Four Novel SARS-CoV-2 Infected Feral American Mink (Neovison Vison) Among 60 Individuals Caught in the Wild

 , , , , , , ,
, , , , , , ,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
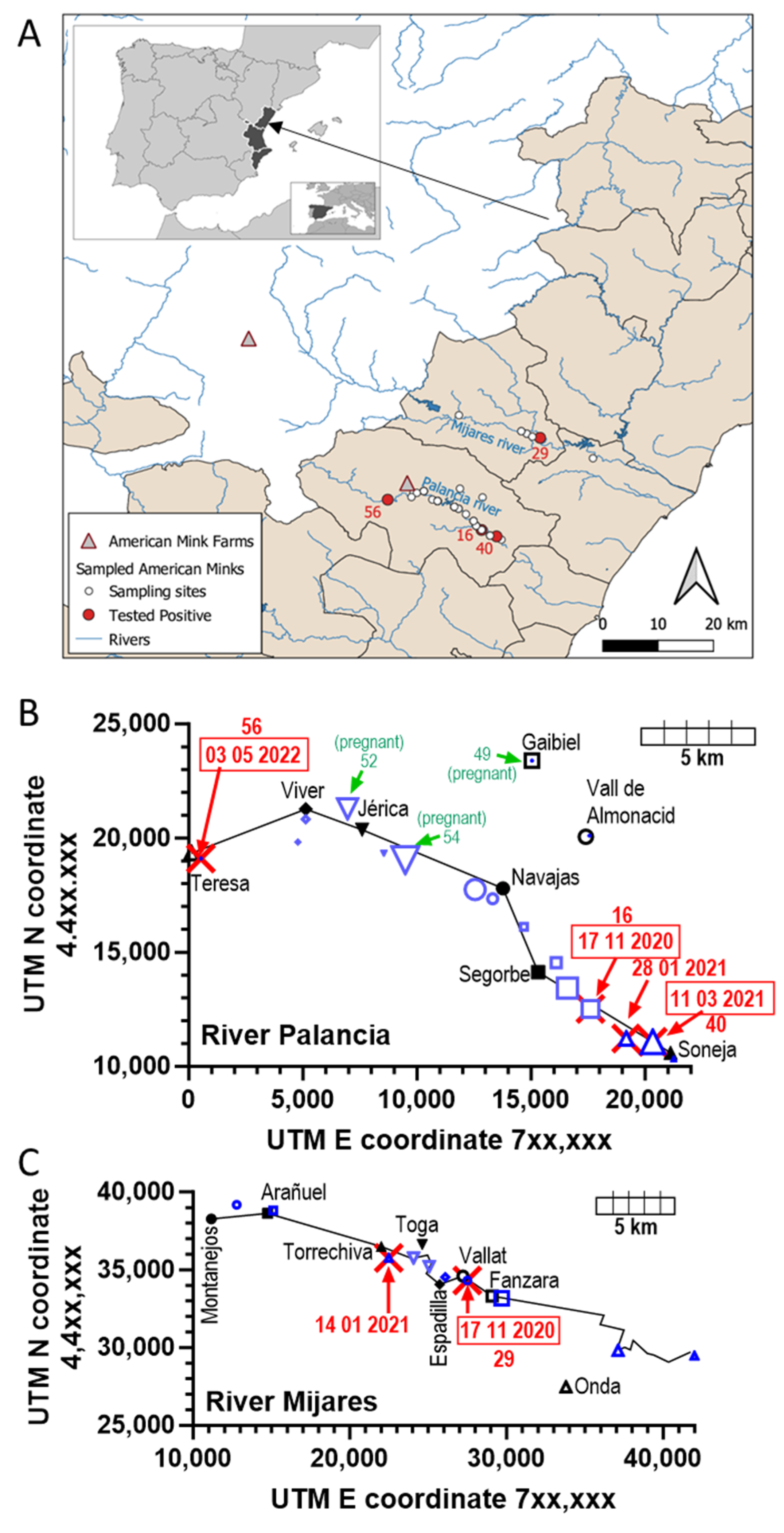
2.1. Study Area, and Procurement of the Samples
2.2. Viral Detection and Molecular Analyses
2.3. Sanger Sequencing and Phylogenetic Analyses
3. Results and Discussion
3.1. Animals Trapped
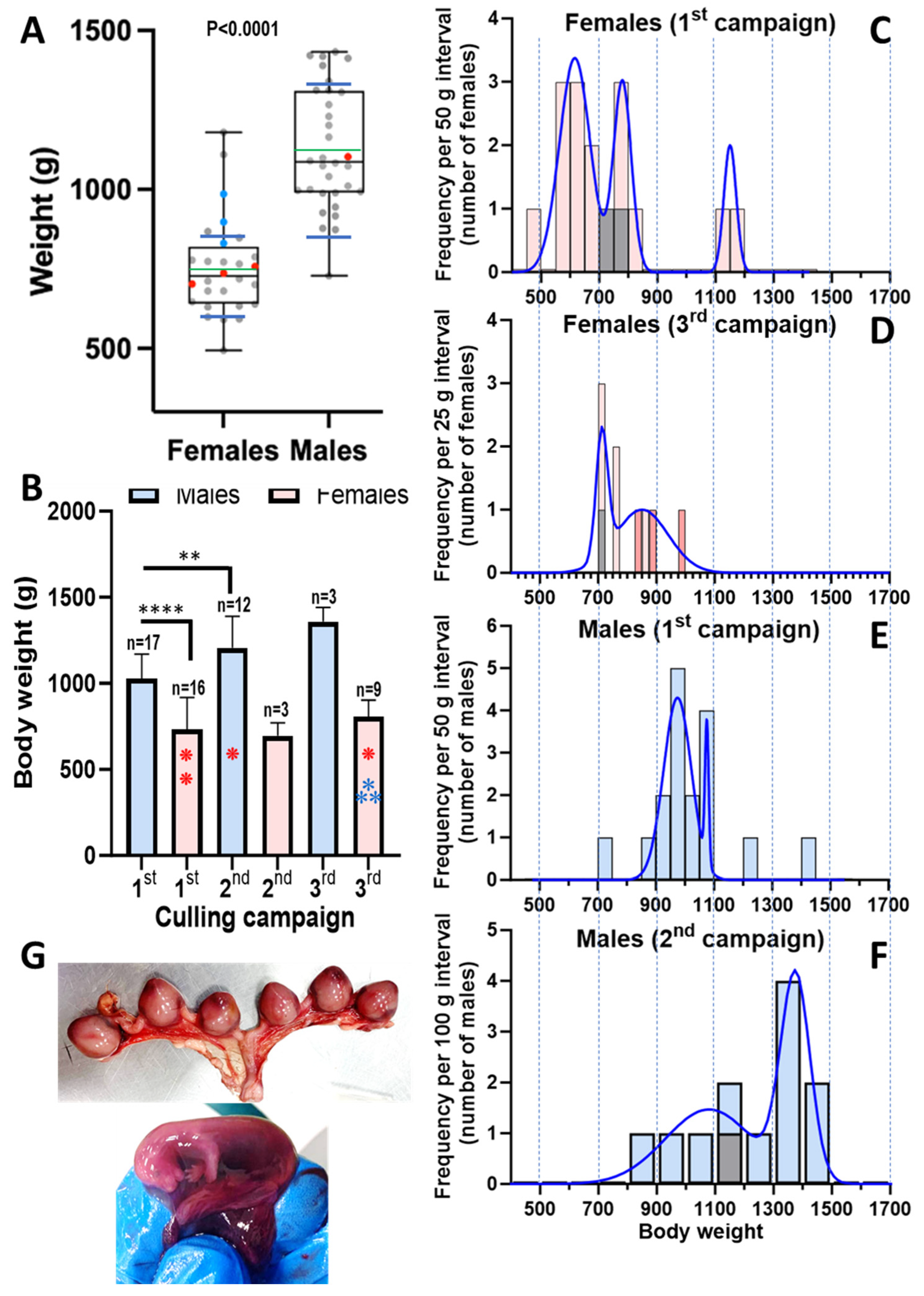
3.2. The Body Weights of the Animals Provide Insight into the Local Population of Feral Mink
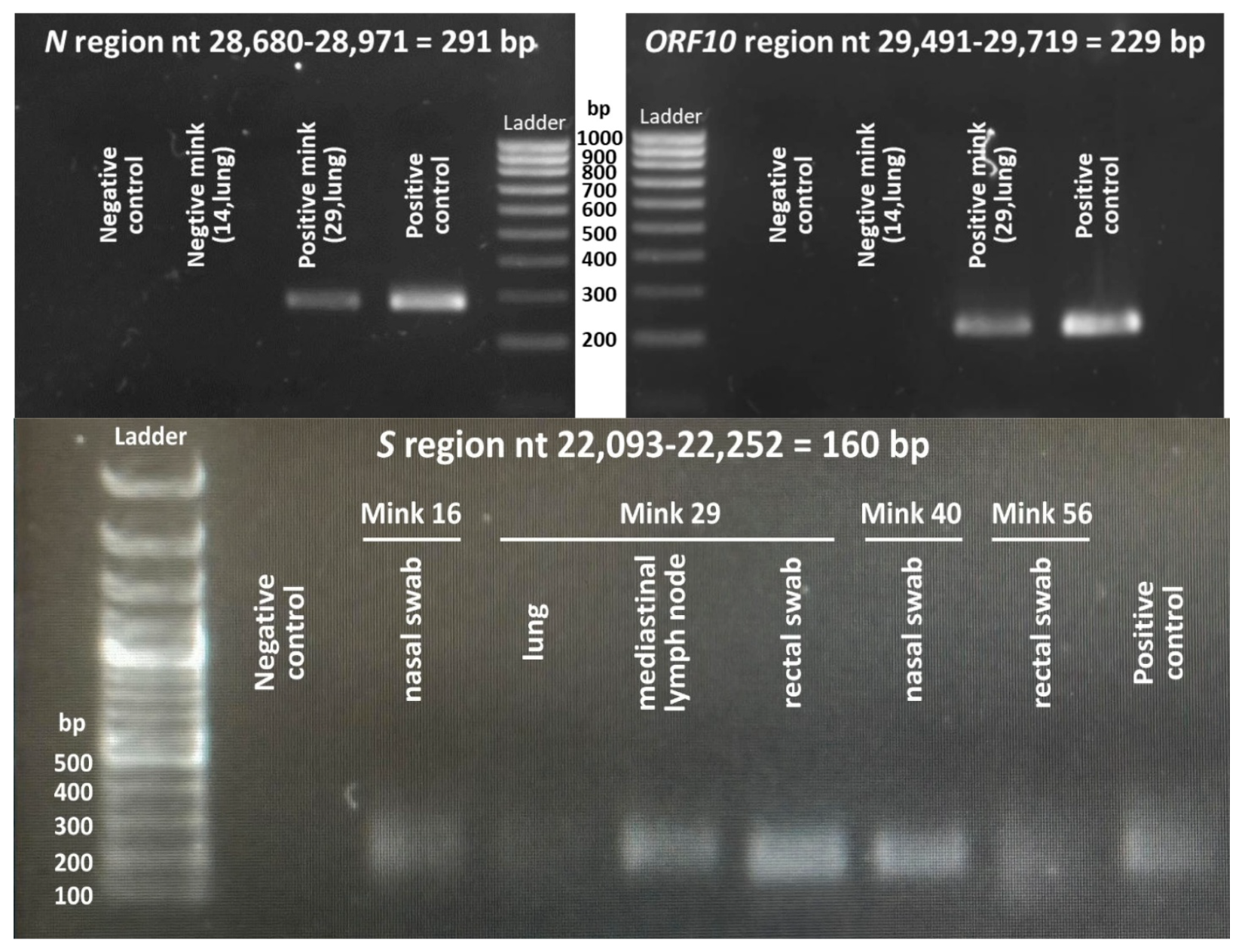
3.3. Four SARS-CoV-2-Positive Animals Were Detected Among the Sixty Animals Trapped
3.4. Partial Gene Sequencing Reveals Common Traits with the B.1.177 and Alpha Variants of SARS-CoV-2
3.5. Final Considerations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SARS-CoV-2: | Severe Acute Respiratory Syndrome Coronavirus 2 |
References
- Hempel, S. The Atlas of Disease: Mapping Deadly Epidemics and Contagion from the Plague to the Zika Virus; White Lion Publishing: London, UK, 2018. [Google Scholar]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- González-Candelas, F.; Shaw, M.A.; Phan, T.; Kulkarni-Kale, U.; Paraskevis, D.; Luciani, F.; Kimura, H.; Sironi, M. One year into the pandemic: Short-term evolution of SARS-CoV-2 and emergence of new lineages. Infect. Genet. Evol. 2021, 92, 104869. [Google Scholar] [CrossRef] [PubMed]
- Bonotti, M.; Zech, S.T. (Eds.) The Human, Economic, Social, and Political Costs of COVID-19. In Recovering Civility During COVID-19; Springer: Singapore, 2021; pp. 1–36. [Google Scholar]
- Nerpel, A.; Käsbohrer, A.; Walzer, C.; Desvars-Larrive, A. Data on SARS-CoV-2 events in animals: Mind the gap! One Health 2023, 1, 100653. [Google Scholar] [CrossRef] [PubMed]
- Jahid, M.J.; Bowman, A.S.; Nolting, J.M. SARS-CoV-2 outbreaks on mink farms-A review of current knowledge on virus infection, spread, spillover, and containment. Viruses 2024, 16, 81. [Google Scholar] [CrossRef]
- Aguiló-Gisbert, J.; Padilla-Blanco, M.; Lizana, V.; Maiques, E.; Muñoz-Baquero, M.; Chillida-Martínez, E.; Cardells, J.; Rubio-Guerri, C. First description of SARS-CoV-2 infection in two feral American mink (Neovison vison) caught in the wild. Animals 2021, 11, 1422. [Google Scholar] [CrossRef]
- Padilla-Blanco, M.; Aguiló-Gisbert, J.; Rubio, V.; Lizana, V.; Chillida-Martínez, E.; Cardells, J.; Maiques, E.; Rubio-Guerri, C. The finding of the severe acute respiratory syndrome coronavirus (SARS-CoV-2) in a wild Eurasian river otter (Lutra lutra) highlights the need for viral surveillance in wild mustelids. Front. Vet. Sci. 2022, 9, 826991. [Google Scholar] [CrossRef]
- Meekins, D.A.; Gaudreault, N.N.; Richt, J.A. Natural and experimental SARS-CoV-2 infection in domestic and wild animals. Viruses 2021, 13, 1993. [Google Scholar] [CrossRef]
- McAloose, D.; Laverack, M.; Wang, L.; Killian, M.L.; Caserta, L.C.; Yuan, F.; Mitchell, P.K.; Queen, K.; Mauldin, M.R.; Cronk, B.D.; et al. From People to Panthera: Natural SARS-CoV-2 Infection in Tigers and Lions at the Bronx Zoo. mBio 2020, 11, e02220-20. [Google Scholar] [CrossRef]
- Nagy, A.; Stará, M.; Vodička, R.; Černíková, L.; Jiřincová, H.; Křivda, V.; Sedlák, K. Reverse-zoonotic transmission of SARS-CoV-2 lineage alpha (B.1.1.7) to great apes and exotic felids in a zoo in the Czech Republic. Arch. Virol. 2022, 167, 1681–1685. [Google Scholar] [CrossRef]
- Feng, A.; Bevins, S.; Chandler, J.; DeLiberto, T.J.; Ghai, R.; Lantz, K.; Lenoch, J.; Retchless, A.; Shriner, S.; Tang, C.Y.; et al. Transmission of SARS-CoV-2 in free-ranging white-tailed deer in the United States. Nat. Commun. 2023, 14, 4078. [Google Scholar] [CrossRef]
- Gómez, J.C.; Cano-Terriza, D.; Segalés, J.; Vergara-Alert, J.; Zorrilla, I.; del Rey, T.; Paniagua, J.; Gonzálvez, M.; Fernández-Bastit, L.; Nájera, F.; et al. Exposure to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the endangered Iberian lynx (Lynx pardinus). Vet. Microbiol. 2024, 290, 110001. [Google Scholar] [CrossRef]
- Manes, C.; Gollakner, R.; Capua, I. Could mustelids spur COVID-19 into a panzootic? Vet. Ital. 2020, 56, 65–66. [Google Scholar] [PubMed]
- Fang, R.; Yang, X.; Guo, Y.; Peng, B.; Dong, R.; Li, S.; Xu, S. SARS-CoV-2 infection in animals: Patterns, transmission routes, and drivers. Eco Environ. Health 2023, 3, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Fenollar, F.; Mediannikov, O.; Maurin, M.; Devaux, C.; Colson, P.; Levasseur, A.; Fournier, P.E.; Raoult, D. Mink, SARS-CoV-2, and the human-animal interface. Front. Microbiol. 2021, 12, 663815. [Google Scholar] [CrossRef]
- Boklund, A.; Hammer, A.S.; Quaade, M.L.; Rasmussen, T.B.; Lohse, L.; Strandbygaard, B.; Jørgensen, C.S.; Olesen, A.S.; Hjerpe, F.B.; Petersen, H.H.; et al. SARS-CoV-2 in Danish Mink Farms: Course of the Epidemic and a Descriptive Analysis of the Outbreaks in 2020. Animals 2021, 11, 164. [Google Scholar] [CrossRef]
- Bayarri-Olmos, R.; Rosbjerg, A.; Johnsen, L.B.; Helgstrand, C.; Bak-Thomsen, T.; Garred, P.; Skjoedt, M.O. The SARS-CoV-2 Y453F mink variant displays a pronounced increase in ACE-2 affinity but does not challenge antibody neutralization. J. Biol. Chem. 2021, 296, 100536. [Google Scholar] [CrossRef]
- Hammer, A.S.; Quaade, M.L.; Rasmussen, T.B.; Fonager, J.; Rasmussen, M.; Mundbjerg, K.; Lohse, L.; Strandbygaard, B.; Jørgensen, C.S.; Alfaro-Núñez, A.; et al. SARS-CoV-2 transmission between mink (Neovison vison) and humans, Denmark. Emerg. Infect. Dis. 2021, 27, 547–551. [Google Scholar] [CrossRef]
- Dip, S.D.; Sarkar, S.L.; Setu, M.A.A.; Das, P.K.; Pramanik, M.H.A.; Ul Alam, A.S.M.R.; Al-Emran, H.M.; Hossain, M.A.; Jahid, I.K. Evaluation of RT-PCR assays for detection of SARS-CoV-2 variants of concern. Sci. Rep. 2023, 13, 2342. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Pagh, S.; Pertoldi, C.; Petersen, H.H.; Jensen, T.H.; Hansen, M.S.; Madsen, S.; Kraft, D.C.E.; Iversen, N.; Roslev, P.; Chriel, M. Methods for the identification of farm escapees in feral mink (Neovison vison) populations. PLoS ONE 2019, 14, e0224559. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Ginex, T.; Marco-Marín, C.; Wieczór, M.; Mata, C.P.; Krieger, J.; Ruiz-Rodriguez, P.; López-Redondo, M.L.; Francés-Gómez, C.; Melero, R.; Sánchez-Sorzano, C.Ó.; et al. The structural role of SARS-CoV-2 genetic background in the emergence and success of spike mutations: The case of the spike A222V mutation. PLoS Pathog. 2022, 18, e1010631. [Google Scholar]
- Rasmussen, T.B.; Qvesel, A.G.; Pedersen, A.G.; Olesen, A.S.; Fonager, J.; Rasmussen, M.; Sieber, R.N.; Stegger, M.; Calvo-Artavia, F.F.; Goedknegt, M.J.F.; et al. Emergence and spread of SARS-CoV-2 variants from farmed mink to humans and back during the epidemic in Denmark, June–November 2020. PLoS Pathog. 2024, 20, e1012039. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Lelli, D.; Trogu, T.; Lavazza, A.; Barbieri, I.; Boniotti, M.; Pezzoni, G.; Salogni, C.; Giovannini, S.; Alborali, G.; et al. SARS-CoV-2 in a Mink Farm in Italy: Case Description, Molecular and Serological Diagnosis by Comparing Different Tests. Viruses 2022, 14, 1738. [Google Scholar] [CrossRef]
- Mistry, P.; Barmania, F.; Mellet, J.; Peta, K.; Strydom, A.; Viljoen, I.M.; James, W.; Gordon, S.; Pepper, M.S. SARS-CoV-2 variants, vaccines, and host immunity. Front. Immunol. 2022, 12, 809244. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O‘Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Murphy, H.L.; Ly, H. Understanding the prevalence of SARS-CoV-2 (COVID-19) exposure in companion, captive, wild, and farmed animals. Virulence 2021, 12, 2777–2786. [Google Scholar] [CrossRef]
- Heneghan, C.J.; Spencer, E.A.; Brassey, J.; Plüddemann, A.; Onakpoya, I.J.; Evans, D.H.; Conly, J.M.; Jefferson, T. SARS-CoV-2 and the role of orofecal transmission: A systematic review. F1000Res 2021, 10, 231. [Google Scholar] [CrossRef]
- Barberá-Riera, M.; de Llanos, R.; Barneo-Muñoz, M.; Bijlsma, L.; Celma, A.; Comas, I.; Gomila, B.; González-Candelas, F.; Goterris-Cerisuelo, R.; Martinez-Garcia, F.; et al. Wastewater monitoring of a community COVID-19 outbreak in a Spanish municipality. J. Environ. Expo. Assess. 2023, 2, 16. [Google Scholar] [CrossRef]
- La Rosa, G.; Iaconelli, M.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Bonadonna, L.; Lucentini, L.; Suffredini, E. First detection of SARS-CoV-2 in untreated wastewaters in Italy. Sci. Total Environ. 2020, 736, 139652. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.E.; Caliendo, A.M.; Arias, C.A.; Englund, J.A.; Lee, M.J.; Loeb, M.; Patel, R.; El Alayli, A.; Kalot, M.A.; Falck-Ytter, Y.; et al. Infectious Diseases Society of America Guidelines on the Diagnosis of COVID-19 (June 2020). Clin. Infect. Dis. 2024, 78, e106–e132. [Google Scholar] [CrossRef] [PubMed]
- Shuai, L.; Zhong, G.; Yuan, Q.; Wen, Z.; Wang, C.; He, X.; Liu, R.; Wang, J.; Zhao, Q.; Liu, Y.; et al. Replication, pathogenicity, and transmission of SARS-CoV-2 in minks. Natl. Sci. Rev. 2020, 8, nwaa291. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Bain, W.; Naqvi, A.; Staines, B.; Castanha, P.M.S.; Yang, H.; Boltz, V.F.; Barratt-Boyes, S.; A Marques, E.T.; Mitchell, S.L.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 Viremia Is Associated with Coronavirus Disease 2019 Severity and Predicts Clinical Outcomes. Clin. Infect. Dis. 2022, 74, 1525–1533. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal | ID | Trapping Date | Riverbed | Belongs to | Coordinates | Weight (g) | Sex | |
|---|---|---|---|---|---|---|---|---|
| E: | N: | |||||||
| 1 | 4618 | 17 November 2020 | Palancia | Navajas | 712,550 | 4,417,756 | 630 | F |
| 2 | 4619 | 17 November 2020 | Mijares | Onda | 737,126 | 4,429,832 | 944 | M |
| 3 | 4620 | 17 November 2020 | Mijares | Toga | 725,070 | 4,435,212 | 771 | F |
| 4 | 4621 | 17 November 2020 | Palancia | Segorbe | 717,603 | 4,412,528 | 1010 | M |
| 5 | 4622 | 17 November 2020 | Palancia | Soneja | 720,334 | 4,411,080 | 1109 | F |
| 6 | 4623 | 17 November 2020 | Palancia | Soneja | 719,168 | 4,411,224 | 1412 | M |
| 7 | 4624 | 17 November 2020 | Palancia | Soneja | 721,249 | 4,410,329 | 700 | F |
| 8 | 4625 | 17 November 2020 | Palancia | Vall de Almonacid | 717,559 | 4,420,103 | 590 | F |
| 9 | 4626 | 17 November 2020 | Palancia | Segorbe | 716,595 | 4,413,536 | 989 | M |
| 10 | 4627 | 17 November 2020 | Palancia | Segorbe | 716,102 | 4,414,547 | 926 | M |
| 11 | 4628 | 17 November 2020 | Palancia | Segorbe | 716,595 | 4,413,436 | 1098 | M |
| 12 | 4629 | 17 November 2020 | Palancia | Segorbe | 716,102 | 4,414,547 | 787 | F |
| 13 | 4630 | 17 November 2020 | Palancia | Jérica | 709,496 | 4,419,036 | 1083 | M |
| 14 | 4631 | 17 November 2020 | Palancia | Navajas | 713,317 | 4,417,348 | 1201 | M |
| 15 | 4632 | 17 November 2020 | Palancia | Navajas | 712,550 | 4,417,756 | 877 | M |
| 16 | 4633 | 17 November 2020 | Palancia | Segorbe | 717,603 | 4,412,528 | 757 | F |
| 17 | 4634 | 17 November 2020 | Palancia | Jérica | 709,496 | 4,419,036 | 1179 | F |
| 18 | 4635 | 17 November 2020 | Palancia | Navajas | 712,550 | 4,417,756 | 993 | M |
| 19 | 4636 | 17 November 2020 | Palancia | Navajas | 712,550 | 4,417,756 | 493 | F |
| 20 | 4637 | 17 November 2020 | Palancia | Soneja | 720,334 | 4,411,080 | 646 | F |
| 21 | 4638 | 17 November 2020 | Palancia | Jérica | 709,496 | 4,419,036 | 1089 | M |
| 22 | 4639 | 17 November 2020 | Palancia | Segorbe | 716,595 | 4,413,436 | 1074 | M |
| 23 | 4640 | 17 November 2020 | Palancia | Jérica | 708,547 | 4,419,355 | 1040 | M |
| 24 | 4641 | 17 November 2020 | Palancia | Segorbe | 717,603 | 4,412,528 | 680 | F |
| 25 | 4642 | 17 November 2020 | Palancia | Soneja | 719,168 | 4,411,224 | 728 | M |
| 26 | 4643 | 17 November 2020 | Palancia | Viver | 705,722 | 4,420,834 | 598 | F |
| 27 | 4644 | 17 November 2020 | Palancia | Jérica | 706,960 | 4,421,281 | 592 | F |
| 28 | 4645 | 17 November 2020 | Palancia | Segorbe | 717,603 | 4,412,528 | 996 | M |
| 29 | 4646 | 17 November 2020 | Mijares | Vallat | 727,495 | 4,434,312 | 736 | F |
| 30 | 4647 | 17 November 2020 | Palancia | Jérica | 709,496 | 4,419,036 | 988 | M |
| 31 | 4648 | 17 November 2020 | Palancia | Viver | 705,122 | 4,420,834 | 999 | M |
| 32 | 4649 | 17 November 2020 | Palancia | Jérica | 706,960 | 4,421,281 | 639 | F |
| 33 | 4650 | 17 November 2020 | Mijares | Espadilla | 726,089 | 4,434,535 | 847 | F |
| 34 | 323 | 11 March 2021 | Palancia | Navajas | 712,550 | 4,417,756 | 916 | M |
| 35 | 324 | 11 March 2021 | Palancia | Soneja | 720,334 | 4,411,080 | 1073 | M |
| 36 | 325 | 11 March 2021 | Palancia | Segorbe | 716,102 | 4,414,547 | 1305 | M |
| 37 | 326 | 11 March 2021 | Palancia | Jérica | 709,496 | 4,419,036 | 1432 | M |
| 38 | 327 | 11 March 2021 | Palancia | Soneja | 720,334 | 4,411,080 | 1418 | M |
| 39 | 328 | 11 March 2021 | Palancia | Navajas | 713,317 | 4,417,348 | 633 | F |
| 40 | 329 | 11 March 2021 | Palancia | Soneja | 720,334 | 4,411,080 | 1102 | M |
| 41 | 330 | 11 March 2021 | Palancia | Navajas | 712,550 | 4,417,756 | 777 | F |
| 42 | 331 | 11 March 2021 | Palancia | Viver | 704,779 | 4,419,830 | 1229 | M |
| 43 | 332 | 11 March 2021 | Palancia | Segorbe | 716.595 | 4,413,436 | 873 | M |
| 44 | 333 | 11 March 2021 | Mijares | Toga | 724,069 | 4,435,762 | 1311 | M |
| 45 | 334 | 11 March 2021 | Palancia | Soneja | 719,168 | 4,411,224 | 1164 | M |
| 46 | 335 | 11 March 2021 | Palancia | Soneja | 720,334 | 4,411,080 | 1340 | M |
| 47 | 336 | 11 March 2021 | Palancia | Jérica | 706,960 | 4,421,281 | 680 | F |
| 48 | 337 | 11 March 2021 | Palancia | Navajas | 713,317 | 4,417,348 | 1311 | M |
| 49 a | 1000 | 3 May 2022 | Palancia | Gaibiel | 715,043 b | 4,423,390 b | 985 | F |
| 50 | 1007 | 3 May 2022 | Palancia | Jérica | 706,960 | 4,421,281 | 765 | F |
| 51 | 1008 | 3 May 2022 | Palancia | Segorbe | 717,603 | 4,412,528 | 867 | F |
| 52 a | 1009 | 3 May 2022 | Palancia | Jérica | 706,960 | 4,421,281 | 830 | F |
| 53 | 1010 | 3 May 2022 | Palancia | Jérica | 709,496 | 4,419,036 | 1421 | M |
| 54 a | 1013 | 3 May 2022 | Palancia | Jérica | 709,496 | 4,419,036 | 897 | F |
| 55 | 1014 | 3 May 2022 | Palancia | Jérica | 709,496 | 4,419,036 | 1389 | M |
| 56 | 1022 | 3 May 2022 | Palancia | Teresa | 700,530 | 4,419,107 | 702 | F |
| 57 | 1023 | 3 May 2022 | Mijares | Montanejos | 712,777 | 4,439,177 | 773 | F |
| 58 | 1024 | 3 May 2022 | Mijares | Toga | 724,069 | 4,435,762 | 1266 | M |
| 59 | 1025 | 3 May 2022 | Palancia | Segorbe | 714,680 | 4,416,113 | 718 | F |
| 60 | 1026 | 3 May 2022 | Mijares | Toga | 725,070 | 4,435,212 | 711 | F |
| Positive Animal a | Sample | qPCR Results as Ct Values Change in the Coding Sequence of the Gene Change in the Amino Acid Sequence of the Protein b | ||
|---|---|---|---|---|
| S c | N | ORF10 | ||
| 16 | Swab (nasal) | 31.3 | 32.1 N:659C > T N: A220V | 29.1 ORF10: 88G > T ORF10: V30L |
| 29 | Swab (rectal) | 29.4 | 32.3 N: 608–610 GGG > AAC N: R203K/G204R | 20.2 ORF10: 88G > T ORF10: V30L |
| Lymph node (mediastinal) | 30.9 | 32.1 N: 608–610 GGG > AAC N: R203K/G204R | 22.8 ORF10: 88G > T ORF10: V30L | |
| Lung tissue | 30.9 | 32.1 N: 608–610 GGG > AAC N: R203K/G204R | 21.3 ORF10: 88G > T ORF10: V30L | |
| 40 | Swab (nasal) | 32.9 | 32.7 N:659C > T N: A220V | 29.3 ORF10: 88G > T ORF10: V30L |
| 56 | Swab (rectal) | 29.0 | 34.4 N: 608–610 GGG > AAC N: R203K/G204R | 24.9 ORF10: 88G > T ORF10: V30L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suita, F.; Padilla-Blanco, M.; Aguiló-Gisbert, J.; Lorenzo-Bermejo, T.; Ballester, B.; Cardells, J.; Maiques, E.; Rubio, V.; Lizana, V.; Rubio-Guerri, C. Four Novel SARS-CoV-2 Infected Feral American Mink (Neovison Vison) Among 60 Individuals Caught in the Wild. Animals 2025, 15, 1636. https://doi.org/10.3390/ani15111636
Suita F, Padilla-Blanco M, Aguiló-Gisbert J, Lorenzo-Bermejo T, Ballester B, Cardells J, Maiques E, Rubio V, Lizana V, Rubio-Guerri C. Four Novel SARS-CoV-2 Infected Feral American Mink (Neovison Vison) Among 60 Individuals Caught in the Wild. Animals. 2025; 15(11):1636. https://doi.org/10.3390/ani15111636
Chicago/Turabian StyleSuita, Francesca, Miguel Padilla-Blanco, Jordi Aguiló-Gisbert, Teresa Lorenzo-Bermejo, Beatriz Ballester, Jesús Cardells, Elisa Maiques, Vicente Rubio, Víctor Lizana, and Consuelo Rubio-Guerri. 2025. "Four Novel SARS-CoV-2 Infected Feral American Mink (Neovison Vison) Among 60 Individuals Caught in the Wild" Animals 15, no. 11: 1636. https://doi.org/10.3390/ani15111636
APA StyleSuita, F., Padilla-Blanco, M., Aguiló-Gisbert, J., Lorenzo-Bermejo, T., Ballester, B., Cardells, J., Maiques, E., Rubio, V., Lizana, V., & Rubio-Guerri, C. (2025). Four Novel SARS-CoV-2 Infected Feral American Mink (Neovison Vison) Among 60 Individuals Caught in the Wild. Animals, 15(11), 1636. https://doi.org/10.3390/ani15111636