1. Introduction
Avian leukosis (AL) is a viral infectious disease in poultry caused by the avian leukosis virus (ALV), characterized by severe immunosuppression and tumorigenic potential. Livestock production, particularly poultry farming, plays a pivotal role in the socio-economic development of many countries worldwide. Among poultry species, the domestic chicken (Gallus gallus domesticus) stands out due to its short generation interval and adaptability to a wide range of agro-ecological environments, making it one of the most widely distributed avian species globally [
1]. As the most extensively raised livestock species, chickens serve as a vital source of high-quality protein and supplemental income for rural households, especially in resource-limited regions. Their popularity is largely attributed to their advantageous traits, including robust disease resistance, adaptability to harsh environments, and efficient utilization of low-quality feed resources [
2].
ALV belongs to the C-type retrovirus family and possesses a lipid envelope [
3,
4]. It is classified into 11 subgroups (A–K) based on the gp85 envelope glycoprotein [
5], among which the J subgroup (ALV-J) exhibits the highest pathogenicity [
6], with widespread prevalence and substantial economic consequences globally. ALV transmission occurs both vertically and horizontally [
7], complicating eradication efforts. Clinically, ALV infection leads to tumor formation, immune suppression, and increased mortality in poultry [
8], particularly under intensive farming conditions that facilitate viral spread. Although p27 antigen-based diagnostic assays have been applied in eradication programs, current control strategies remain challenged by the virus’s long incubation period and limitations in detection methods [
9].
In recent years, genome-wide association studies (GWASs) have made remarkable contributions to disease-resistance research. By detecting linkage disequilibrium between genetic variants and traits of interest, GWAS enables the identification of significant single nucleotide polymorphisms (SNPs) correlated with phenotypic variation, providing powerful insights into the genetic architecture of disease resistance [
10,
11,
12]. For example, Wossenie Mebratie et al. employed a mixed linear model to identify 11 quantitative trait loci (QTLs) and numerous SNPs associated with body weight and feed conversion efficiency in poultry [
13]. Similar GWAS approaches have been applied to traits such as egg quality and disease resistance [
14]. In studies of avian influenza resistance, Anna Wolc and colleagues utilized SNP arrays to identify resistance-associated genomic regions, offering key targets for selective breeding, although causal genes remain elusive due to the polygenic nature of the trait [
15]. In another study, Xiao et al. identified significantly enriched SNPs on chromosome 5 associated with Salmonella resistance, laying the groundwork for breeding against pullorum disease [
16]. Collectively, these findings underscore the utility of GWAS in poultry genetic improvement and disease resistance and provide a valuable reference for future investigations into ALV resistance loci and candidate genes [
17,
18].
ALV infection is known to reduce egg production, induce tumorigenesis, and suppress the immune system, thereby posing a serious threat to poultry health. Although it is known that ALV enters host cells via specific receptors such as Tva, Tvb, Tvc, and chNHE1, the genetic basis of host resistance remains poorly understood, with limited studies addressing resistance loci and genes associated with ALV-J. The indigenous Chengkou mountain chicken, a local Chinese breed, exhibits strong adaptability and disease resistance. Preliminary long-term AL eradication programs involving multiple generations of birds revealed a significant variation in infection rates among different populations. Notably, some groups exhibited markedly lower infection rates after several rounds of purification, while others showed no obvious improvement, suggesting a potential genetic basis for resistance. In this context, the present study aims to identify resistance-associated loci against ALV-J through whole-genome resequencing and GWAS, thereby providing a genetic foundation for resistance breeding in poultry.
3. Results
3.1. Genotypic and Phenotypic Data
Phenotypic Data
In this study, families with significant differences in ALV infection status were selected from populations that had undergone multiple generations of avian leukosis purification. A total of 1050 chickens were sampled for cloacal swabs, egg white, and serum. Using ELISA to detect the ALV p27 antigen, 500 individuals were selected for whole-genome resequencing, comprising 325 ALV+ and 175 ALV– individuals, yielding an ALV–/ALV+ ratio of approximately 2:1 (
Table A1).
3.2. SNP Detection
3.2.1. Alignment to the Reference Genome
Genomic DNA was extracted from a total of 500 chickens and subjected to whole-genome sequencing. The DNA quality assessment results are presented in
Table A2. The chicken reference genome size was 1,049,948,333 base pairs. The alignment rate of the population samples ranged from 98.44% to 99.64%, with an average sequencing depth of 12.08× and an average genome coverage of 98.50%.
3.2.2. SNP Detection and Annotation
A total of 20,959,220 raw SNPs were initially detected, and after filtering, 12,644,463 high-quality SNPs were retained. A remarkable accumulation of SNPs was observed around the 5 Mb region on chromosome 6 (Chr6). Annotation revealed that most of the filtered SNPs were enriched in intronic and intergenic regions, while a smaller proportion were located in upstream, exonic, and downstream regions of genes. A further analysis showed that the majority of SNP variations were caused by base transitions (
Table A3).
3.3. Population Genetic Diversity Analysis
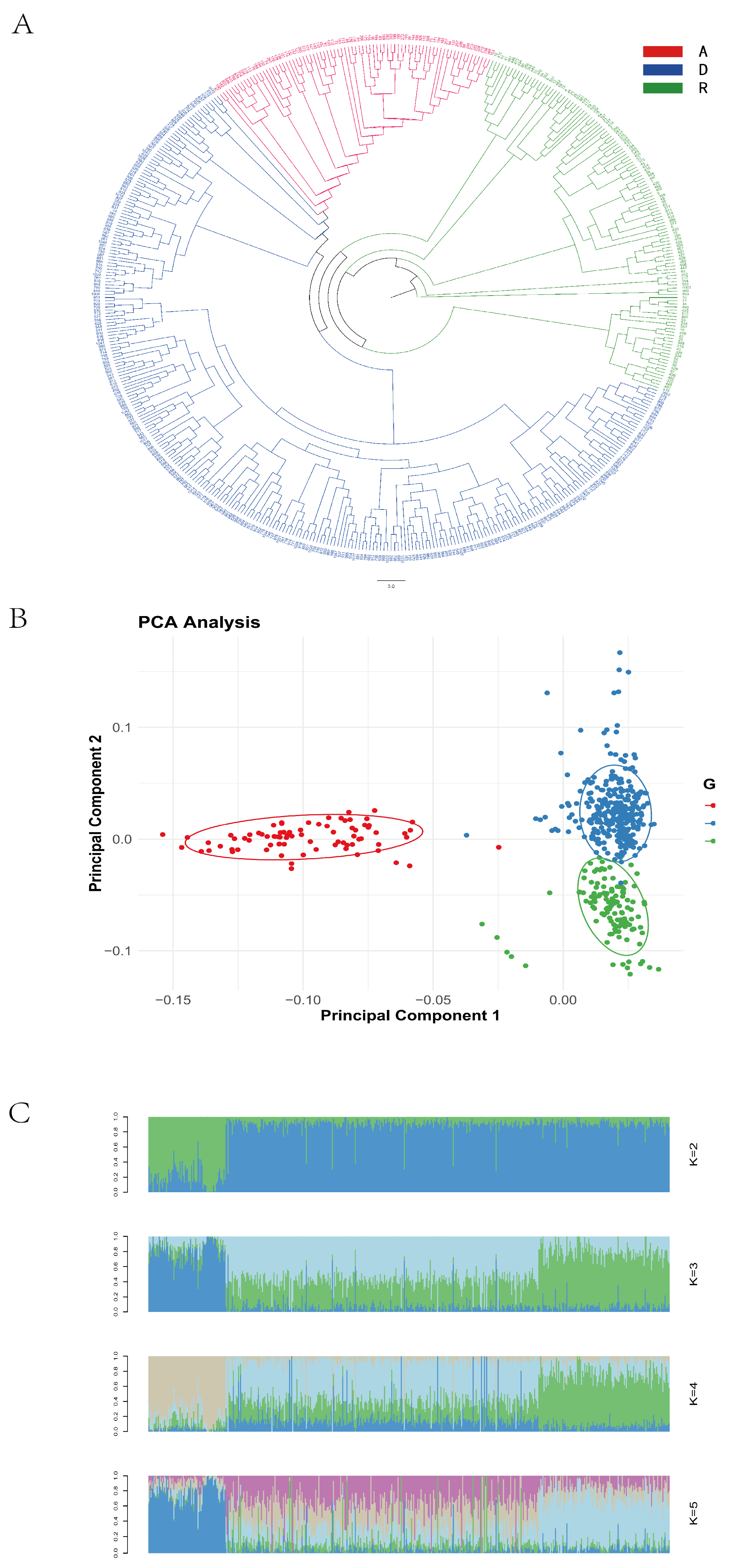
The phylogenetic tree revealed clear clustering patterns consistent with the line-specific classifications (Lines A, R, and D), as recorded during the sample collection (
Figure 1A). A principal component analysis (PCA) showed that some individuals from Line D clustered with Lines A and R, while, overall, the three lines demonstrated good intra-line clustering and clear inter-line separation (
Figure 1B). When the number of ancestral populations was set to three, each individual’s genome exhibited clear structural grouping (
Figure 1C), indicating that the 500 resequenced samples originated from three ancestral populations.
Based on the results of the population diversity analyses, the sequenced samples were determined to originate from three ancestral populations. This finding is consistent with the three lines recorded during sample collection, confirming the accuracy of population classification and validating the feasibility of conducting a line-specific genome-wide association analysis in the subsequent steps.
3.4. Genome-Wide Association Analysis
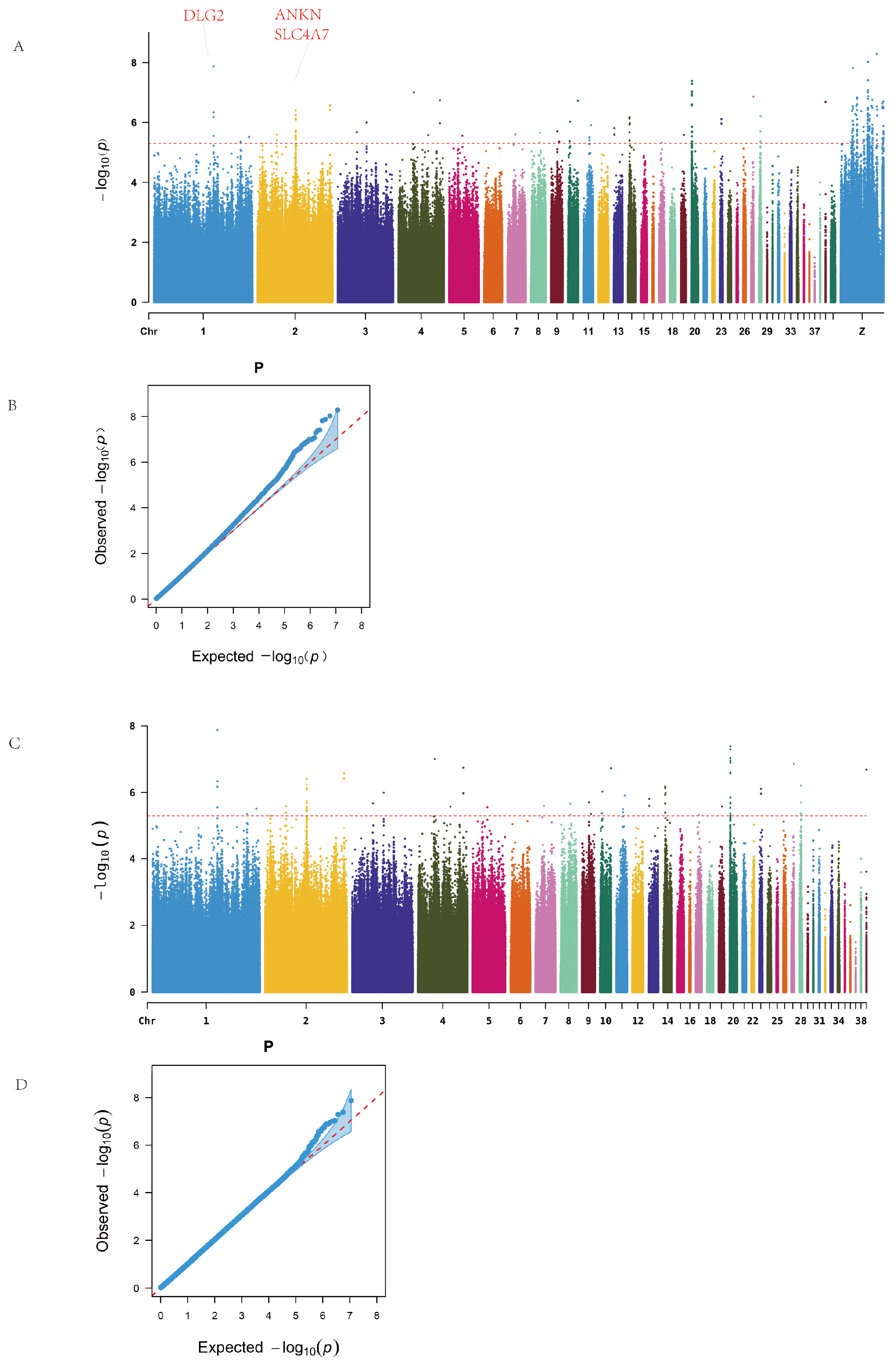
Genome-wide association analysis revealed significant loci on chromosomes 1, 2, 4, 14, 20, 28, and the Z chromosome, as illustrated in the Manhattan plot (
Figure 2A). No continuous regions of significance were observed on the Z sex chromosome. A total of 218 SNPs surpassing the genome-wide significance threshold were identified and mapped to 49 candidate genes. Functional annotations showed that two SNPs were located in exonic regions, including one synonymous mutation and one non-synonymous mutation (on chromosome Z). Additionally, 76 SNPs were located in intergenic regions (
Table A4).
Table 1 lists the top 20 SNPs and their corresponding genes, including a synonymous SNP on chromosome 1 with the most significant
p-value.
Among the significantly enriched genes, the Z chromosome harbored the largest number of SNPs, with BNC2 and NRG1 annotated with 15 and 13 SNPs, respectively. On the autosomes, ANKH on chromosome 2 and EIF6 on chromosome 20 exhibited substantial SNP enrichment. Additionally, genes such as FBXL7 and SLC4A7 were also significantly enriched (
Table A5).
Since no continuous high-signal SNP regions were observed on the Z chromosome, and to avoid the dilution of potential association signals by the high mutation load on the sex chromosome, we excluded the Z chromosome data from the subsequent analyses. This adjustment also aimed to better explore intra- and inter-line differences among chicken populations. Therefore, follow-up genome-wide association analyses for ALV resistance were conducted both within individual lines and across the entire population to further identify potential associated loci and candidate genes.
After removing sex chromosome data, the resulting Manhattan plot revealed continuous high-signal regions on chromosomes 1, 2, and 20 (
Figure 2B). A total of 78 significant SNPs were identified, spanning 17 intergenic regions and 21 genes, including one synonymous mutation located in an exon. Notable candidate genes included ANKH (12 associated SNPs), EIF6 (11 SNPs), and SLC4A7, DLG2, and FBXL7, which are widely implicated in cellular inflammation, immune response, and tumor development.
Further within-line analysis revealed the following findings:
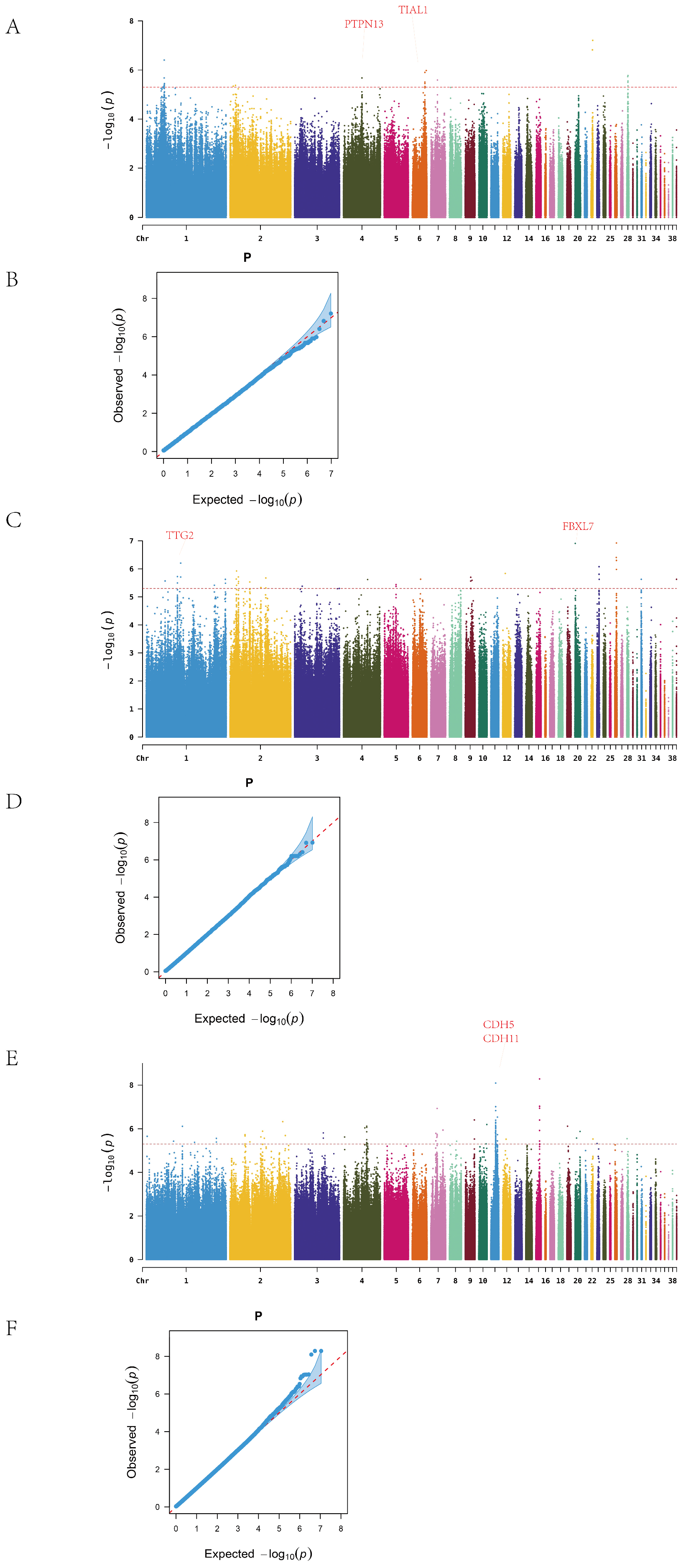
In Line A, a total of 26 ALV-negative and 48 ALV-positive individuals were analyzed. After excluding SNPs on the sex chromosome, genome-wide association analysis identified continuous clusters of significant variants on chromosomes 1, 4, 6, and 28 (
Figure 3A). In total, 34 significant SNPs were detected, mapped to 12 candidate genes. The most significant SNP on chromosome 22 was located in an intergenic region, while 11 SNPs on chromosome 1 were concentrated in a single intergenic region. Twelve SNPs were located within gene regions, corresponding to nine genes, among which PTPN13 and TIAL1 are associated with cancer and immune function (
Table A6).
In Line R, after excluding sex chromosome data, the Manhattan plot revealed significant SNPs on chromosomes 1, 2, 9, 23, and 26 (
Figure 3B). A total of 46 SNPs were identified, distributed across 13 chromosomes and mapped to 20 candidate genes. The most significant SNP on chromosome 26 was located between NGF and FANCE; NGF is known to be involved in nerve growth, while FANCE plays a key role in the Fanconi anemia pathway. On chromosome 1, 14 SNPs were detected, with 7 mapping to the TTF2 gene, which is associated with thyroid development.
In Line D, 92 significant SNPs were identified across 16 chromosomes after removal of sex-linked loci, and these were mapped to 21 candidate genes. High-density SNP regions were detected on chromosomes 1, 4, 7, 11, and 15 (
Figure 3C). The most significant SNP on chromosome 15 was located in the YWHAH gene, which has been implicated in viral encephalitis and tumor progression. On chromosome 11, 30 SNPs were concentrated in the intergenic region between CDH5 and CDH11, both members of the cadherin family, which are involved in cancer and vascular disease. In addition, the identification of SLC5A1, a member of the solute carrier (SLC) family, supports the relevance of this gene family in ALV resistance, consistent with previous findings related to SLC4A7.
Q–Q plots confirmed that the observed p-value distribution closely followed the expected uniform distribution, indicating no systematic bias in the GWAS results.
3.5. GO and KEGG Enrichment Analyses
To comprehensively evaluate the functional roles of candidate genes annotated from significant SNPs, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed under the threshold of p < 0.05.
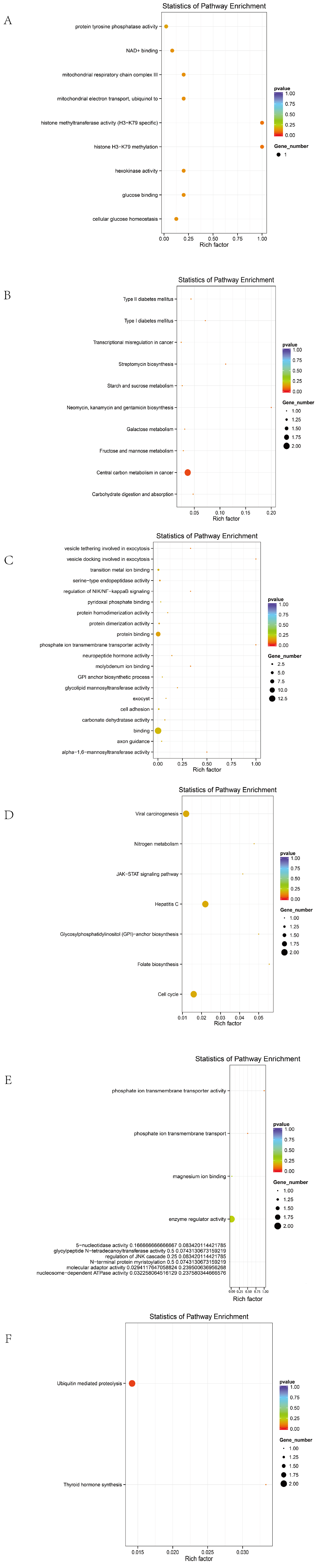
Before excluding sex chromosomes, the enriched terms included 9 biological processes, 3 cellular components, 15 molecular functions, and 4 KEGG pathways (
Figure 4A,B). The top 20 GO terms with the highest enrichment scores are shown in the figure.
After excluding sex chromosomes, an enrichment analysis revealed 16 biological processes, 1 cellular component, 14 molecular functions, and 9 KEGG pathways (
Figure 4C,D).
Line-specific analyses yielded the following results:
In Line A, 26 biological processes, 10 cellular components, 16 molecular functions, and 10 KEGG pathways were enriched (
Figure 5A,B).
In Line R, 25 biological processes, 10 molecular functions, and 2 KEGG pathways were identified (
Figure 5C,D).
In Line D, 39 biological processes, 3 cellular components, 22 molecular functions, and 7 KEGG pathways were significantly enriched (
Figure 5E,F).
Candidate genes were significantly enriched in membrane-associated signaling pathways and tumorigenesis-related mechanisms, including membrane transporter activity, transmembrane transport, cellular metabolism, endoplasmic reticulum function, ion transport, and cancer-related functions. Representative genes included SLC4A7, SLC5A1, ANKH, EIF6, DLG2, FBXL7, and CDH5.
A KEGG analysis further indicated that these genes are involved in the JAK/STAT signaling pathway, ECM–receptor interaction, glycosaminoglycan biosynthesis, folate-mediated one-carbon metabolism, and other pathways, which are potentially associated with the tumorigenic mechanisms in ALV-infected individuals.
3.6. Identification of Candidate Genes by RNA-Seq Analysis
To validate the potential functions of candidate genes identified through GWAS, we integrated RNA-Seq data and performed an intersection analysis between differentially expressed genes and a randomly selected subset of three GWAS candidate genes (ANKH, CDH11, and SLC5A1). All three genes exhibited significant differential expression in the RNA-Seq analysis (
p < 0.05) (
Table A7).
3.7. qRT-PCR Validation of Candidate Gene Expression
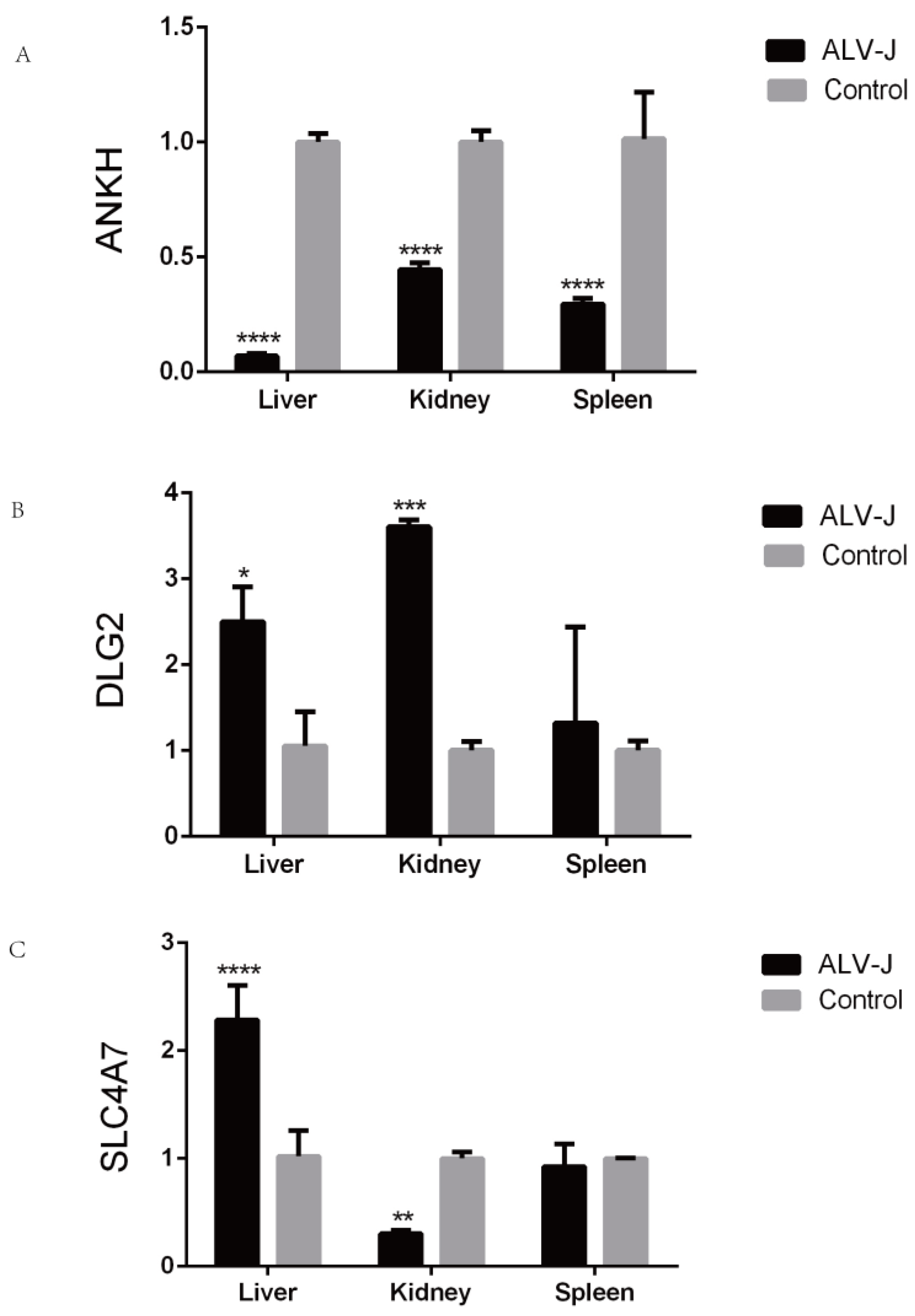
Three candidate genes (ANKH, DLG2, and SLC4A7) were randomly selected for qRT-PCR validation (
Figure 6). Under ALV-J infection, ANKH exhibited significant differential expression in the liver, spleen, and kidney tissues. Both DLG2 and SLC4A7 showed significant changes in expression in the liver and kidney tissues.
4. Discussion
Avian leukosis virus subgroup J (ALV-J) is a highly contagious disease in poultry, characterized by rapid transmission and the lack of effective vaccines, resulting in substantial economic losses to the poultry industry. Although continuous screening and culling strategies have helped to control the spread of ALV-J, these approaches have not completely eliminated the virus from poultry populations. In 2006, researchers identified the sodium–hydrogen exchanger 1 (NHE1) as a cellular receptor for ALV-J [
32]. The study demonstrated that quails and certain wild birds exhibited resistance to ALV-J, while turkeys and some domestic chicken breeds were susceptible. However, the specific genetic loci associated with ALV-J resistance remain largely unknown. In this study, a genome-wide association study (GWAS) was used to identify genetic variants associated with ALV-J susceptibility, providing insights into the host’s genetic determinants of infection.
GWAS revealed several loci significantly associated with ALV-J infection, particularly within genes related to immune response, cancer progression, and membrane function. Among these,
PTPN13, a known tumor suppressor, showed a strong association with ALV-J susceptibility.
PTPN13 is involved in regulating cell death and migration, and its dysregulation in various cancers suggests that it may influence ALV-J infection by modulating immune responses and intracellular signaling pathways [
33,
34,
35].
Another notable gene,
TTF2, a thyroid-specific transcription factor, has been implicated in the pathogenesis of multiple cancers, including thyroid carcinoma [
36,
37,
38]. ALV integrates into the host genome and can activate or disrupt proto-oncogene expression, leading to abnormal cell proliferation such as lymphoma. Given ALV’s reliance on host transcriptional machinery,
TTF2 may affect the efficiency of viral replication or integration, thereby influencing susceptibility to ALV-J.
TIAL1, a gene involved in RNA splicing and DNA repair, plays a critical role in B cell development. Mutations in
TIAL1 may impair B cell maturation, contributing to immune tolerance and persistent ALV-J infection [
39]. In addition, genes such as
DLG2 [
40,
41,
42] and
FBXL7 [
43] are involved in tumor progression and immune regulation. ALV-J infection has been shown to suppress JAK-STAT signaling by upregulating SOCS3 (suppressor of cytokine signaling 3), thereby inhibiting innate immune responses and promoting viral replication [
44]. The enrichment of these genes in the JAK-STAT pathway suggests that they may influence ALV-J susceptibility by modulating host immunity.
As ALV-J initiates infection by binding to specific receptors on the host cell membrane, genes involved in membrane structure and function are critical for viral entry and propagation. In this study, several candidate genes associated with membrane function were identified, including ANKH, SLC4A7, SLC5A1, and members of the cadherin (CDH) family. These genes are essential for ion transport, cell adhesion, and maintenance of cellular homeostasis—processes that are vital for viral infection.
ANKH encodes a transmembrane protein responsible for inorganic pyrophosphate transport [
45]. Mutations in
ANKH have been linked to ion transport disorders and inflammatory conditions, such as arthritis [
46,
47]. Our findings suggest that
ANKH may alter ionic homeostasis at the cell surface, thereby influencing ALV-J binding and entry.
SLC4A7 and
SLC5A1, members of the solute carrier family, are involved in bicarbonate and glucose transport, respectively. Enrichment of these genes in pathways related to ion transport and cancer metabolism suggests that their dysfunction may alter membrane permeability, facilitating viral invasion and disease progression [
48,
49,
50,
51,
52,
53,
54,
55,
56,
57]. Furthermore, their association with cancer-related pathways supports the notion that changes in membrane function influence both viral infection and tumorigenesis.
The cadherin family genes
CDH5 and
CDH11 are crucial for cell–cell adhesion and tissue integrity. These proteins are known to participate in tumor immune responses and can enhance antitumor immunity. Notably,
CDH5 and
CDH11 were enriched in the JAK-STAT signaling pathway, which has been associated with immune regulation. High expression of
CDH5 can enhance CD8
+ T cell activity, thereby suppressing tumor growth. Their enrichment in this pathway suggests a role in modulating host susceptibility to ALV-J by influencing immune signaling [
58,
59,
60].
In addition to identifying candidate genes, this study highlights several key biological pathways involved in ALV-J infection and immune suppression. The JAK-STAT signaling pathway, crucial for immune cell signal transduction, was significantly enriched. This pathway has been linked to various immune disorders and cancers [
61,
62,
63,
64]. In chicken macrophages and DF-1 cells, ALV-J infection has been shown to reduce STAT1 phosphorylation, impairing interferon responses and facilitating immune evasion [
65]. Thus, ALV-J may promote tumorigenesis by disrupting JAK-STAT signaling and weakening host antiviral defenses.
Enrichment of the extracellular matrix (ECM)–receptor interaction pathway was also observed. This pathway plays a pivotal role in cell adhesion, migration, and proliferation during tumor development [
66], and its involvement in cancers such as prostate and gastric cancer is well-documented [
67,
68,
69]. ALV may disrupt ECM-receptor interactions, thereby promoting viral entry, immune evasion, and tumorigenesis. Additional enriched pathways, including glycosaminoglycan biosynthesis and folate-mediated one-carbon metabolism, are involved in cell proliferation, migration, and survival—processes often dysregulated in cancer [
70,
71,
72,
73,
74].
Although this study successfully identified potential loci associated with ALV-J resistance in Chengkou mountain chickens via GWAS, several limitations should be acknowledged. Firstly, despite applying Bonferroni correction, false positives may still arise due to multiple testing and residual population stratification. Secondly, phenotypic misclassification may affect the strength of association signals. While RNA-Seq and qPCR validations were partially conducted, further functional genomics studies and cross-population validations are necessary to confirm the biological relevance of these candidate loci.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}