1. Introduction
Hemp is one of the oldest cultivated plants in the world, as many parts of the plant can be used in a variety of applications. Currently, the stalks are deployed for textile and industrial fibre production; the seeds can be used for food production, and the flowers for the extraction of cannabinoids. The leaves of hemp plants can used to prepare tea but also fed to various animal species [
1,
2,
3].
Due to its noteworthy crude fibre and crude protein contents [
4,
5], leaves attached to the whole aboveground industrial hemp plant have recently been tested as feed in dairy cattle nutrition [
6,
7,
8]. The inclusion of 13% (on a DM basis) spent hemp biomass (SHB), the whole plant residue remaining after alcoholic extraction, or 7.4% dried hemp leaves in the ration of dairy cows decreased dry matter intake (DMI) but not dry matter (DM) digestibility [
7].
Apart from macronutrients, hemp leaves contain several bioactive, ethanol-soluble cannabinoids, and many of them modulate oxidative stress, inhibit the release of excitotoxic amino acids and cytokines, or exert anti-inflammatory properties [
9,
10,
11]. The anti-inflammatory properties are, among others, attributable to the influence of cannabinoids, such as tetrahydrocannabinol (THC), cannabidiol (CBD), and cannabigerol (CBG). These cannabinoids can affect cellular signalling pathways, including the phosphorylation of NF-κB and the subsequent inhibition of the transcription of pro-inflammatory cytokines such as interleukin-1beta (IL-1ß) and tumour necrosis factor alpha (TNF-α) [
12,
13].
Although the total CBD concentration of SHB is much lower than in non-extracted hemp plants, the inclusion of 6 to 11% SHB in the diet of dairy cows reduced their plasma IL-1ß but not TNF-α concentrations [
14]. However, previous research showed that cannabinoids can be transferred into the milk and can exceed the acute THC (tetrahydrocannabinol) reference dose for humans. For example, feeding a 0.92 kg of DM hemp silage containing 58.3 mg/kg of DM ∆9-THC resulted in the exceedance of the acute reference dose by a factor of 1.5 in infants with high milk consumption, whereas feeding 1.68 kg hemp silage per cow containing 1254.7 mg/kg of DM ∆
9-THC led to the acute reference dose being exceeded in all consumer groups [
6]. Furthermore, feeding industrial hemp silage to dairy cows, even in small amounts, altered animal behaviour and impaired animal health [
6]. In order to still make use of the positive effects of non-THC cannabinoids, only hemp varieties containing no or very low THC concentrations (e.g., 1/100 of the threshold of 0.3% of DM, valid in the European Union) come into question as nutritional supplements. The variety Santhica 27 contains particularly low THC concentrations [
15] and thus may serve as a feed supplement with potential antioxidant, anti-inflammatory and amino acid-modulating effects in dairy cattle.
In addition to cannabinoids, industrial hemp leaves are a source of phenolic and flavonoid compounds, some of them being soluble in ethanol. Phenolic and flavonoid compounds exhibit antioxidant properties, which could improve the antioxidant capacity of cows alike [
16]. However, feeding SHB to ruminants did not improve the antioxidant capacity in plasma, the activity of enzymes combating oxidative stress or parameters related to immune function [
7,
14,
17], likely because only limited amounts of phenolic, flavonoids and further biologically active substances remain in SHB after extraction. Thus, the effect of feeding industrial hemp leaves with a very low THC content on the antioxidative capacity of dairy cows still remains to be investigated.
The SHB, similar to soya, further contains minor n-3 but even more n-6 polyunsaturated fatty acids (PUFA) concentrations with an n-6/n-3 ratio of about 6:1 [
7]. When SHB was supplemented to a total mixed ration (TMR), resulting in an n-6/n-3 ratio of 2.4:1, the percentage of milk
C18:0 and the percentage of de novo milk fatty acids were significantly reduced, which led to improved nutritional indices of the milk for human nutrition, e.g., the hypocholesterolemic/hypercholesterolemic (h/H) index or atherogenic index (AI) [
7]. Yet, it remains to be elucidated how the feeding of THC-free industrial hemp leaves, not depleted of lipids, affects milk fatty acid concentrations and nutritional indices.
The aim of this study was, therefore, to investigate the effects of feeding dried Santhica 27 leaves compared to soybean meal on performance traits, the antioxidant capacity and inflammatory parameters, plasma cannabinoid and amino acid concentrations, as well as milk fatty and amino acid profiles of dairy cows. We hypothesised that the supplementation of dried Santhica 27 hemp leaves to the diet of dairy cows would improve the antioxidant capacity, reduce the abundance of excitatory amino acids and pro-inflammatory cytokines, and modulate the milk amino and fatty acid profiles.
2. Materials and Methods
2.1. Ethical Considerations
The experimental protocol was evaluated by the ethical committee and approved by the Federal Office of Agriculture, Food Security and Fishery Mecklenburg—Western Pomerania, Rostock, Germany (LALLF, permission no. 7221.3-1-028/22) and conducted in accordance with the ARRIVE guidelines (
https://arriveguidelines.org/), the European Directive 2010/63/EU and the German Animal Welfare Act. Persons involved in sample preparation and analyses were blinded. Persons who collected blood samples, prepared the feed, and fed the cows were not blinded.
2.2. Animals and Experimental Design
For this study, 12 first-lactating German Holstein dairy cows were randomly selected from the herd of the Experimental Farm for Cattle (FBN, Dummerstorf, Germany) and fed two isoenergetic and isonitrogenous formulated diets in a cross-over design. The experimental design consisted of 3 blocks of 4 animals each.
At the beginning of the experimental trial, cows were 227 ± 29 days in milk (DIM) and 116 ± 45 days in gestation (mean ± SD). In experimental period 1, cows were fed ad libitum a TMR supplemented with 7.4% (on dry matter (DM) basis) dried industrial hemp leaves (HEMP) of the variety “Santhica 27” containing 154.17 g/kg of DM crude protein and 11.45 MJ/kg of DM metabolisable energy (ME) or a TMR containing 3.5% soybean meal (CON) containing 152.17 g/kg of DM crude protein and 11.45 MJ/kg of DM ME for 21 days. During the 2-week washout period, all cows were fed a hemp- and soy-free TMR. In experimental period 2, cows were fed the opposite diet for ad libitum intake for a further 21 days. All cows had ad libitum access to feed and water throughout the trail. Cows were fed at 0730 h and 1430 h.
Groups were housed in the free-ranging barn of the experimental facilities at FBN to measure the individual daily feed intake using the Roughage Intake Control system (RIC, Insentec B.V., Repelweg, The Netherlands) in the first two weeks of each experimental period. Feed samples were taken twice a week, dried at 60 °C for 24 h, ground (1 mm particle size) and further dried at 103 °C for 4 h to determine feed DM according to the methodological guidelines of the Association of German Agricultural Analytic and Research Institutes (VDLUFA) [
18]. Dried feed samples were sent to the Landwirtschaftliche Untersuchungs- und Forschungsanstalt (LUFA GmbH, Rostock, Germany) for the analyses of nutrient composition, including crude ash, crude protein, crude fat, crude fibre, acid detergent fibre based on organic matter basis, neutral detergent fibre based on organic matter basis and starch. Based on the analysis results, the metabolisable energy (ME) of feed was calculated according to the Society of Nutrition Physiology (GfE, 2001) [
19]:
including ether extract (EE), crude protein (CP) and crude fibre (CF) in g/kg of DM of feed.
Cows were milked, and milk yield was recorded with a tandem milking parlour equipped with a milk meter (DeLaval GmbH, Glinde, Germany) at 0630 h and 1700 h. Twice a week, a pooled milk sample of the evening and the subsequent morning milking was taken in the first two weeks of each experimental period. Milk constitutes were analysed by infrared spectroscopy, and the somatic cells were counted fluoro-optically at the State Inspection Association for Performance and Quality Testing Mecklenburg—Western Pomerania e.V. (LKV Güstrow, Germany). The energy-corrected milk yield (ECM) was calculated using the following equation:
The body weight (BW) of the cows was determined after each milking using a walk-through scale. The average BW and the average metabolic BW (mBW = BW0.75) were calculated for the first two weeks of each experimental period. The rectal temperature was measured daily before the morning feeding to monitor animal health. Blood samples were drawn from the jugular vein at 0700 h in 9 mL ethylenediaminetetraacetic acid (EDTA)-containing tubes (Sarstedt, Nümbrecht, Germany) on days 0, 7 and 14. The obtained blood samples were immediately placed on ice and subsequently centrifuged at 1.570× g and 4 °C for 20 min. The plasma was separated, the buffy coat was mixed with lysis buffer DL (MACHEREY-NAGEL, Düren, Germany), and samples were stored at −80 °C until further analysis.
2.3. Amino Acid Analysis
For the free amino acid analysis in whey, milk samples were centrifuged twice at 50,000× g and 4 °C for 20 min, and the obtained clear whey was diluted fivefold with ultrapure water. Plasma samples were tenfold diluted with ultrapure water.
The amino acid analysis was performed as described by Krömer (2006) [
20]. In brief, total amino acids in plasma and whey were separated after pre-column derivatisation with ortho-phthalaldehyde/3-mercaptopropionic acid (primary amino acids) and 9-fluorenylmethoxycarbonyl chloride (secondary amino acids) and prior blocking of SH groups with iodoacetic acid after cleavage of sulphur bridges with 3-mercaptopropionic acid using a high-performance liquid chromatography (HPLC) column at 40 °C (250 × 4.6 mm Gemini
® 5 µm
C18 110 Å with 4 × 3 mm pre-column (both Phenomenex, Aschaffenburg, Germany) and detected by a fluorescence detector.
Chromatographic separation was performed using a gradient of 40 mM phosphate buffer (pH 7.45) and acetonitrile/methanol/water (45:45:10) ranging from 7 to 100% at a flow rate of 0.8 mL/min in 48 min. Ortho-phthalaldehyde derivatives were detected at 340 nm excitation and 450 nm emission, and the 9-fluorenylmethoxycarbonyl derivatives at 266 nm excitation and 305 nm emission. All amino acids and their metabolites were quantified against multipoint calibrated external standards.
2.4. Fatty Acids Analysis
Fatty acid analysis of the diets was performed according to the procedure recently described by Tadesse et al. [
21]. The feed samples were finely ground to 1 mm, 2 g weighed in 10 mL screw-capped Pyrex tubes, and 2.0 mL of nonadecanoic acid (4.0 mg) was added as an internal standard. For fatty acid extraction, 3 mL of 5% methanolic hydrogen chloride (HCl) was added and vortexed in tightly closed Pyrex tubes for 2 h at 60 °C in a water bath. After cooling, the sample solution was treated with 10 mL 6% potassium carbonate (K
2CO
3) solution and vortexed. The solutions were centrifuged at 4 °C, 1200×
g for 5 min, and finally, the fatty acid methyl esters (FAMEs) were extracted two times with 2 mL of n-hexane. After being dried with 1 g sodium sulphate and cleaned with activated charcoal of the organic phase as required, the extracts were filtrated and evaporated using a vacuum centrifuge at 438×
g, 30 °C, 30 min. Finally, the extracts were stored at −18 °C until gas chromatography (GC) analysis.
Fatty acids were extracted using a fatty acid extraction kit (MAK174, low standard, Sigma-Aldrich, St. Louis, MO, USA). According to the kit protocol, the milk samples were stirred, and 200 mg milk was treated with 3 mL extraction solvent consisting of chloroform/methanol (2:1, vol/vol) containing the ethyl ester of nonadecanoic acid (C19:0) as internal standard.
After vortexing and a reaction time of 4 min, 0.5 mL of an aqueous buffer (kit buffer solution) was added, and the sample was vortexed again. Subsequently, the milk extraction solution was transferred into a syringe system containing a filter (provided in the kit). Finally, the eluted solvent contained the chloroform phase with total milk lipids. To complete the extraction, the solutions were incubated overnight at 4 °C.
Then, the solvents of the extracted lipids were evaporated with a gentle stream of nitrogen, and the amounts of total lipids were weighed. The milk lipid extracts were dissolved in toluene for methyl ester derivatisation. Next, 0.5 M sodium methoxide in methanol was added to the samples, which were shaken in a 60 °C water bath for 10 min.
Subsequently, 14% boron trifluoride in methanol was added to the mixture, which was then shaken for an additional 10 min at 60 °C. The fatty acid methyl esters (FAMEs) were extracted three times in 2 mL of n-hexane. The FAMEs were stored at −18 °C until used for GC analysis.
The GC analysis of fatty acids in feed and milk samples was performed as described before [
22]. Briefly, separation and quantification of the fatty acid methyl esters were conducted using a fused silica capillary column (100 m × 0.25 mm, Agilent, Santa Clara, CA, USA) on a PerkinElmer Clarus 680 gas chromatograph equipped with an autosampler and flame ionisation detector. For calibration, the methyl esters of
cis-vaccenic acid (
C18:1
cis-11), docosapentaenoic acid (
C22:5n-3), rumenic acid (
C18:2
cis-9, trans-11), docosatetraenoic acid (
C22:4n-6) and stearidonic acid (
C18:4n-3) of the “Sigma FAME” standard were used. The five-point calibration of single fatty acids ranged from 16 to 415 µg/mL.
To evaluate the nutritional value of milk for human nutrition, the thrombogenic index (TI) and atherogenic index (AI) were calculated according to Irawan et al. (2024) [
7] using the following equations:
in which
C12:0 is lauric acid,
C14:0 is myristic acid,
C16:0 is palmitic acid,
C18:0 is stearic acid
, UFA is unsaturated fatty acids, MUFA is monounsaturated fatty acids, and PUFA is polyunsaturated fatty acid.
In addition, the hypocholesterolemic/hypercholesterolemic ratio (h/H) was calculated according to Chen and Liu (2020) [
23]:
in which
cis-
C18:1 is oleic acid,
C12:0 is lauric acid,
C14:0 is myristic acid,
C16:0 is palmitic acid and PUFA is polyunsaturated fatty acid.
Furthermore, the health-promoting index (HPI) was calculated according to Chen et al. (2004) [
24] as follows:
in which
C12:0 is lauric acid,
C14:0 is myristic acid,
C16:0 is palmitic acid and UFAs are unsaturated fatty acids.
2.5. Analysis of Secondary Plant Metabolites
For the analysis of total tannins, condensed tannins, and total phenol concentrations, an extract was prepared from the feedstuff according to Cork and Krockenberger (1990) with some changes [
25]. Briefly, 50 mg of dried and ground (0.7 mm) TMR or individual feed components was mixed with 10 mL 50% acetone and incubated on the shaker for 24 h. Subsequently, the extract was centrifuged at 2500×
g and 4 °C for 15 min.
The total phenol and total tannin concentrations were determined as described by the Joint FAO/IAEA Division of Nuclear Techniques in Food and Agriculture with some modifications [
26]. In brief, 0.5 mL of the supernatant was mixed with 0.25 mL Follin–Ciocalteau reagent (1:1 diluted with aqua dest. Carl Roth, Karlsruhe, Germany) and incubated for 1 min at room temperature. Subsequently, 1.25 mL of 20% Na
2CO
3 was added. The mixture was vortexed and incubated for 40 min at room temperature in the dark. The absorption was measured at 700 nm wavelength. Tannic acid (Merck, Darmstadt, Germany) was used to create a standard curve ranging from 0 to 10 µg.
For the analysis of total tannin concentrations, 100 mg polyvinylpyrrolidone (Merck, Darmstadt, Germany) was mixed with 1 mL distilled water and 1 mL of the acetonic feed extract (1:10 diluted with water). The mixture was vortexed and incubated at 4 °C for 15 min. After additional vortex, the mixture was centrifuged at 2.500× g for 15 min. Subsequently, 0.5 mL of the supernatant was mixed with 0.25 mL Follin–Ciocalteau reagent (1:1 diluted in water) and incubated for 1 min at room temperature. After adding 1.25 mL of 20% Na2CO3, the mixture was vortexed and incubated for 40 min in the dark. Subsequently, the mixture was centrifuged at 16,100× g for 5 min at room temperature, and the absorbance was measured at 700 nm. To calculate the total tannin content, the measured absorbance was subtracted from the absorbance of the total phenol content measurement (see above).
Condensed tannin concentrations were analysed according to Porter et al. (1986) and recommended previously [
26,
27]. In brief, for the analysis of the condensed tannin concentration, 100 µL of the acetonic feed extract was mixed with 600 µL butanol–HCl reagent (butanol–HCl 95:5
v/
v) and 20 µL ferric reagent (2% ferric ammonium sulphate in 2N HCl). The mixture was vortexed and incubated at 70 °C for 4 h. After cooling, the absorbance was measured at 550 nm wavelength. Samples for the determination of total tannin, condensed tannin, and total phenol concentrations were analysed in triplicate. The intra-assay coefficient of variation (CV) was 8.6% for the analyses of condensed tannin in TMR samples and 5.9% for individual feed component samples. For the analyses of the total phenol concentration, intra-assay CV was 5.9% for individual feed component samples and 4.7% for TMR samples. The intra-assay CV was 4.1% for the analyses of total tannin in TMR samples and 1.1% for individual feed component samples.
For the measurement of total flavonoid concentration in TMR, hemp leaves, straw, rapeseed and soybean meal, approximately 1 g of each sample was incubated with 30 mL 75% ethanol in an ultrasonic bath at 60 °C for 30 min. The obtained extract was filtered using a paper filter (MN 614, MACHEREY-NAGEL, Düren, Germany), and the extraction procedure was repeated twice. The combined extracts were diluted with 75% ethanol to 100 mL stored at 4 °C until further analysis.
The total flavonoid concentration was determined using quercetin as standard as described by Ruiz-Reyes et al. (2022) [
28] with the following modifications: A mixture of 200 μL feed extract, 0.8 mL 70% ethanol, and 1 mL aluminium chloride (AlCl
3) solution (20 mg AlCl
3 in 1 mL 70% ethanol and 5% acetic acid) was incubated in the dark for 30 min and subsequently measured at a wavelength of 410 nm using a microplate reader (Sunrise, Tecan Trading, Switzerland). All samples were run in duplicate. Intra-assay CV was 8.5%.
The concentrations of the cannabinoids cannabidivarin (CBDV), cannabidiol (CBD), cannabinol (CBN), Δ9-tetrahydrocannabivarin (Δ9-THCV), Δ8-tetrahydrocannabivarin (Δ8-THC), Δ9-tetrahydrocannabinol (Δ9-THC) and tetrahydrocannabinolic acid (THCA) in Santhica 27 hemp leaves were analysed by the Chemical and Veterinary Federal Office (Münsterland-Emsche-Lippe, Germany) using LC-MS/MS as described by Wagner et al. (2022) [
6]. The detection limit was 0.5 µg/kg and the limit of quantification was 1 µg/kg. The recovery rates ranged between 70 and 120% and were corrected via isotope-labelled ISTs. The expanded measurement uncertainty was 25%. The specificity was verified via the retention time and specific mass transitions.
For the cannabinoid analyses, 0.5 mL of plasma or milk was mixed with the internal standard DHA-ethanolamide-d4 and acidified with citrate buffer. The samples were extracted twice with ethyl acetate and dried under a nitrogen stream. The resulting extract was dissolved in 100 µL methanol/water (60:40), including 1 µg/mL butylated hydroxy toluene. The cannabinoid concentrations in plasma and milk, namely Δ9-THC, THCA, CBD, CBN, cannabichromene (CBC), CBG, cannabigerolic acid (CBGA), and cannabidiolic acid (CBDA) using an HPLC coupled to a triple-quad mass spectrometer with electrospray ionisation (Agilent 6495/1290, Santa Clara, CA, USA) equipped with a Poroshell EC120 column (100 × 2.1 mm, 2.6 µm, Agilent, Santa Clara, CA, USA). Separation was performed using a gradient of 5 mM aqueous ammonium acetate solution and methanol. Cannabinoids were identified on the basis of at least three characteristic mass transitions and the retention time. Authentic standards (Sigma-Aldrich, St. Louis, MO, USA) in relation to the internal standard were used for the quantification. The calibration curves were linear in the range of 0.01 to 10 ng/mL (r2 > 0.99). Sample preparation and concentration measurement of the cannabinoids were performed by Lipidomix GmbH (Berlin, Germany). The detection limit was 0.003 ng/mL, and the limit of quantification was 0.01 ng/mL. The extraction yields ranged between 80 and 110% and were taken into account in the calculations.
2.6. Determination of the Antioxidative Capacity
To measure the antioxidant capacity of the feed, an extract from a 1 g sample was prepared as described for the measurement of total flavonoid concentration. For the determination of the ferric-reducing antioxidant power (FRAP), a working reagent (0.3 M acetate buffer, 0.01 M 2,4,6-Tri(2-pyridyl)-1,3,5-triazine (TPTZ) in 0.04 M HCl and 0.02 M FeCl
3 × 6H
2O, 10:1:1
v/
v/
v) was prepared as described by Wojtunik-Kulesza (2020) [
29]. For the analysis of FRAP, 10 μL of the feed extract was mixed with 90 μL working reagent solution, and the absorbance was measured at 595 nm at room temperature after 10 min. Analyses were run in duplicate. The intra-assay CV was 9.6% for TMR samples and 2.3% for individual feed component samples.
The ferric-reducing ability of plasma was determined as described by Benzie and Strain (1996) with minor modifications [
30]. After thawing on ice, 100 µL of plasma was mixed with 100 µL 75% ethanol and centrifuged at 13,000×
g for 10 min, while rumen fluid was centrifuged without prior ethanol addition. Ten microlitres of the respective supernatant was mixed with ninety microlitres of FRAP working reagent, and the absorbance was measured as described above. Analyses were run in duplicate. The intra-assay CV was 2.5%.
To analyse FRAP in whey, milk samples were thawed, and 1 mL was centrifuged at 50,380× g for 20 min at 4 °C. Subsequently, 400 µL of the aqueous phase was taken and mixed with 75% ethanol and again centrifuged. Ten microlitres of the supernatant was mixed with 90 microlitres of the FRAP working reagent, and the absorbance was measured as described above. Analyses were run in duplicate. The intra-assay CV was 3.9%.
In addition, a 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) decolourisation assay was used to assess antioxidant capacity in feed and plasma adapted from Re et al. (1999) with some modifications [
31]. In brief, the ABTS stock solution was prepared containing 900 µM ABTS (Merck, Darmstadt, Germany) and 450 µM potassium persulfate. Plasma samples were thawed on ice and 1:3 diluted with phosphate-buffered saline +0.5 mM EDTA. Ten microlitres of the respective feed extract or diluted plasma samples was mixed with two hundred microlitres of ABTS stock solution, and the absorbance was measured at 700 nm at room temperature after 5 min incubation in the dark. A Trolox solution diluted with ethanol was used to create a standard curve between 0 and 1200 µM. Plasma samples were run in triplicate, and feed samples in duplicate. Intra-assay CV was 2.0% for plasma samples, 0.7% for individual feed components and 0.4% for TMR samples.
The antioxidant activity in feed samples was further assessed by a 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay, according to Brand-Williams et al. (1995) [
32]. Briefly, 50 µL of the feed extract was mixed with 100 µM DPPH in ethanol and incubated for 30 min in the dark. Absorbance was measured at 520 nm wavelength. All DPPH analyses were run in triplicate. The intra-assay CV was 5.0% for TMR samples and 5.1% for individual feed components. The antioxidant capacity determined by ABTS and DPPH assays is expressed as Trolox-equivalent antioxidant capacity (TEAC).
2.7. Analysis of Thiobarbituric Acid Reactive Substances
The thiobarbituric acid reactive substances (TBARS) in plasma samples obtained on day 14 were determined using a TBARS Parameter Assay Kit (KGE013, R&D Systems Europe, Abingdon, United Kingdom) according to the manufacturer’s instructions. Absorbance was measured using a microplate reader (Sunrise, Tecan Trading, Männedorf, Switzerland). Samples were analysed in duplicates. The intra-assay CV was 4.5%.
2.8. Gene Expression in White Blood Cells
Total RNA was extracted from 400 µL buffy coat using the NucleoSpin RNA Blood Kit (MACHEREY-NAGEL, Düren, Germany) according to the manufacturer’s instructions. The RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA, USA). RNA integrity numbers (RIN) between 5 and 9.4 (mean: 7.7, SD: 1.1) were yielded. The cDNA synthesis and qPCR were performed as described recently with the following modification [
33].
The PCR contained 2 µL cDNA (10 ng/µL), 1 µL H2O, 0.5 µL of each primer (4 µM), and 6 µL 2 × Puffer SensiFAST SYBR No-ROX mix (Meridian Bioscience, Cincinnati, OH, USA). Data were quantified using qbase software (Biogazelle, Gent, Belgium).
Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation
(YWHAZ), ribosomal protein S9 (
RPS9), and succinate dehydrogenase complex flavoprotein subunit A1 (
SDHA) were used as reference genes (M-value: 0.298, CV-value: 0.119). The primer sequences are shown in
Supplementary Table S1.
The efficiency of amplification was calculated using LinReg-PCR software (v.2014.4, Academic Medical Centre, Amsterdam, The Netherlands). Only 6 samples per group could be analysed because the RNA concentrations of the remaining samples were insufficient.
2.9. Tumour Necrosis Factor Alpha Concentrations
Plasma tumour necrosis factor alpha (TNF-α) concentration in samples obtained on day 14 of each period was measured using a bovine ELISA kit (Bovine TNF Alpha ELISA Kit—LS-F5014, LifeSpan BioScience, Washington, DC, USA) in accordance with the manufacturer’s instructions. Absorbance was measured using a microplate reader (Sunrise, Tecan Trading, Männedorf, Switzerland). All samples were analysed in duplicates. The intra-assay CV was 5.6%. Three samples (2 HEMP, 1 CON) had to be excluded from the statistical analysis because the CV exceeded the cut-off value of 15% that was set prior to the experiment.
2.10. Statistical Analysis
The required sample size to demonstrate the equivalence of parameters was calculated using ‘sampleN. TOST()’ from the “PowerTOST” package [
34] in R Statistical Software (version 4.4.2) [
35]. A coefficient of variation of 15%, a ratio of the means of 0.9, a lower and upper equivalence limit of 0.75 and 1.33, respectively, and a probability for the type I error of α = 0.05 and for the type II error of β = 0.2 were assumed. Additionally, assuming Cohen’s d = 1.0 and a type I error probability of α = 0.05 and power = 0.8, the sample size to detect differences between two parameters was calculated using the function pwr.t.test()’ from the R package ‘pwr’ [
36].
Statistical analysis was performed using R Statistical Software (v4.3.1; R Core Team 2021, R Foundation for Statistical Computing, Vienna, Austria) [
35]. Visual inspection of boxplots was used to check datasets for the presence of outliers.
Data were analysed with a linear mixed model (LMM, lmerTest package, v3.1-3; [
37] including group (level: HEMP and CON), period (level: period 1 and period 2), sequence (level: HEMP-CON or CON-HEMP) and block (levels: Block 1, Block 2, and Block 3) as fixed effects and the animal ID as random effect.
where ϒ
ijkl is the dependent variable for
i-th group,
j-th period, the
k-th sequence, and the
l-th block;
μ is the overall mean;
αi is the fixed effect of the
i-th group;
βj is the fixed effect of the
j-th period;
γk is the fixed effect of the
k-th sequence;
δl is the fixed effect of the
l-th block;
εijkl is the random error term;
υm is the random effect of the
m-th animals.
For the statistical analysis of the antioxidant ability in feed extracts (2 groups with n = 3), a linear model including group and block as fixed effects was used.
where ϒ
ij is value of the dependent variable for the
i-th group and the
j-th block,
µ is the mean,
αi is the effect of the
i-th group,
βj is the effect of the
j-th block, and
εij is the residual.
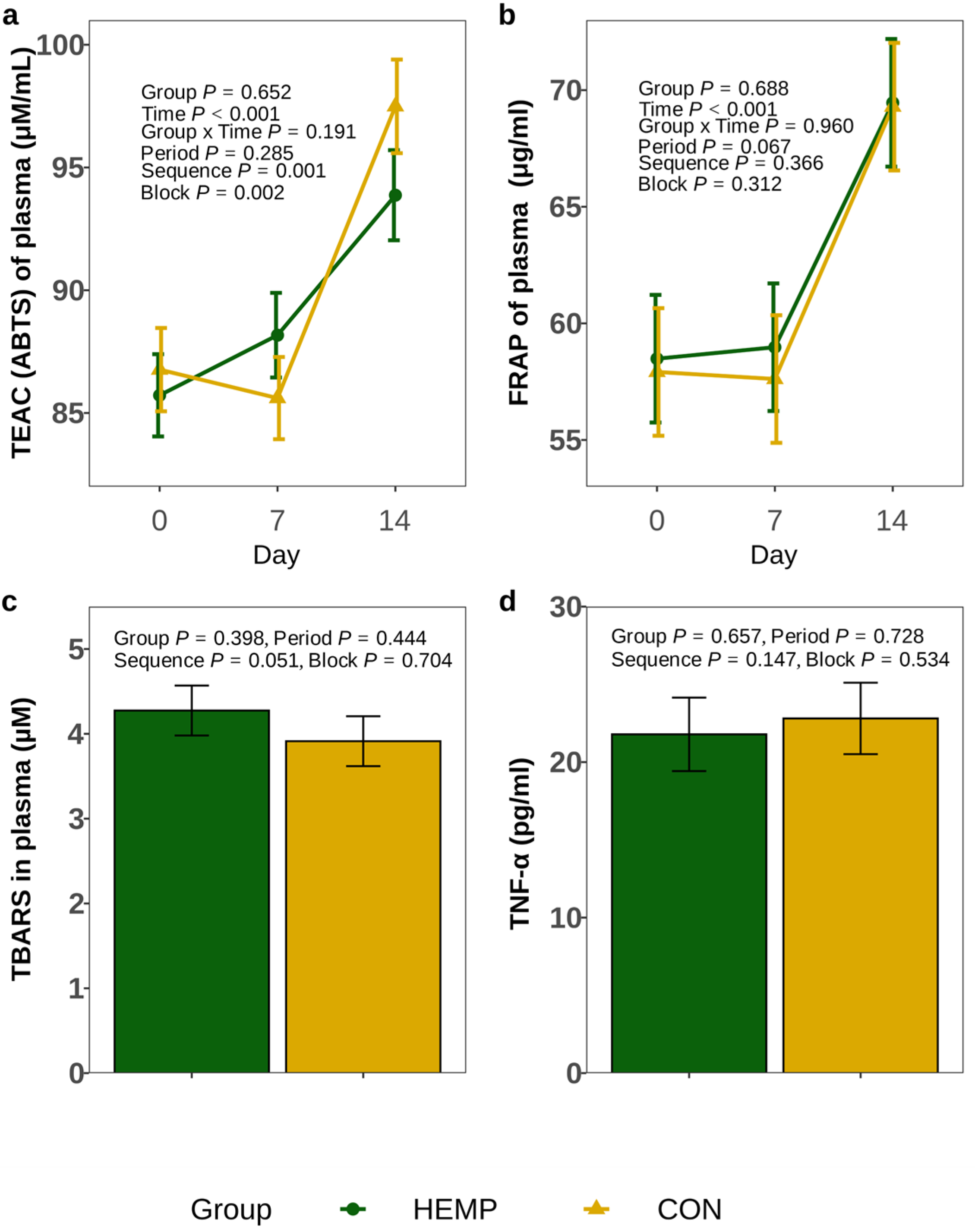
For datasets containing multiple measurements (TEAC and FRAP of plasma) within a feeding period, the time point (day, 3 timepoints) and the interaction with the group (group × time point) were included as additional fixed effects.
where ϒ
ijklm is the dependent variable for
i-th group,
j-th period, the
k-th sequence,
l-th block and the
m-th timepoint;
μ is the overall mean; α
i is the fixed effect of the
i-th group;
βj is the fixed effect of the
j-th period;
γk is the fixed effect of the
k-th sequence;
δl is the fixed effect of the
l-th block;
τm is the fixed effect of the
m-th timepoint; (
ατ) is the interaction between
i-th group and
m-th timepoint;
εijklm is the random error term;
υn is the random effect of the
n-th animals.
The residuals of the models were checked for normal distribution using the check_normality function and singularity using the check_singularity function of the performance package (v0.13.0 [
38]). If the assumption of normality was violated, data were log2 transformed and re-evaluated. If singularity was detected, data were analysed using a linear model including group (level: HEMP and CON), period (level: period 1 and period 2), sequence (level: HEMP-CON or CON-HEMP) and the block (levels: Block 1, Block 2 and Block 3) as fixed effects. Thus, two treatments (HEMP and CON) in three replicates (Block 1 to 3) were evaluated.
Pairwise differences were tested by using the Tukey–Kramer test. For the fixed effect of interest, estimated marginal means (EMMs) and their standard errors (SE) were estimated. Effects and differences were considered significant at p < 0.05 and considered as a trend at p < 0.10.
To test the correlation between cannabinoid concentrations in milk and plasma, Kendall’s τ coefficient was computed, as samples were not normally distributed.







{kind=link}
{kind=link}
{kind=link}