Construction of Coexpression Networks Affecting Litter Size in Goats Based on Transcriptome Analysis

 , , , , , , and
, , , , , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Test Animals and Sample Collection
2.2. RNA Extraction and Library Construction
2.3. Differential Expression and Enrichment Analysis
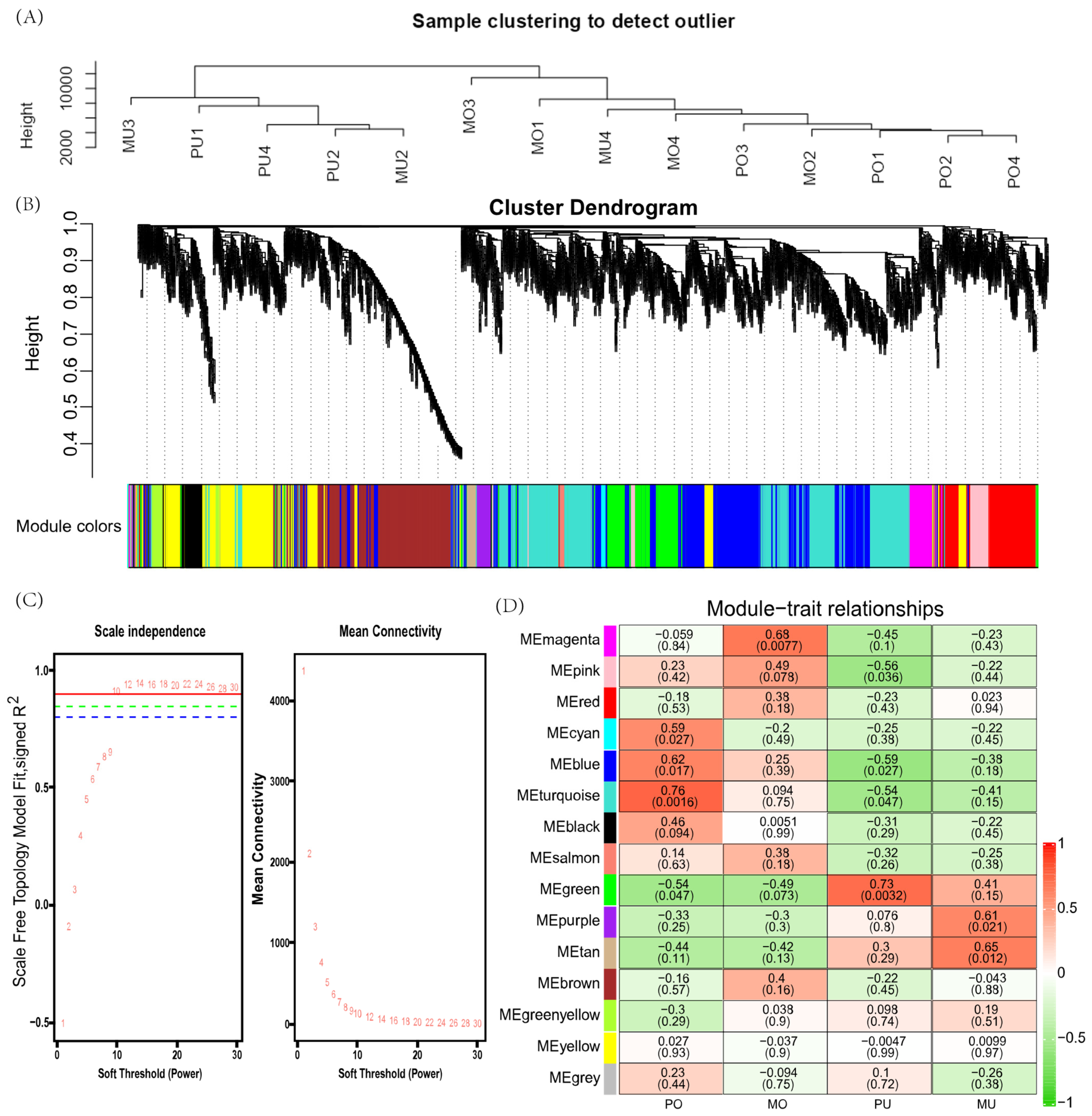
2.4. Weighted Gene Coexpression Network Analysis (WGCNA)
2.5. Identification of Pivotal Genes Associated with Ovarian and Uterine Litter Sizes
3. Results
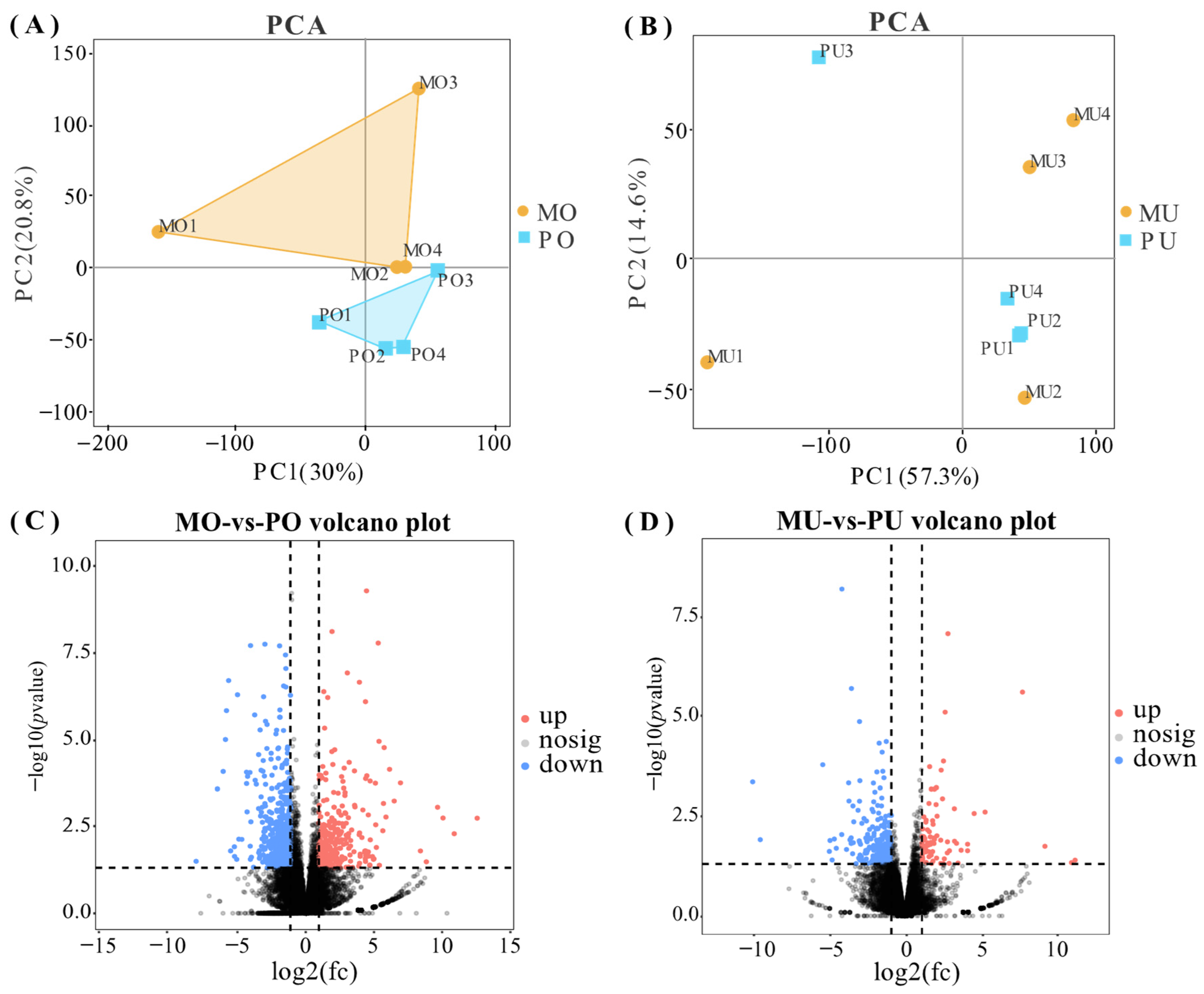
3.1. Differential Gene Screening
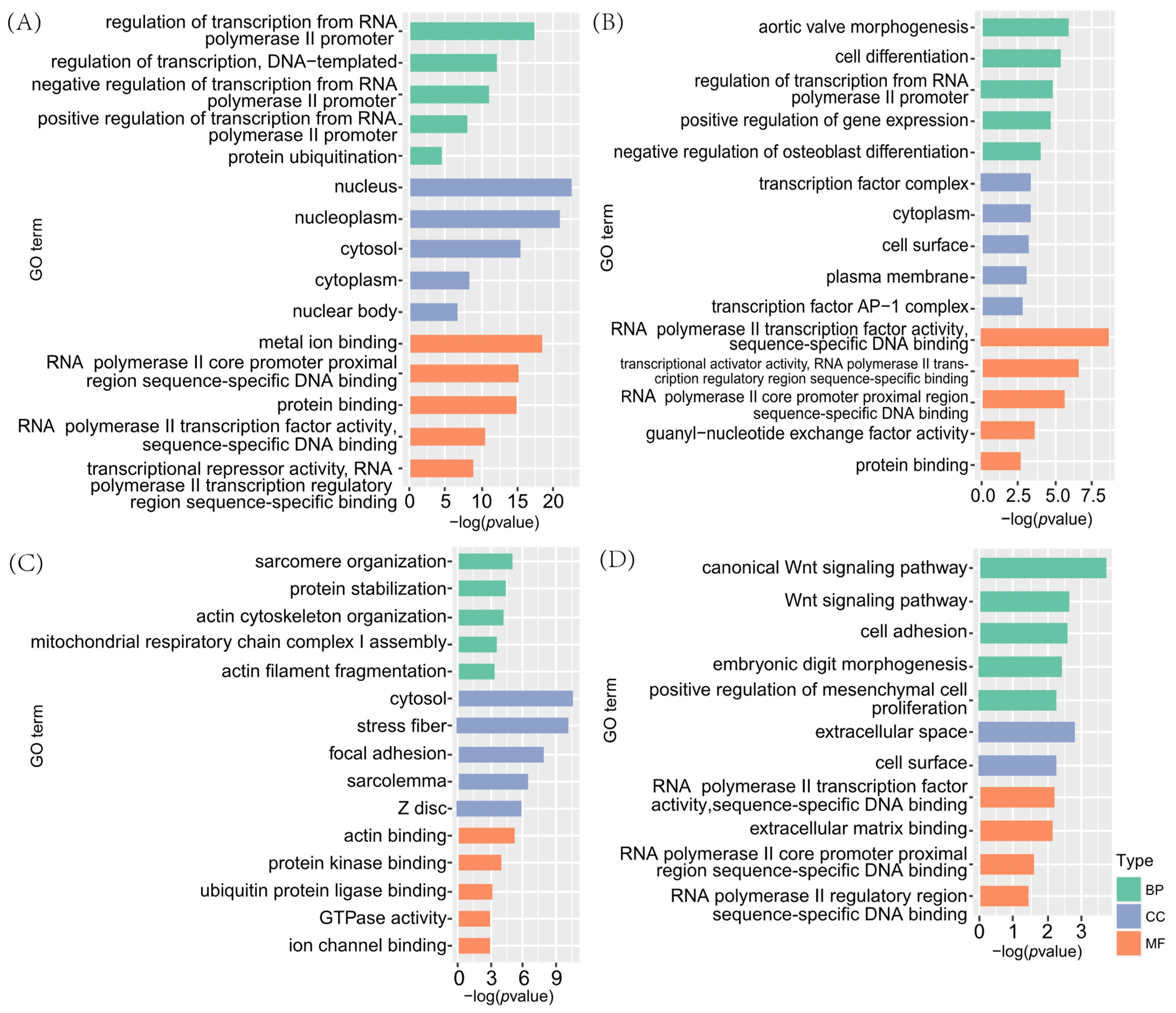
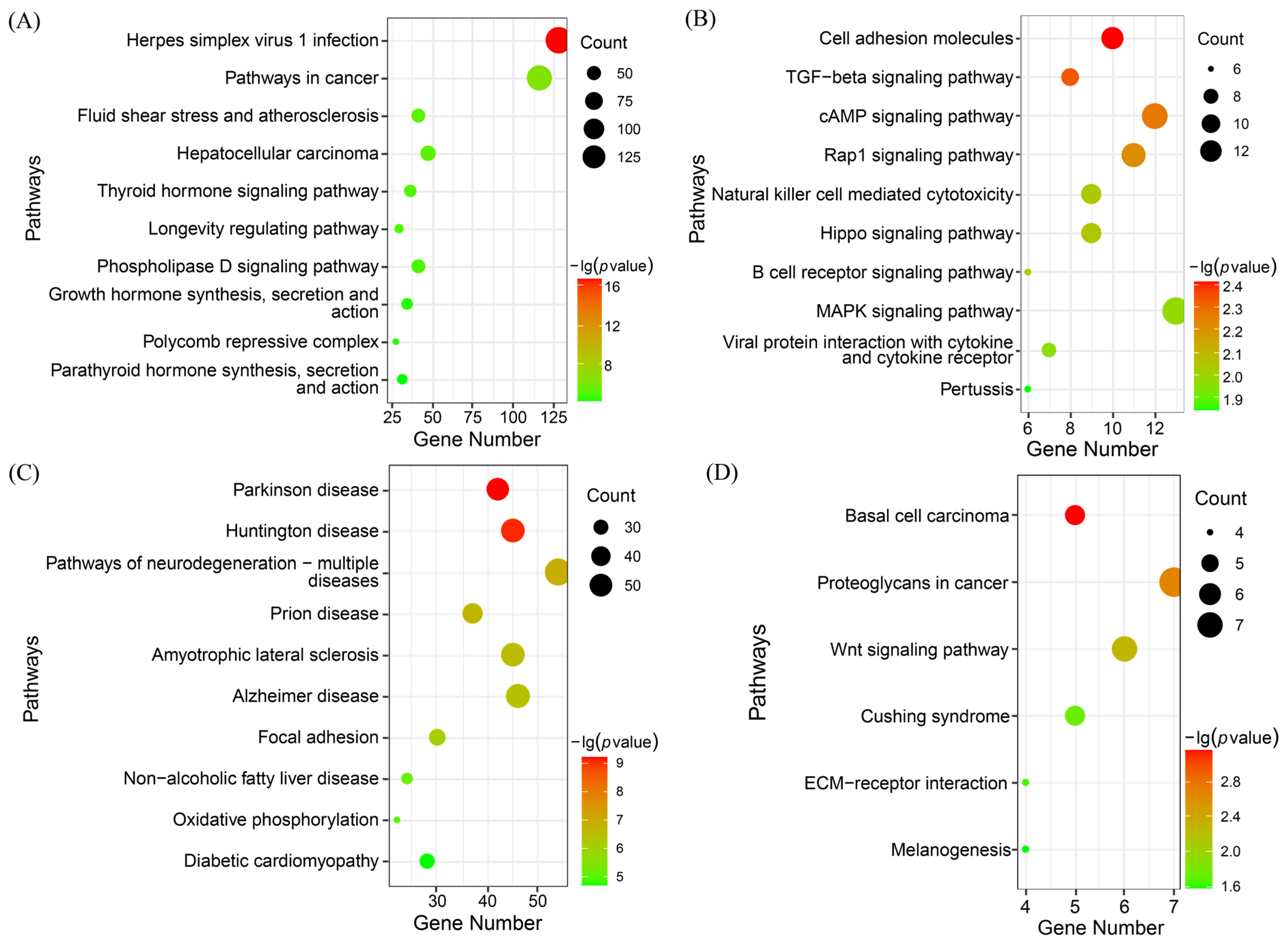
3.2. Functional Enrichment Analysis of DEGs
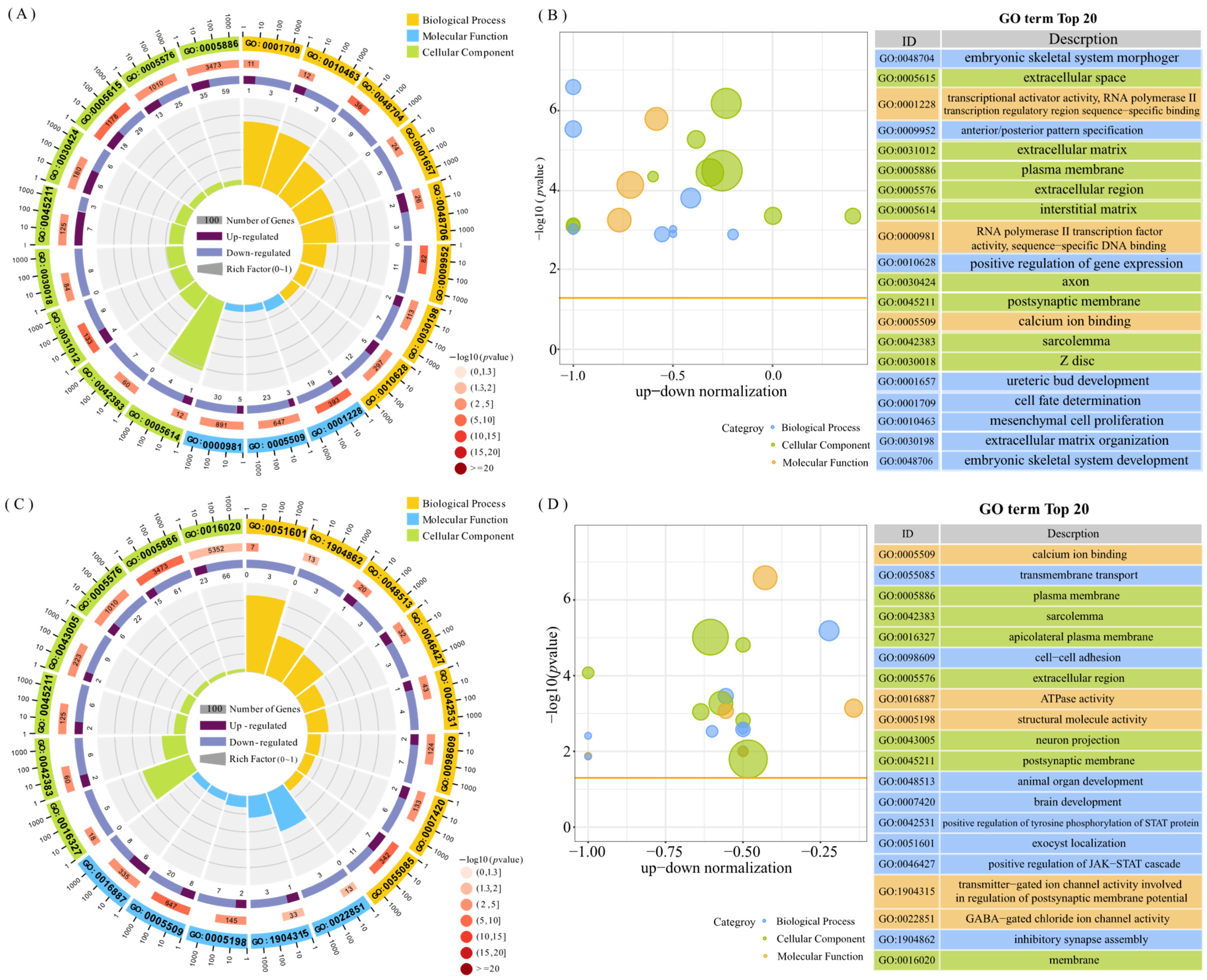
3.2.1. GO Enrichment Analysis
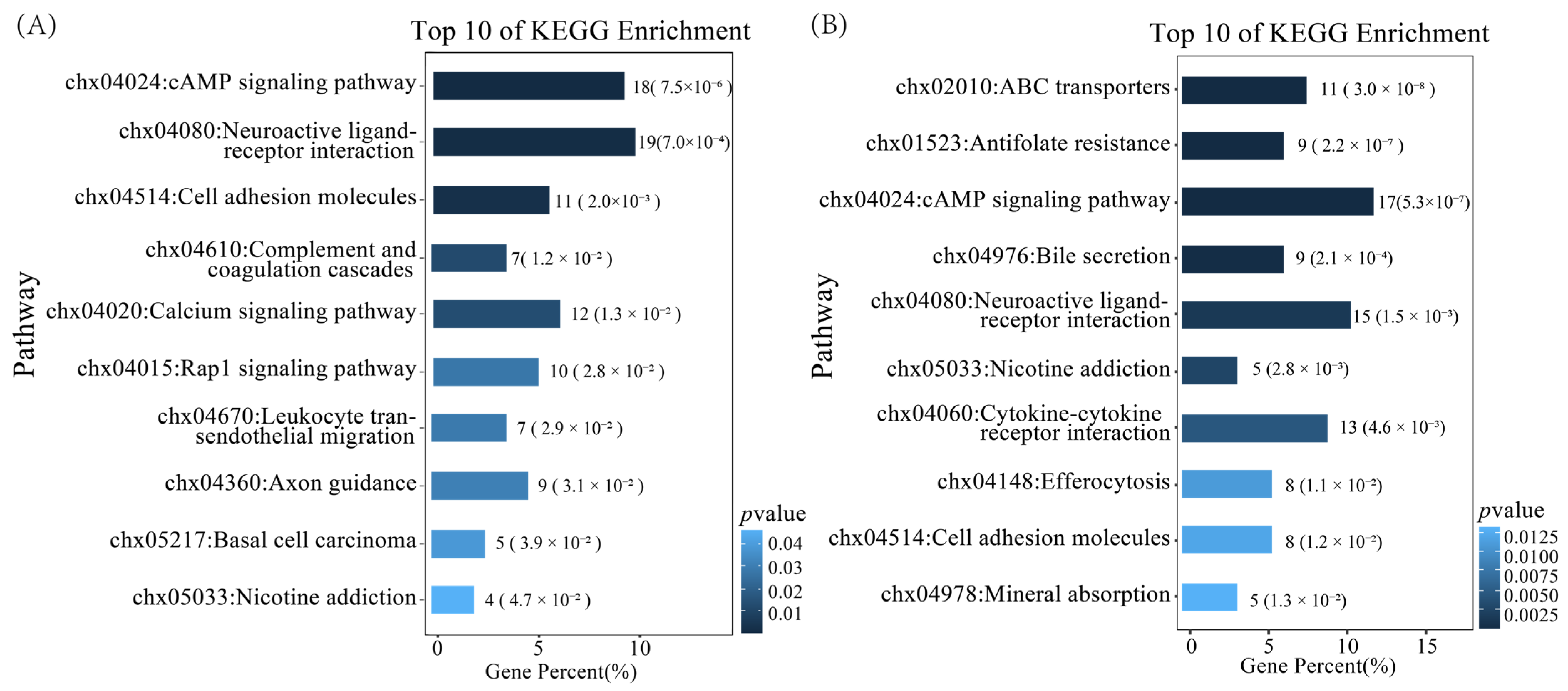
3.2.2. KEGG Enrichment Analysis
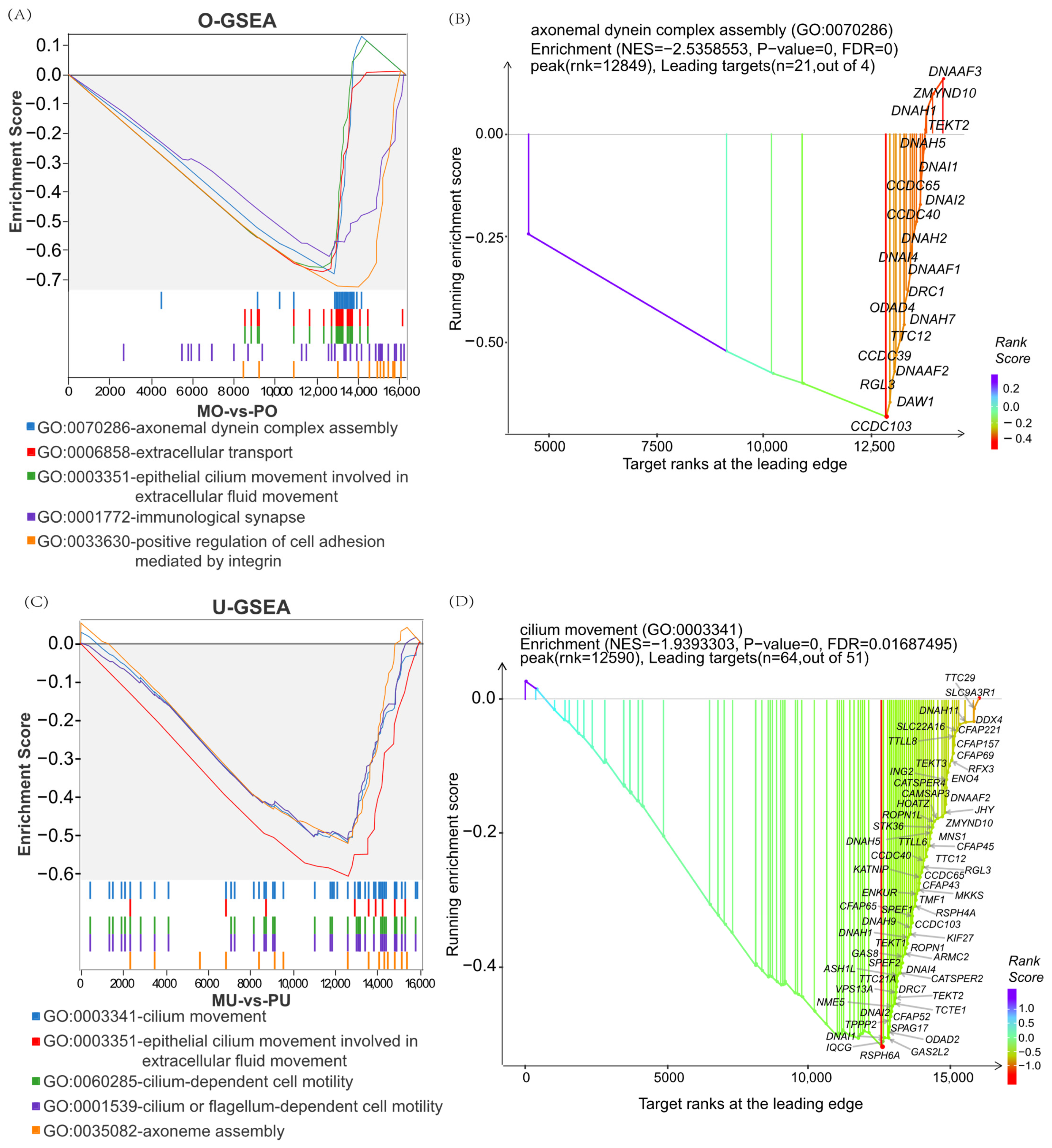
3.2.3. GSEA Enrichment Analysis
3.3. Weighted Gene Coexpression Network Construction
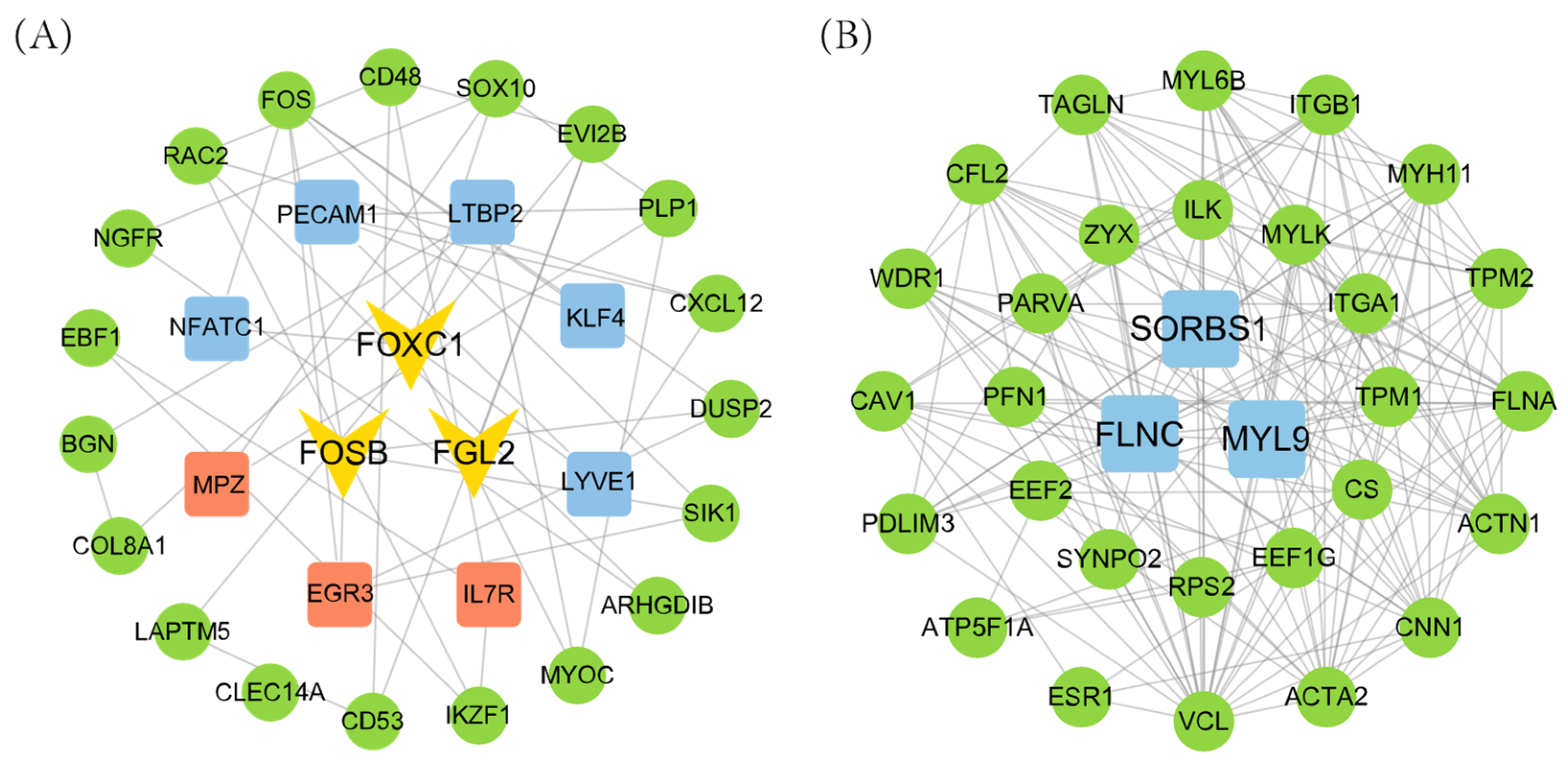
3.4. Hub Gene Identification
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gawat, M.; Boland, M.; Singh, J.; Kaur, L. Goat Meat: Production and Quality Attributes. Foods 2023, 12, 3130. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Tang, J.; Zhang, Z.; Tang, Q.; Yan, Y.; Wang, P.; Wang, X.; Liu, Q.; Guo, X.; Jin, M.; et al. Polymorphisms in the ASMT and ADAMTS1 gene may increase litter size in goats. Vet. Med. Sci. 2020, 6, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.H.; Montgomery, G.W.; Allison, A.J.; Kelly, R.W.; Bray, A.R. Segregation of a major gene influencing fecundity in progeny of Booroola sheep. N. Z. J. Agric. Res. 1982, 25, 525–529. [Google Scholar] [CrossRef]
- Ji, X.; Cao, Z.; Hao, Q.; He, M.; Cang, M.; Yu, H.; Ma, Q.; Li, X.; Bao, S.; Wang, J.; et al. Effects of New Mutations in BMPRIB, GDF9, BMP15, LEPR, and B4GALNT2 Genes on Litter Size in Sheep. Vet. Sci. 2023, 10, 258. [Google Scholar] [CrossRef]
- Yang, J.; Tang, J.; He, X.; Di, R.; Zhang, X.; Zhang, J.; Guo, X.; Hu, W.; Chu, M. Key mRNAs and lncRNAs of pituitary that affect the reproduction of FecB + + small tail han sheep. BMC Genom. 2024, 25, 392. [Google Scholar] [CrossRef]
- Zhong, T.; Hou, D.; Zhao, Q.; Zhan, S.; Wang, L.; Li, L.; Zhang, H.; Zhao, W.; Yang, S.; Niu, L. Comparative whole-genome resequencing to uncover selection signatures linked to litter size in Hu Sheep and five other breeds. BMC Genom. 2024, 25, 480. [Google Scholar] [CrossRef]
- Guo, C.; Ye, J.; Liu, J.; Li, Z.; Deng, M.; Guo, Y.; Liu, G.; Sun, B.; Li, Y.; Liu, D. Whole-genome sequencing identified candidate genes associated with high and low litter size in Chuanzhong black goats. Front. Vet. Sci. 2024, 11, 1420164. [Google Scholar] [CrossRef]
- Wang, S.Z.; E, G.X.; Zeng, Y.; Han, Y.G.; Huang, Y.F.; Na, R.S. Three SNPs within exons of INHA and ACVR2B genes are significantly associated with litter size in Dazu black goats. Reprod. Domest. Anim. 2021, 56, 936–941. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, X.; Li, D.; Tong, X.; Min, L.; Chen, W.; Ju, X.; Xu, B. The Identification of RPL4 as a Hub Gene Associated with Goat Litter Size via Weighted Gene Co-Expression Network Analysis. Animals 2024, 14, 1470. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Liu, Y.; Cao, G.; Di, R.; Wang, J.; Chu, M. Polymorphism and expression of GLUD1 in relation to reproductive performance in Jining Grey goats. Arch. Anim. Breed. 2023, 66, 411–419. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, L.; Dai, J.; Lv, Y.; Liao, R.; Shen, X.; Gao, J. Characterization and Comparative Analysis of Whole-Transcriptome Sequencing in High- and Low-Fecundity Chongming White Goat Ovaries during the Estrus Phase. Animals 2024, 14, 988. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, X.; Gao, T.; Zheng, Z.; Liu, A.; Tian, S. Differential expression and functional prediction of mRNA in the ovaries of Hanper sheep of high and low fecundity. Reprod. Domest. Anim. 2022, 57, 1623–1635. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Zeng, X. Identification of hub genes associated with follicle development in multiple births sheep by WGCNA. Front. Vet. Sci. 2022, 9, 1057282. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Zhang, Y.; Jiang, S.; Zeng, X.; Shen, H. Comprehensive Analysis of Differentially Expressed CircRNAs in the Ovaries of Low- and High-Fertility Sheep. Animals 2023, 13, 236. [Google Scholar] [CrossRef]
- Xu, S.S.; Gao, L.; Xie, X.L.; Ren, Y.L.; Shen, Z.Q.; Wang, F.; Shen, M.; Eyϸórsdóttir, E.; Hallsson, J.H.; Kiseleva, T.; et al. Genome-Wide Association Analyses Highlight the Potential for Different Genetic Mechanisms for Litter Size Among Sheep Breeds. Front. Genet. 2018, 9, 118. [Google Scholar] [CrossRef]
- Filant, J.; Spencer, T.E. Uterine glands: Biological roles in conceptus implantation, uterine receptivity and decidualization. Int. J. Dev. Biol. 2014, 58, 107–116. [Google Scholar] [CrossRef]
- Du, X.; Liu, Y.; He, X.; Tao, L.; Fang, M.; Chu, M. Identification and expression profile analysis of circRNAs associated with goat uterus with different fecundity during estrous cycle. BMC Genom. 2025, 26, 349. [Google Scholar] [CrossRef]
- Shah, A.M.; Cai, Y.; Zou, H.; Zhang, X.; Wang, L.; Xue, B.; Wang, Z.; Peng, Q. Effects of Supplementation of Branches and Leaves Trimmed from Tea Plant on Growth Performance, Rumen Fermentation and Meat Composition of Nanjiang Yellow Goats. Animals 2019, 9, 590. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Liu, Z.; Tan, X.; Jin, Q.; Zhan, W.; Liu, G.; Cui, X.; Wang, J.; Meng, X.; Zhu, R.; Wang, K. Multiomics analyses of Jining Grey goat and Boer goat reveal genomic regions associated with fatty acid and amino acid metabolism and muscle development. Anim. Biosci. 2024, 37, 982–992. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, L.; Lv, Y.; Liao, R.; Zhang, K.; Zhou, J.; Zhang, S.; Xu, J.; He, M.; Wu, C.; et al. Transcriptomic and metabolomic dissection of skeletal muscle of crossbred Chongming white goats with different meat production performance. BMC Genom. 2024, 25, 443. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, Y.; He, X.; Di, R.; Wang, X.; Ren, C.; Zhang, Z.; Chu, M. Integrative Proteomics and Transcriptomics Profiles of the Oviduct Reveal the Prolificacy-Related Candidate Biomarkers of Goats (Capra hircus) in Estrous Periods. Int. J. Mol. Sci. 2022, 23, 14888. [Google Scholar] [CrossRef] [PubMed]
- NY/T 1236-2006; Technical Specifications for Sheep and Goat Stud Productivity Testing. Ministry of Agriculture and Rural Affairs (MARA) of the People’s Republic of China: Beijing, China, 2006.
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, Y.; Gao, X.; Song, Y.; Huang, Y.; Jiang, Q. Unveiling the Ovarian Cell Characteristics and Molecular Mechanism of Prolificacy in Goats via Single-Nucleus Transcriptomics Data Analysis. Curr. Issues Mol. Biol. 2024, 46, 2301–2319. [Google Scholar] [CrossRef]
- Gao, K.; Chen, Y.; Wang, P.; Chang, W.; Cao, B.; Luo, L. GATA4: Regulation of expression and functions in goat granulosa cells. Domest. Anim. Endocrinol. 2024, 89, 106859. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Genome-wide analysis of miRNAs in the ovaries of Jining Grey and Laiwu Black goats to explore the regulation of fecundity. Sci. Rep. 2016, 6, 37983. [Google Scholar] [CrossRef]
- Kelleher, A.M.; DeMayo, F.J.; Spencer, T.E. Uterine Glands: Developmental Biology and Functional Roles in Pregnancy. Endocr. Rev. 2019, 40, 1424–1445. [Google Scholar] [CrossRef]
- Bourdon, M.; Maget, A.S.; Jeljeli, M.; Doridot, L.; Marcellin, L.; Thomas, M.; Chêne, C.; Chouzenoux, S.; Batteux, F.; Chapron, C.; et al. Reduced fertility in an adenomyosis mouse model is associated with an altered immune profile in the uterus during the implantation period. Hum. Reprod. 2024, 39, 119–129. [Google Scholar] [CrossRef]
- La, Y.; Tang, J.; Guo, X.; Zhang, L.; Gan, S.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Proteomic analysis of sheep uterus reveals its role in prolificacy. J. Proteom. 2020, 210, 103526. [Google Scholar] [CrossRef]
- Liu, B.; Xu, Q.; Wang, Q.; Feng, S.; Lai, F.; Wang, P.; Zheng, F.; Xiang, Y.; Wu, J.; Nie, J.; et al. The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 2020, 587, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, B.; Zhang, X.; Shen, X.; Ouyang, H.; Wu, Z.; Tian, Y.; Fang, L.; Huang, Y. Comparative transcriptomics in the hypothalamic-pituitary-gonad axis of mammals and poultry. Genomics 2022, 114, 110396. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wen, D.; Cen, J.; Mu, R. Hypothalamic transcriptome profile from laying period to incubation period of Changshun green-shell laying hens. Poult. Sci. 2024, 103, 103950. [Google Scholar] [CrossRef]
- Pei, S.; Wang, Z.; Liu, Y.; Xu, Y.; Bai, J.; Li, W.; Li, F.; Yue, X. Transcriptomic analysis of the HPG axis-related tissues reveals potential candidate genes and regulatory pathways associated with testicular size in Hu sheep. Theriogenology 2024, 216, 168–176. [Google Scholar] [CrossRef]
- Yang, B.; An, Y.; Yang, Y.; Zhao, Y.; Yu, K.; Weng, Y.; Du, C.; Li, H.; Yu, B. The ERβ-cAMP signaling pathway regulates estradiol-induced ovine oocyte meiotic arrest. Theriogenology 2024, 214, 81–88. [Google Scholar] [CrossRef]
- Zou, X.; Lu, T.; Zhao, Z.; Liu, G.; Lian, Z.; Guo, Y.; Sun, B.; Liu, D.; Li, Y. Comprehensive analysis of mRNAs and miRNAs in the ovarian follicles of uniparous and multiple goats at estrus phase. BMC Genom. 2020, 21, 267. [Google Scholar] [CrossRef]
- Stival, C.; Puga Molina Ldel, C.; Paudel, B.; Buffone, M.G.; Visconti, P.E.; Krapf, D. Sperm Capacitation and Acrosome Reaction in Mammalian Sperm. Adv. Anat. Embryol. Cell Biol. 2016, 220, 93–106. [Google Scholar] [CrossRef]
- Austin, C.R. Observations on the penetration of the sperm in the mammalian egg. Aust. J. Sci. Res. B 1951, 4, 581–596. [Google Scholar] [CrossRef]
- Schier, A.C.; Taatjes, D.J. Structure and mechanism of the RNA polymerase II transcription machinery. Genes. Dev. 2020, 34, 465–488. [Google Scholar] [CrossRef]
- Alexander, A.K.; Rice, E.J.; Lujic, J.; Simon, L.E.; Tanis, S.; Barshad, G.; Zhu, L.; Lama, J.; Cohen, P.E.; Danko, C.G. A-MYB and BRDT-dependent RNA Polymerase II pause release orchestrates transcriptional regulation in mammalian meiosis. Nat. Commun. 2023, 14, 1753. [Google Scholar] [CrossRef]
- Wu, D.; Dean, J. Reduced female fertility due to sequestration of RNA Pol II by pervasive transcription in exosome RNase-depleted oocytes. Cell Rep. 2023, 42, 113247. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Chen, F.; Stein, P.; Wang, J.; Zhou, Z.; Wang, L.; Zhao, Q.; Lin, Z.; Liu, B.; Xu, K.; et al. OBOX regulates mouse zygotic genome activation and early development. Nature 2023, 620, 1047–1053. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, Y.; He, Y.; Ye, M.; Yuan, J.; Ren, C.; Wang, X.; Wang, S.; Guo, Y.; Cao, Q.; et al. Maternal KLF17 controls zygotic genome activation by acting as a messenger for RNA Pol II recruitment in mouse embryos. Dev. Cell 2024, 59, 613–626.e6. [Google Scholar] [CrossRef]
- Abuhashem, A.; Chivu, A.G.; Zhao, Y.; Rice, E.J.; Siepel, A.; Danko, C.G.; Hadjantonakis, A.K. RNA Pol II pausing facilitates phased pluripotency transitions by buffering transcription. Genes. Dev. 2022, 36, 770–789. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, Z.; Zhao, W.; Zhang, Z.; Xiang, Y.; Wang, Q.; Pan, Y.; Guo, X.; Wang, Z. Genome-wide epistatic interactions of litter size at birth in Chinese indigenous pigs. Anim. Genet. 2021, 52, 739–743. [Google Scholar] [CrossRef]
- Xin, H.; Li, B.; Meng, F.; Hu, B.; Wang, S.; Wang, Y.; Li, J. Quantitative proteomic analysis and verification identify global protein profiling dynamics in pig during the estrous cycle. Front. Vet. Sci. 2023, 10, 1247561. [Google Scholar] [CrossRef]
- Qin, P.; Pan, Z.; Zhang, W.; Wang, R.; Li, X.; Lu, J.; Xu, S.; Gong, X.; Ye, J.; Yan, X.; et al. Integrative proteomic and transcriptomic analysis in the female goat ovary to explore the onset of puberty. J. Proteom. 2024, 301, 105183. [Google Scholar] [CrossRef]
- He, Z.; Chen, Q.; Ouyang, Q.; Hu, J.; Shen, Z.; Hu, B.; Hu, S.; He, H.; Li, L.; Liu, H.; et al. Transcriptomic analysis of the thyroid and ovarian stroma reveals key pathways and potential candidate genes associated with egg production in ducks. Poult. Sci. 2023, 102, 102292. [Google Scholar] [CrossRef]
- Bates, M.D.; Conn, P.M. Calcium Mobilization in the Pituitary Gonadotrope: Relative Roles of intra- and Extracellular Sources*. Endocrinology 1984, 115, 1380–1385. [Google Scholar] [CrossRef]
- Jasoni, C.L.; Romanò, N.; Constantin, S.; Lee, K.; Herbison, A.E. Calcium dynamics in gonadotropin-releasing hormone neurons. Front. Neuroendocrinol. 2010, 31, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.F.J. Tuning exocytosis for speed: Fast and slow modes. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2003, 1641, 157–165. [Google Scholar] [CrossRef]
- Sitarska, E.; Diz-Muñoz, A. Pay attention to membrane tension: Mechanobiology of the cell surface. Curr. Opin. Cell Biol. 2020, 66, 11–18. [Google Scholar] [CrossRef]
- Albanese, J.; Dainiak, N. Modulation of intercellular communication mediated at the cell surface and on extracellular, plasma membrane-derived vesicles by ionizing radiation. Exp. Hematol. 2003, 31, 455–464. [Google Scholar] [CrossRef]
- Stadler, T.; Pybus, O.G.; Stumpf, M.P.H. Phylodynamics for cell biologists. Science 2021, 371, eaah6266. [Google Scholar] [CrossRef]
- Kim, Y.Y.; Kim, S.W.; Kim, E.; Kim, Y.J.; Kang, B.C.; Ku, S.Y. Transcriptomic Profiling of Reproductive Age Marmoset Monkey Ovaries. Reprod. Sci. 2024, 31, 81–95. [Google Scholar] [CrossRef]
- Kaestner, K.H.; Knochel, W.; Martinez, D.E. Unified nomenclature for the winged helix/forkhead transcription factors. Genes. Dev. 2000, 14, 142–146. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, X.; An, J.; Zhang, Y.; He, M.; Tang, L. Genotype-phenotype association of PITX2 and FOXC1 in Axenfeld-Rieger syndrome. Exp. Eye Res. 2023, 226, 109307. [Google Scholar] [CrossRef]
- Motojima, M.; Tanaka, M.; Kume, T. Foxc1 and Foxc2 are indispensable for the maintenance of nephron and stromal progenitors in the developing kidney. J. Cell Sci. 2022, 135, jcs260356. [Google Scholar] [CrossRef]
- Yue, Y.; Jiang, M.; He, L.; Zhang, Z.; Zhang, Q.; Gu, C.; Liu, M.; Li, N.; Zhao, Q. The transcription factor Foxc1a in zebrafish directly regulates expression of nkx2.5, encoding a transcriptional regulator of cardiac progenitor cells. J. Biol. Chem. 2018, 293, 638–650. [Google Scholar] [CrossRef]
- Uhlenhaut, N.H.; Treier, M. Forkhead transcription factors in ovarian function. Reproduction 2011, 142, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Mattiske, D.; Kume, T.; Hogan, B.L. The mouse forkhead gene Foxc1 is required for primordial germ cell migration and antral follicle development. Dev. Biol. 2006, 290, 447–458. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Song, Y.; Hou, J.; Zhang, Y.; Chen, K.; Ma, H.; Zhao, X.; Li, G.; Gao, K.; Wang, S.; et al. Chi-miR-4110 promotes granulosa cell apoptosis by targeting Sma- and Mad-related protein 2 (Smad2) in the caprine ovary. PLoS ONE 2017, 12, e0181162. [Google Scholar] [CrossRef]
- Schuermann, M.; Jooss, K.; Müller, R. fosB is a transforming gene encoding a transcriptional activator. Oncogene 1991, 6, 567–576. [Google Scholar]
- Kataoka, F.; Tsuda, H.; Arao, T.; Nishimura, S.; Tanaka, H.; Nomura, H.; Chiyoda, T.; Hirasawa, A.; Akahane, T.; Nishio, H.; et al. EGRI and FOSB gene expressions in cancer stroma are independent prognostic indicators for epithelial ovarian cancer receiving standard therapy. Genes. Chromosomes Cancer 2012, 51, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Li, X.; Zhao, Z.; Cao, Y.; Liu, X.; Liu, Z.; Ma, H.; Lu, W. Melatonin Protects the Apoptosis of Sheep Granulosa Cells by Suppressing Oxidative Stress via MAP3K8 and FOS Pathway. Genes 2023, 14, 1067. [Google Scholar] [CrossRef]
- Lappas, M.; Riley, C.; Lim, R.; Barker, G.; Rice, G.E.; Menon, R.; Permezel, M. MAPK and AP-1 proteins are increased in term pre-labour fetal membranes overlying the cervix: Regulation of enzymes involved in the degradation of fetal membranes. Placenta 2011, 32, 1016–1025. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, Z.; Xu, C.; Yan, Y.; Cao, X.; Zhang, F.; Shen, B. FGL2 as a predictive biomarker for prognosis and immunotherapy in bladder cancer. Int. J. Med. Sci. 2024, 21, 1447–1460. [Google Scholar] [CrossRef]
- Feng, Y.; Guo, C.; Wang, H.; Zhao, L.; Wang, W.; Wang, T.; Feng, Y.; Yuan, K.; Huang, G. Fibrinogen-Like Protein 2 (FGL2) is a Novel Biomarker for Clinical Prediction of Human Breast Cancer. Med. Sci. Monit. 2020, 26, e923531. [Google Scholar] [CrossRef]
- Wang, Y.; Hua, R.; Xue, S.; Li, W.; Wu, L.; Kang, T.; Lei, M. mRNA/lncRNA expression patterns and the function of fibrinogen-like protein 2 in Meishan pig endometrium during the preimplantation phases. Mol. Reprod. Dev. 2019, 86, 354–369. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, M.; Zhu, M.; Song, Y.; Zhang, J.; Zheng, C. Cuyun Recipe ameliorates pregnancy loss by regulating macrophage polarization and hypercoagulable state during the peri-implantation period in an ovarian hyperstimulation mouse model. Phytomedicine 2023, 119, 154974. [Google Scholar] [CrossRef]
- Mu, J.; Qu, D.; Bartczak, A.; Phillips, M.J.; Manuel, J.; He, W.; Koscik, C.; Mendicino, M.; Zhang, L.; Clark, D.A.; et al. Fgl2 deficiency causes neonatal death and cardiac dysfunction during embryonic and postnatal development in mice. Physiol. Genom. 2007, 31, 53–62. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | ID | Age | Body Weight/kg | Body Heigh/cm | Body Length/cm | Chest Circum- Ference/cm | Cannon Bone Circumference/cm | Litter Size | ||
|---|---|---|---|---|---|---|---|---|---|---|
| First Parity | Second Parity | Third Parity | ||||||||
| Polytocous group (P) | P1 | 3–4 | 28.5 | 66 | 55 | 78 | 7 | 3 | 5 | 4 |
| P2 | 3–4 | 32 | 82 | 56 | 83 | 9 | 4 | 5 | 3 | |
| P3 | 3–4 | 30.7 | 74 | 53 | 84 | 8 | 4 | 5 | 4 | |
| P4 | 3–4 | 29.5 | 67 | 55 | 77 | 8 | 4 | 4 | 3 | |
| Monotocous group (M) | M1 | 3–4 | 28.6 | 67 | 52 | 75 | 7.5 | 1 | ||
| M2 | 3–4 | 28.3 | 73 | 52 | 82 | 8 | ||||
| M3 | 3–4 | 30.5 | 68 | 56 | 76 | 8 | ||||
| M4 | 3–4 | 31.5 | 73 | 52 | 82 | 8 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Y.; He, J.; Liu, G.; Wei, C.; Li, X.; Mao, J.; Zhang, G.; Zhang, W.; Long, L.; Wang, M.; et al. Construction of Coexpression Networks Affecting Litter Size in Goats Based on Transcriptome Analysis. Animals 2025, 15, 1505. https://doi.org/10.3390/ani15111505
Ren Y, He J, Liu G, Wei C, Li X, Mao J, Zhang G, Zhang W, Long L, Wang M, et al. Construction of Coexpression Networks Affecting Litter Size in Goats Based on Transcriptome Analysis. Animals. 2025; 15(11):1505. https://doi.org/10.3390/ani15111505
Chicago/Turabian StyleRen, Yifan, Junmin He, Guifen Liu, Chen Wei, Xue Li, Jingyi Mao, Guoping Zhang, Wenhao Zhang, Li Long, Ming Wang, and et al. 2025. "Construction of Coexpression Networks Affecting Litter Size in Goats Based on Transcriptome Analysis" Animals 15, no. 11: 1505. https://doi.org/10.3390/ani15111505
APA StyleRen, Y., He, J., Liu, G., Wei, C., Li, X., Mao, J., Zhang, G., Zhang, W., Long, L., Wang, M., Tian, K., & Huang, X. (2025). Construction of Coexpression Networks Affecting Litter Size in Goats Based on Transcriptome Analysis. Animals, 15(11), 1505. https://doi.org/10.3390/ani15111505