gunny, A.A.N.; Teoh, Y.P.; Shuit, S.H.; Ng, Q.H.; Hoo, P.Y. Isolation and Partial Characterisation of Thermophilic Cellulolytic Bacteria from North Malaysian Tropical Mangrove Soil. Trop. Life Sci. Res. 2019, 30, 123–147. [Google Scholar] [CrossRef]
- Khosravi, F.; Khaleghi, M.; Naghavi, H. Screening and identification of cellulose-degrading bacteria from soil and leaves at Kerman province, Iran. Arch. Microbiol. 2021, 204, 88. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Li, C.; Cao, P.; Li, T.; Du, D.; Wang, X.; Zhao, J.; Xiang, W. Massilia cellulosiltytica sp. nov., a novel cellulose-degrading bacterium isolated from rhizosphere soil of rice (Oryza sativa L.) and its whole genome analysis. Antonie Van Leeuwenhoek 2021, 114, 1529–1540. [Google Scholar] [CrossRef]
- Lam, M.Q.; Oates, N.C.; Leadbeater, D.R.; Goh, K.M.; Yahya, A.; Md Salleh, M.; Ibrahim, Z.; Bruce, N.C.; Chong, C.S. Genomic Analysis to Elucidate the Lignocellulose Degrading Capability of a New Halophile Robertkochia solimangrovi. Genes 2022, 13, 2135. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Jianzheng, L.; Jun, X.; Yiyang, F.; Furao, W.; Meng, J. Isolation and characterization of a low-temperature and cellulose-degrading fungus Tausonia pullulans LC-6. Environ. Technol. 2025, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Fu, S.; Zhan, H.; Zhan, Y.; Lucia, L.A. Analysis of the chemical composition and morphological structure of banana pseudo-stem. BioResources 2010, 5, 576–585. [Google Scholar] [CrossRef]
- Legodi, L.M.; La Grange, D.; van Rensburg, E.L.J.; Ncube, I. Isolation of Cellulose Degrading Fungi from Decaying Banana Pseudostem and Strelitzia alba. Enzym. Res. 2019, 2019, 1390890. [Google Scholar] [CrossRef]
- Sinza, E.; Mwakilili, A.; Mpinda, C.; Lyantagaye, S. Cellulase-producing bacteria isolated from Mufindi Paper Mill industrial effluent, Iringa Tanzania. Tanzan. J. Sci. 2021, 47, 204–213. [Google Scholar]
- Demissie, M.S.; Legesse, N.H.; Tesema, A.A. Isolation and characterization of cellulase producing bacteria from forest, cow dung, Dashen brewery and agro-industrial waste. PLoS ONE 2024, 19, e0301607. [Google Scholar] [CrossRef]
- Hussain, A.A.; Abdel-Salam, M.S.; Abo-Ghalia, H.H.; Hegazy, W.K.; Hafez, S.S. Optimization and molecular identification of novel cellulose degrading bacteria isolated from Egyptian environment. J. Genet. Eng. Biotechnol. 2017, 15, 77–85. [Google Scholar] [CrossRef]
- Khatri, D.; Chhetri, S.B.B. Reducing Sugar, Total Phenolic Content, and Antioxidant Potential of Nepalese Plants. BioMed Res. Int. 2020, 2020, 7296859. [Google Scholar] [CrossRef] [PubMed]
- Lanjekar, V.B.; Hivarkar, S.S.; Vasudevan, G.; Joshi, A.; Dhakephalkar, P.K.; Dagar, S.S. Actinomyces ruminis sp. nov., an obligately anaerobic bacterium isolated from the rumen of cattle. Arch. Microbiol. 2022, 205, 9. [Google Scholar] [CrossRef]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Walker, A.W.; Roehe, R.; Watson, M. Compendium of 4941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019, 37, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef]
- Kaminski, T.S.; Scheler, O.; Garstecki, P. Droplet microfluidics for microbiology: Techniques, applications and challenges. Lab A Chip 2016, 16, 2168–2187. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.M.; Bloom, R.J.; Silverman, J.D.; Durand, H.K.; Jiang, S.; Wu, A.; Dallow, E.P.; Huang, S.; You, L.; David, L.A. Interindividual Variation in Dietary Carbohydrate Metabolism by Gut Bacteria Revealed with Droplet Microfluidic Culture. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Zehavi, T.; Probst, M.; Mizrahi, I. Insights Into Culturomics of the Rumen Microbiome. Front. Microbiol. 2018, 9, 1999. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, F.; Ma, X.; Li, F.; Wang, Z. Effects of Barley Starch Level in Diet on Fermentation and Microflora in Rumen of Hu Sheep. Animals 2022, 12, 1941. [Google Scholar] [CrossRef]
- Lee, P.; Lee, S.; Hong, S.; Chang, H. Isolation and characterization of a new succinic acid-producing bacterium, Mannheimia succiniciproducens MBEL55E, from bovine rumen. Appl. Microbiol. Biotechnol. 2002, 58, 663–668. [Google Scholar] [CrossRef]
- Yeoman Carl, J.; Fields Christopher, J.; Lepercq, P.; Ruiz, P.; Forano, E.; White Bryan, A.; Mosoni, P. In Vivo Competitions between Fibrobacter succinogenes, Ruminococcus flavefaciens, and Ruminoccus albus in a Gnotobiotic Sheep Model Revealed by Multi-Omic Analyses. mBio 2021, 12. [Google Scholar] [CrossRef]
- Dassa, B.; Borovok, I.; Ruimy-Israeli, V.; Lamed, R.; Flint, H.J.; Duncan, S.H.; Henrissat, B.; Coutinho, P.; Morrison, M.; Mosoni, P.; et al. Rumen Cellulosomics: Divergent Fiber-Degrading Strategies Revealed by Comparative Genome-Wide Analysis of Six Ruminococcal Strains. PLoS ONE 2014, 9, e99221. [Google Scholar] [CrossRef] [PubMed]
- Boonsaen, P.; Poonko, S.; Kanjanapruetipong, J.; Phiriyangkul, P.; Sawanon, S. Isolation and partial characterization of Ruminococcus flavefaciens from the rumen of swamp buffalo. Buffalo Bull. 2019, 38, 311–325. [Google Scholar]
- Odenyo, A.A.; Mackie, R.I.; Fahey, G.C., Jr.; White, B.A. Degradation of wheat straw and alkaline hydrogen peroxide-treated wheat straw by Ruminococcus albus 8 and Ruminococcus flavefaciens FD-1. J. Anim. Sci. 1991, 69, 819–826. [Google Scholar] [CrossRef]
- van Erven, G.; Nayan, N.; Sonnenberg, A.S.M.; Hendriks, W.H.; Cone, J.W.; Kabel, M.A. Mechanistic insight in the selective delignification of wheat straw by three white-rot fungal species through quantitative 13C-IS py-GC–MS and whole cell wall HSQC NMR. Biotechnol. Biofuels 2018, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.S.; Chander, M.; Gill, P.K. Involvement of lignin peroxidase, manganese peroxidase and laccase in degradation and selective ligninolysis of wheat straw. Int. Biodeterior. Biodegrad. 2002, 50, 115–120. [Google Scholar] [CrossRef]
- Tabka, M.G.; Herpoël-Gimbert, I.; Monod, F.; Asther, M.; Sigoillot, J.C. Enzymatic saccharification of wheat straw for bioethanol production by a combined cellulase xylanase and feruloyl esterase treatment. Enzym. Microb. Technol. 2006, 39, 897–902. [Google Scholar] [CrossRef]
- Shrivastava, B.; Thakur, S.; Khasa, Y.P.; Gupte, A.; Puniya, A.K.; Kuhad, R.C. White-rot fungal conversion of wheat straw to energy rich cattle feed. Biodegradation 2011, 22, 823–831. [Google Scholar] [CrossRef]
- van Erven, G.; Wang, J.; Sun, P.; de Waard, P.; van der Putten, J.; Frissen, G.E.; Gosselink, R.J.A.; Zinovyev, G.; Potthast, A.; van Berkel, W.J.H.; et al. Structural Motifs of Wheat Straw Lignin Differ in Susceptibility to Degradation by the White-Rot Fungus Ceriporiopsis subvermispora. ACS Sustain. Chem. Eng. 2019, 7, 20032–20042. [Google Scholar] [CrossRef]
- Arantes, V.; Jellison, J.; Goodell, B. Peculiarities of brown-rot fungi and biochemical Fenton reaction with regard to their potential as a model for bioprocessing biomass. Appl. Microbiol. Biotechnol. 2012, 94, 323–338. [Google Scholar] [CrossRef]
- Eastwood, D.C.; Floudas, D.; Binder, M.; Majcherczyk, A.; Schneider, P.; Aerts, A.; Asiegbu, F.O.; Baker, S.E.; Barry, K.; Bendiksby, M.; et al. The Plant Cell Wall–Decomposing Machinery Underlies the Functional Diversity of Forest Fungi. Science 2011, 333, 762–765. [Google Scholar] [CrossRef]
- Gaskell, J.; Blanchette, R.A.; Stewart, P.E.; BonDurant, S.S.; Adams, M.; Sabat, G.; Kersten, P.; Cullen, D. Transcriptome and Secretome Analyses of the Wood Decay Fungus Wolfiporia cocos Support Alternative Mechanisms of Lignocellulose Conversion. Appl. Environ. Microbiol. 2016, 82, 3979–3987. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhang, X.; Zhou, Y.; Zhang, C.; Wen, J.; Deng, S.; Luo, B.; Fan, M.; Xia, Y. Selectively enzymatic conversion of wood constituents with white and brown rot fungi. Ind. Crops Prod. 2023, 199, 116703. [Google Scholar] [CrossRef]
- Cömert, M.; Şayan, Y.; Özelçam, H.; Baykal, G.Y. Effects of Saccharomyces cerevisiae Supplementation and Anhydrous Ammonia Treatment of Wheat Straw on In-situ Degradability and, Rumen Fermentation and Growth Performance of Yearling Lambs. Asian-Australas J. Anim. Sci. 2015, 28, 639–646. [Google Scholar] [CrossRef]
- Ben Saïd, S.; Jabri, J.; Amiri, S.; Aroua, M.; Najjar, A.; Khaldi, S.; Maalaoui, Z.; Kammoun, M.; Mahouachi, M. Effect of Saccharomyces cerevisiae Supplementation on Reproductive Performance and Ruminal Digestibility of Queue Fine de l’Ouest Adult Rams Fed a Wheat Straw-Based Diet. Agriculture 2022, 12, 1268. [Google Scholar] [CrossRef]
- Ding, G.; Chang, Y.; Zhao, L.; Zhou, Z.; Ren, L.; Meng, Q. Effect of Saccharomyces cerevisiae on alfalfa nutrient degradation characteristics and rumen microbial populations of steers fed diets with different concentrate-to-forage ratios. J. Anim. Sci. Biotechnol. 2014, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.-P.; Lin, X.-Y.; Hu, Y.-Q.; Chen, Z.-Z.; Chen, L.; Huang, Y.-F.; Huang, X.-H.; Li, J. Metagenomics approach to identify lignocellulose-degrading enzymes in the gut microbiota of the Chinese bamboo rat cecum. Electron. J. Biotechnol. 2021, 50, 29–36. [Google Scholar] [CrossRef]
- Chai, S.; Zhang, X.; Jia, Z.; Xu, X.; Zhang, Y.; Wang, S.; Feng, Z. Identification and characterization of a novel bifunctional cellulase/hemicellulase from a soil metagenomic library. Appl. Microbiol. Biotechnol. 2020, 104, 7563–7572. [Google Scholar] [CrossRef]
- Wang, L.; Hatem, A.; Catalyurek, U.V.; Morrison, M.; Yu, Z. Metagenomic Insights into the Carbohydrate-Active Enzymes Carried by the Microorganisms Adhering to Solid Digesta in the Rumen of Cows. PLoS ONE 2013, 8, e78507. [Google Scholar] [CrossRef]
- He, B.; Jin, S.; Cao, J.; Mi, L.; Wang, J. Metatranscriptomics of the Hu sheep rumen microbiome reveals novel cellulases. Biotechnol. Biofuels 2019, 12, 153. [Google Scholar] [CrossRef]
- Yu, W.; Wu, Y.; Li, D. Oxidative cleavage of cellulose by fungi in the termite gut. Int. J. Biol. Macromol. 2025, 284, 138222. [Google Scholar] [CrossRef]
- Xie, M.; An, F.; Wu, J.; Liu, Y.; Shi, H.; Wu, R. Meta-omics reveal microbial assortments and key enzymes in bean sauce mash, a traditional fermented soybean product. J. Sci. Food Agric. 2019, 99, 6522–6534. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Whon, T.W.; Roh, S.W.; Jeon, C.O. Unraveling microbial fermentation features in kimchi: From classical to meta-omics approaches. Appl. Microbiol. Biotechnol. 2020, 104, 7731–7744. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, A.; Iio, W.; Mitsumori, M.; Minato, H. Isolation and Identification of Cellulose-Binding Proteins from Sheep Rumen Contents. Appl. Environ. Microbiol. 2009, 75, 1667–1673. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, R.; Li, J.; Lin, L.; Zhao, J.; Sun, W.; Tian, C. Development of a genome-editing CRISPR/Cas9 system in thermophilic fungal Myceliophthora species and its application to hyper-cellulase production strain engineering. Biotechnol. Biofuels 2017, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gong, H.; Tang, H.; Meng, Z.; Wang, Z.; Cui, W.; Zhang, K.; Chen, Y.; Yang, Y. Enhanced lignocellulose degradation in Bacillus subtilis RLI2019 through CRISPR/Cas9-mediated chromosomal integration of ternary cellulase genes. Int. J. Biol. Macromol. 2025, 306, 141727. [Google Scholar] [CrossRef]
- Zhou, C.; Zhou, H.; Li, D.; Zhang, H.; Wang, H.; Lu, F. Optimized expression and enhanced production of alkaline protease by genetically modified Bacillus licheniformis 2709. Microb. Cell Factories 2020, 19, 45. [Google Scholar] [CrossRef]


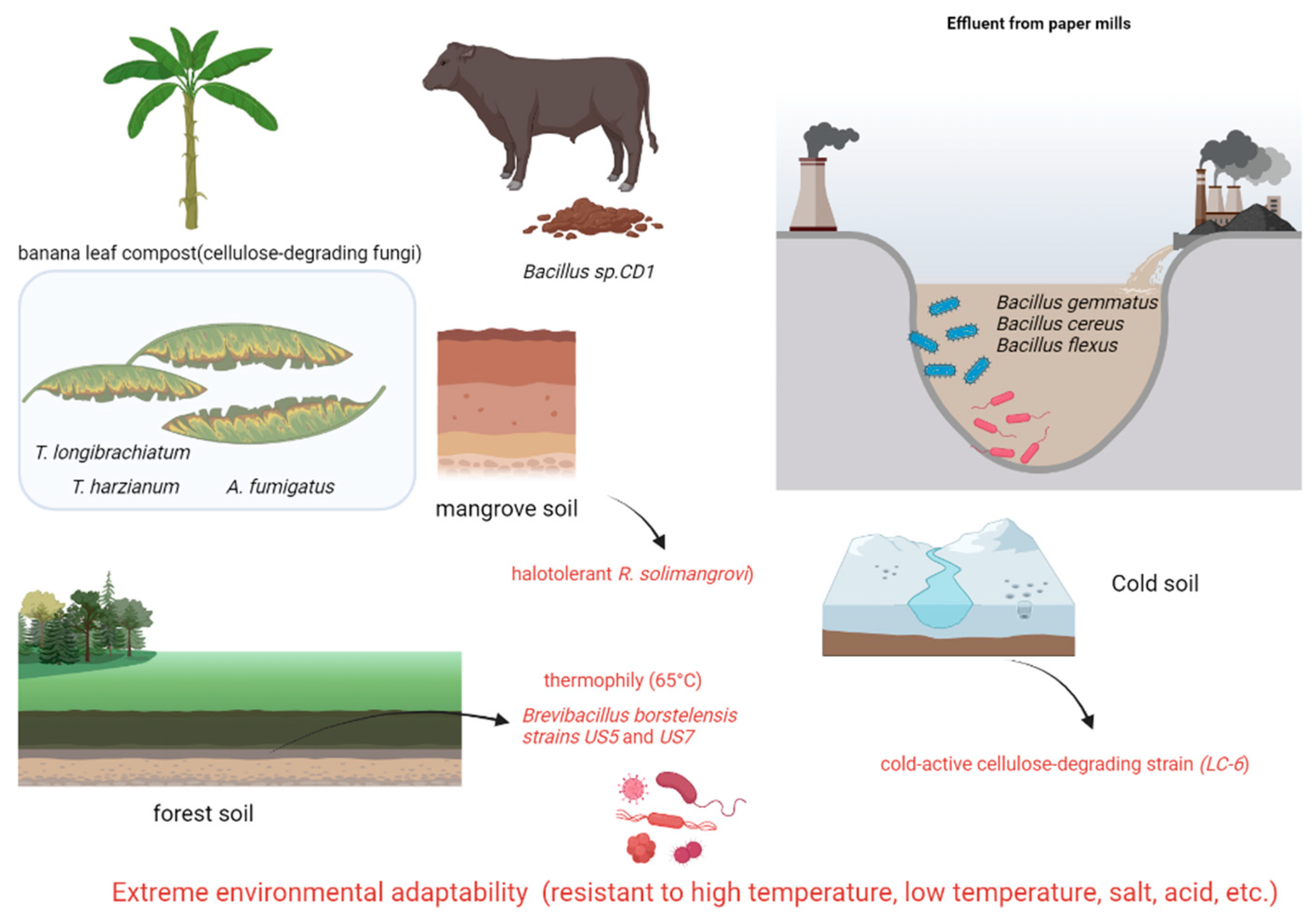
| Year | Source of Flora | Isolated Cellulose-Degrading Bacteria | Microbe Phylum, Genus | References |
|---|---|---|---|---|
| 2018 | Canadian Campus Ginkgo Grove | Bacillus sp. MKAL6 | Firmicutes | [39] |
| 2020 | Qinling Mountain deadwood | Bacillus methylotrophicus 1EJ7 | Proteobacteria | [40] |
| 2019 | Tropical Forest of Malaysia | Anoxybacillus sp. UniMAP-KB06 | Firmicutes | [41] |
| 2021 | Kerman Forest, Iran | Brevibacillus borstelensis US5 | Firmicutes | [42] |
| 2021 | Rice rhizosphere soil | Massilia sp. NEAU-DD11T | Proteobacteria Massilia | [43] |
| 2024 | Soil | Rhodococcus wratislaviensis YZ02, Pseudomonas anthosomatis YZ03 | Actinobacteria Proteobacteria | [15] |
| 2022 | Mangrove soil | Robertkochia solimangrovi | Bacteroidetes | [44] |
| Year | Source of Flora | Isolated Cellulose-Degrading Bacteria | Microbe Phylum, Genus | References |
|---|---|---|---|---|
| 2019 | Banana plantation compost | Trichoderma longibrachiatum Trichoderma harzianum Aspergillus fumigatus | Ascomycota, Trichoderma | [47] |
| 2021 | Paper Mill Wastewater Contaminated Soil (Tanzania) | Bacillus gemmatus Bacillus cereus Bacillus flexus | Firmicutes | [48] |
| 2024 | Cow dung | Bacillus sp. CD1 | Firmicutes | [49] |
| Culture | Key Enzyme Systems | Substrate Specificity | Critical Applications |
|---|---|---|---|
| Fibrobacter succinogenes | Cellulase system (GH family unspecified) pectinase, glucanase, arabinogalactase, xylanase | Cellulose, cellobiose, and glucose preferentially degrade cellulose-hemicellulose-pectin crosslinking network | Industrial production of succinic acid (yield 13.5 g/L) Feed additive (improve the degradation rate of neutral detergent fiber) |
| Ruminococcus Albus | GH5 family enzymes (62.3% of CAZymes) have prominent cellobiase activity | Cellobiose (38% more available than glucose) pH 6.7–5.5 optimum (less than 6.0 growth inhibition) | Complementarity with xanthococcus to maintain rumen microecological balance (proton power 60 mV) |
| Ruminococcus flavus | GH9 family enzymes (51.8% CAZymes). Complex enzyme system (cellulose, hemicellulose, pectin degrading enzymes) | Alkaline hydrogen peroxide treatment of wheat straw (AHPWS degradation rate 6.1 mg/d) needs phenylacetic acid/phenylacetic acid synergism | Co-culture improves fiber digestibility (e.g., OS14 +S137 strains) and promotes acetic acid and propionic acid production |
| Culture | Key Enzyme Systems | Substrate Specificity | Critical Applications |
|---|---|---|---|
| White-rot fungi | Lignin-degrading enzymes (lignin peroxidase, manganese peroxidase, laccase); cellulases, hemicellulases, esterases | Preferential degradation of lignin (β-O-4′ aryl ether bond) significantly reduced lignin (p < 0.05) in wheat straw treated for 30 days | Feed pretreatment (increase crude protein content, metabolic energy); industrial delignification (selective degradation) |
| Brown-rot fungi | Simplified enzyme system (GH family hemicellulases, chitinases), hydroxyl radical mediated oxidative depolymerization of cellulose | Rapid depolymerization of cellulose/hemicellulose in Pinus yunnanensis preferential degradation of polysaccharides | Biomass pretreatment (to increase wood porosity) and white-rot fungi synergize to enhance digestibility |
| Yeast | Indirect regulation (promotion of cellulase secretion by rumen microorganisms), metabolites (e.g., short chain fatty acids) | Free sugar promotes fiber decomposition-Digestibility of crude fiber in wheat straw diet increased by 24% (p < 0.05) | Feed additive (increased dry matter digestibility 7.3%) improved rumen fermentation (increased TVFA, NH3-N) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Qiao, Z.; Chen, C.; Hua, J.; Zhou, C. Exploration of Multi-Source Lignocellulose-Degrading Microbial Resources and Bioaugmentation Strategies: Implications for Rumen Efficiency. Animals 2025, 15, 1920. https://doi.org/10.3390/ani15131920
Lv X, Qiao Z, Chen C, Hua J, Zhou C. Exploration of Multi-Source Lignocellulose-Degrading Microbial Resources and Bioaugmentation Strategies: Implications for Rumen Efficiency. Animals. 2025; 15(13):1920. https://doi.org/10.3390/ani15131920
Chicago/Turabian StyleLv, Xiaokang, Zhanhong Qiao, Chao Chen, Jinling Hua, and Chuanshe Zhou. 2025. "Exploration of Multi-Source Lignocellulose-Degrading Microbial Resources and Bioaugmentation Strategies: Implications for Rumen Efficiency" Animals 15, no. 13: 1920. https://doi.org/10.3390/ani15131920
APA StyleLv, X., Qiao, Z., Chen, C., Hua, J., & Zhou, C. (2025). Exploration of Multi-Source Lignocellulose-Degrading Microbial Resources and Bioaugmentation Strategies: Implications for Rumen Efficiency. Animals, 15(13), 1920. https://doi.org/10.3390/ani15131920