

Correlated Responses to Selection for Intramuscular Fat on the Gut Microbiome in Rabbits
 ,
,  , , ,
, , , {kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Material and Sampling
2.2. Microbial Abundance Measurements
2.3. Effect of Selection on Alpha Diversity
2.4. Effect of Selection on the Microbial Abundance
2.4.1. Microbial Abundances Transformations
2.4.2. Microbiability of IMF
2.4.3. Correlated Response to Selection for IMF on the Microbial Abundances
2.5. Search of Microbial Biomarkers for Prediction of IMF
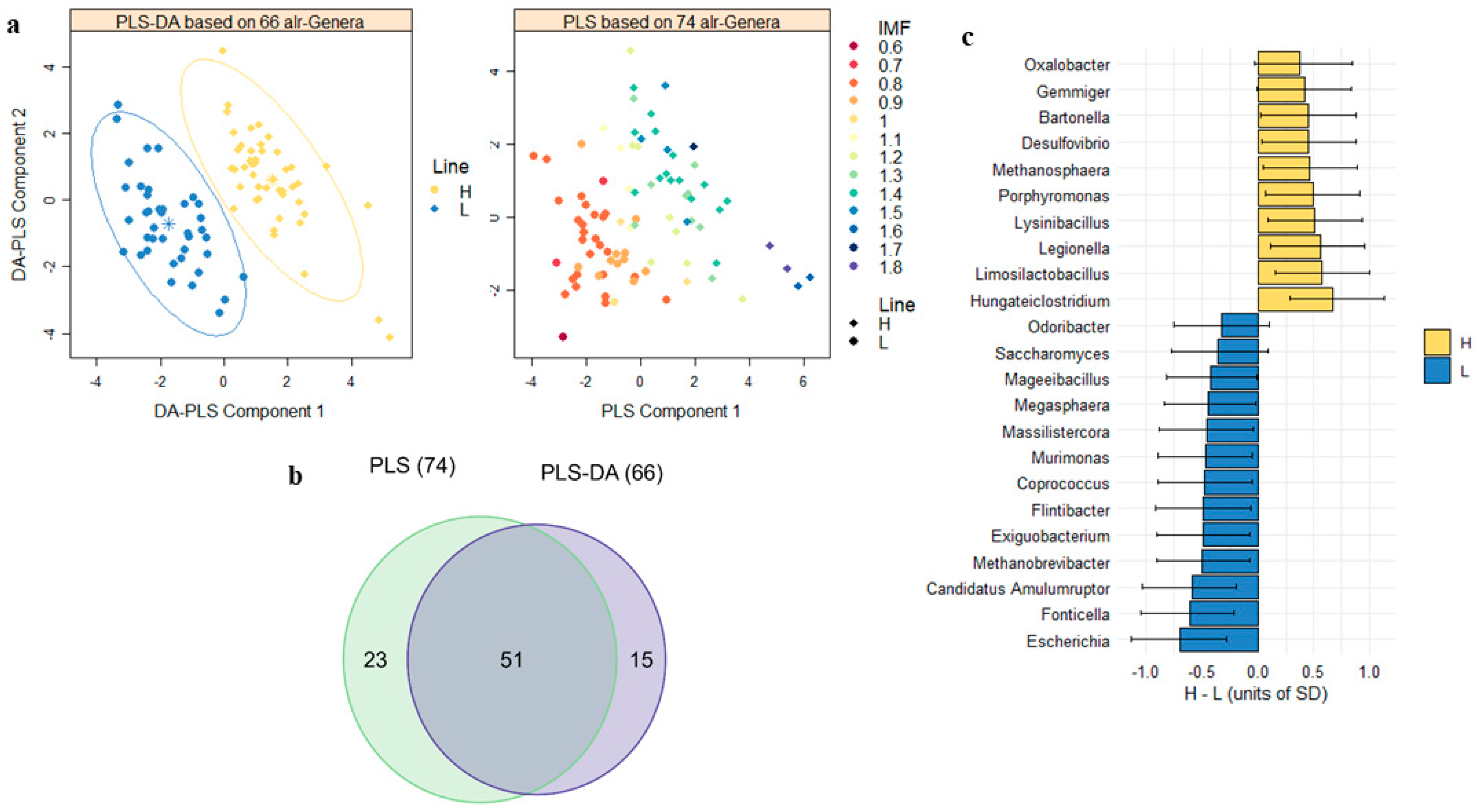
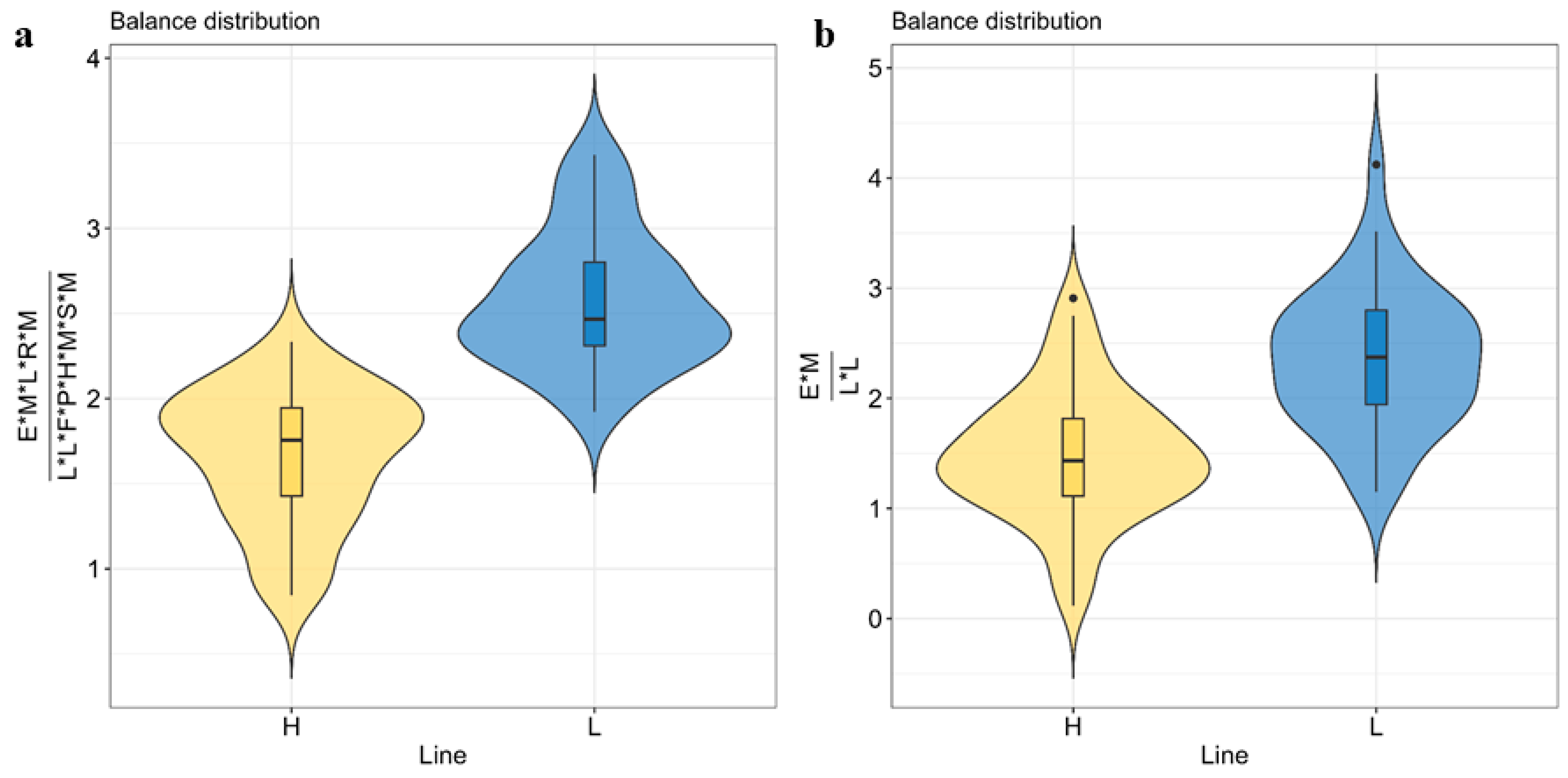
3. Results
3.1. Cecum Microbiome Composition in Rabbits
3.2. Microbiability of IMF
3.3. Divergences between IMF Lines in the Gut Microbiome as a Response to Selection
3.4. Microbial Biomarkers to Predict Host IMF Genetic Background
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wood, J.D.; Enser, M.; Fisher, A.V.; Nute, G.R.; Sheard, P.R.; Richardson, R.I.; Hughes, S.I.; Whittington, F.M. Fat deposition, fatty acid composition and meat quality: A review. Meat Sci. 2008, 78, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Theriault, R.; Watkins, S.C.; Kelley, D.E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism 2000, 49, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, M.; Bentham, J.; Stevens, G.A.; Zhou, B.; Danaei, G.; Lu, Y.; Bixby, H.; Cowan, M.J.; Riley, L.M.; Hajifathalian, K. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Martínez-Álvaro, M.; Hernández, P.; Blasco, A. Divergent selection on intramuscular fat in rabbits: Responses to selection and genetic parameters. J. Anim. Sci. 2016, 94, 4993–5003. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.R.; Baas, T.J.; Stalder, K.J. Results from six generations of selection for intramuscular fat in Duroc swine using real-time ultrasound. II. Genetic parameters and trends. J. Anim. Sci. 2010, 88, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.M.; Lindgren, C.M. The genetics of obesity. Curr. Diab. Rep. 2010, 10, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Ringseis, R.; Gessner, D.K.; Eder, K. The Gut-Liver Axis in the Control of Energy Metabolism and Food Intake in Animals. Annu. Rev. Anim. Biosci. 2020, 8, 295–319. [Google Scholar] [CrossRef]
- Lin, H.; Frasetto, A.; Kowalik, E., Jr.; Nawrocki, A.; Lu, M.; Kosinski, J.; Huber, J.A.; Szeto, D.; Yao, X.; Forrest, G. Butyrate and propionate protect agains diet-induced obesity and regulate gut hormones. PLoS ONE 2012, 7, e35240. [Google Scholar]
- Lim, Y.Y.; Lee, Y.S.; Ooi, D.S.Q. Engineering the Gut Microbiome for Treatment of Obesity: A Review of Current Understanding and Progress. Biotechnol. J. 2020, 15, e2000013. [Google Scholar] [CrossRef]
- Xiong, Y.; Miyamoto, N.; Shibata, K.; Valasek, M.A.; Motoike, T.; Kedzierski, R.M.; Yanagisawa, M. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc. Natl. Acad. Sci. USA 2004, 101, 1045–1050. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2020, 19, 55–71. [Google Scholar] [CrossRef]
- Krajmalnik-Brown, R.; Ilhan, Z.-E.E.; Kang, D.-W.W.; DiBaise, J.K. Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Blaser, M.J. Pathways in microbe-induced obesity. Cell Metab. 2013, 17, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.-L.; Guoyao, W.; Zhu, W.-Y. Amino acid metabolism in intestinal bacteria: Links between gut ecology and host health. Front. Biosci. 2011, 16, 1768–1786. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Gou, Y.K.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Davenport, E.R.; Cusanovich, D.A.; Michelini, K.; Barreiro, L.B.; Ober, C.; Gilad, Y. Genome-wide association studies of the human gut microbiota. PLoS ONE 2015, 10, e0140301. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, M.; Venema, K. Does the gut microbiota contribute to obesity? Going beyond the gut feeling. Microorganisms 2015, 3, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1131. [Google Scholar] [CrossRef]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef]
- Koliada, A.; Syzenko, G.; Moseiko, V.; Budovska, L.; Puchkov, K.; Perederiy, V.; Gavalko, Y.; Dorofeyev, A.; Romanenko, M.; Tkach, S. Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol. 2017, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yi, C.; Han, J.; Ming, T.; Zhou, J.; Lu, C.; Li, Y.; Su, X. Novel high-docosahexaenoic-acid tuna oil supplementation modulates gut microbiota and alleviates obesity in high-fat diet mice. Food Sci. Nutr. 2020, 8, 6513–6527. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Zomeño, C.; Blasco, A.; Hernández, P. Divergent selection for intramuscular fat content in rabbits. II. Correlated responses on carcass and meat quality traits. J. Anim. Sci. 2013, 91, 4532–4539. [Google Scholar] [CrossRef]
- Zubiri-Gaitán, A.; Blasco, A.; Ccalta, R.; Satué, K.; Hernández, P. Intramuscular fat selection in rabbits modifies the fatty acid composition of muscle and liver tissues. Animals 2022, 12, 893. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. FAOSTAT. 2022. Available online: https://www.fao.org/faostat/en/#data/QCL (accessed on 8 May 2024).
- Fan, J.; Watanabe, T. Transgenic rabbits as therapeutic protein bioreactors and human disease models. Pharmacol. Ther. 2003, 99, 261–282. [Google Scholar] [CrossRef]
- Zhao, S.; Chu, Y.; Zhang, C.; Lin, Y.; Xu, K.; Yang, P.; Fan, J.; Liu, E. Diet-induced central obesity and insulin resistance in rabbits. J. Anim. Physiol. Anim. Nutr. 2008, 92, 105–111. [Google Scholar] [CrossRef]
- Martínez-Álvaro, M.; Hernández, P.; Agha, S.; Blasco, A. Correlated responses to selection for intramuscular fat in several muscles in rabbits. Meat Sci. 2018, 139, 187–191. [Google Scholar] [CrossRef]
- Martinez-Alvaro, M.; Agha, S.; Blasco, A.; Hernandez, P. Muscle lipid metabolism in two rabbit lines divergently selected for intramuscular fat. J. Anim. Sci. 2017, 95, 2576–2584. [Google Scholar] [CrossRef]
- Martínez-Álvaro, M.; Paucar, Y.; Satué, K.; Blasco, A.; Hernández, P. Liver metabolism traits in two rabbit lines divergently selected for intramuscular fat. Animal 2018, 12, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Álvaro, M.; Blasco, A.; Hernandez, P. Effect of selection for intramuscular fat on the fatty acid composition of rabbit meat. Animal 2018, 12, 2002–2008. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Madrid, B.S.; Hernández, P.; Blasco, A.; Haley, C.S.; Fontanesi, L.; Santacreu, M.A.; Pena, R.N.; Navarro, P.; Ibáñez-Escriche, N. Genomic regions influencing intramuscular fat in divergently selected rabbit lines. Anim. Genet. 2020, 51, 58–69. [Google Scholar] [CrossRef]
- Laghouaouta, H.; Sosa-Madrid, B.S.; Zubiri-Gaitán, A.; Hernández, P.; Blasco, A. Novel genomic regions associated with intramuscular fatty acid composition in rabbits. Animals 2020, 10, 2090. [Google Scholar] [CrossRef] [PubMed]
- Zubiri-Gaitán, A.; Blasco, A.; Hernández, P. Plasma metabolomic profiling in two rabbit lines divergently selected for intramuscular fat content. Commun. Biol. 2023, 6, 893. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Álvaro Zubiri-Gaitán, A.; Ferrer, A.; Blasco, A.; Greenacre, M. Comprehensive functional core microbiome comparison in genetically obese and lean hosts under the same environment. Commun. Biol. 2021, 4, 1246. [Google Scholar] [CrossRef] [PubMed]
- Zomeño, C.; Hernandez, P.; Blasco, A. Divergent selection for intramuscular fat content in rabbits. I. Direct response to selection. J. Anim. Sci. 2013, 91, 4526–4531. [Google Scholar] [CrossRef] [PubMed]
- Zomeño, C.; Hernández, P.; Blasco, A. Use of near infrared spectroscopy for intramuscular fat selection in rabbits. World Rabbit. Sci. 2011, 19, 203–208. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Tamames, J.; Puente-Sánchez, F. SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 2019, 9, 3349. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Veech, J.A. Measuring biodiversity. In Encyclopedia of the Anthropocene; Elsevier Inc.: Amsterdam, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Martín-Fernández, J.A.; Hron, K.; Templ, M.; Filzmoser, P.; Palarea-Albaladejo, J. Bayesian-multiplicative treatment of count zeros in compositional data sets. Stat. Model. 2015, 15, 134–158. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef]
- Greenacre, M.; Martínez-Álvaro, M.; Blasco, A. Compositional data analysis of microbiome and any-omics datasets: A validation of the additive logratio transformation. Front. Microbiol. 2021, 12, 727398. [Google Scholar] [CrossRef]
- Blasco, A. Bayesian Data Analysis for Animal Scientists: The Basics, 1st ed.; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Saborío-Montero, A.; Gutiérrez-Rivas, M.; López-García, A.; García-Rodríguez, A.; Atxaerandio, R.; Goiri, I.; Jiménez-Montero, J.A.; González-Recio, O. Holobiont effect accounts for more methane emission variance than the additive and microbiome effects on dairy cattle. Livest. Sci. 2021, 250, 104538. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. BGLR : A Statistical Package for Whole Genome Regression and Prediction. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Plummer, M.; Best, N.; Cowles, K.; Vines, K. CODA: Convergence diagnosis and output analysis for MCMC. R News 2006, 6, 7–11. [Google Scholar] [CrossRef]
- Kucheryavskiy, S. Mdatools—R package for chemometrics. Chemom. Intell. Lab. Syst. 2020, 198, 103937. [Google Scholar] [CrossRef]
- Rivera-Pinto, J.; Egozcue, J.J.; Pawlowsky-Glahn, V.; Paredes, R.; Noguera-Julian, M.; Calle, M.L. Balances: A new perspective for microbiome analysis. mSystems 2018, 3, e00053-18. [Google Scholar] [CrossRef] [PubMed]
- Difford, G.F.; Lassen, J.; Løvendahl, P. Genes and microbes, the next step in dairy cattle breeding. In Proceedings of the 67th Annual Meeting European Federation of Animal Science, Belfast, UK, 29 August–2 September 2016. [Google Scholar]
- Casto-Rebollo, C.; Argente, M.J.; García, M.L.; Pena, R.N.; Blasco, A.; Ibáñez-Escriche, N. Selection for environmental variance shifted the gut microbiome composition driving animal resilience. Microbiome 2023, 11, 147. [Google Scholar] [CrossRef]
- Khanal, P.; Maltecca, C.; Schwab, C.; Fix, J.; Tiezzi, F. Microbiability of meat quality and carcass composition traits in swine. J. Anim. Breed. Genet. 2020, 138, 223–236. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, P.; Zhou, X.; Fu, M.; Wang, T.; Liu, Z.; Liu, X.; Wang, Z.; Liu, B. Effect of host genetics and gut microbiome on fat deposition traits in pigs. Front. Microbiol. 2022, 13, 925200. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tiezzi, F.; Jiang, J.; Howard, J.; Huang, Y.; Gray, K.; Choi, J.W.; Maltecca, C. Exploring methods to summarize gut microbiota composition for microbiability estimation and phenotypic prediction in swine. J. Anim. Sci. 2022, 100, skac231. [Google Scholar] [CrossRef]
- Cauquil, L.; Beaumont, M.; Liaubet, L.; Lippi, Y.; Gress, L. Coprophagy in rabbit upregulates immune system. In Proceedings of the 12th World Rabbit Congress, Nantes, France, 3–5 November 2021. [Google Scholar]
- Zeng, S.; Wang, S.; Ross, R.P.; Stanton, C. The road not taken: Host genetics in shaping intergenerational microbiomes. Trends Genet. 2022, 38, 1180–1192. [Google Scholar] [CrossRef]
- Velasco-Galilea, M.; Piles, M.; Ramayo-Caldas, Y.; Varona, L.; Sánchez, J.P. Use of Bayes factors to evaluate the effects of host genetics, litter and cage on the rabbit cecal microbiota. Genet. Sel. Evol. 2022, 54, 46. [Google Scholar] [CrossRef] [PubMed]
- Combes, S.; Michelland, R.J.; Monteils, V.; Cauquil, L.; Soulié, V.; Tran, N.U.; Gidenne, T.; Fortun-Lamothe, L. Postnatal development of the rabbit caecal microbiota composition and activity. FEMS Microbiol. Ecol. 2011, 77, 680–689. [Google Scholar] [CrossRef]
- Bian, G.; Ma, S.; Zhu, Z.; Su, Y.; Zoetendal, E.G.; Mackie, R.; Liu, J.; Mu, C.; Huang, R.; Smidt, H. Age, introduction of solid feed and weaning are more important determinants of gut bacterial succession in piglets than breed and nursing mother as revealed by a reciprocal cross-fostering model. Environ. Microbiol. 2016, 18, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Squillario, M.; Bonaretti, C.; Valle ALa Marco EDi Piccolo, G.; Minuto, N.; Patti, G.; Napoli, F.; Bassi, M.; Maghnie, M. Gut-microbiota in obese children and adolescents: Inferred functional analysis and machine-learning algorithms to classify microorganisms. Sci. Rep. 2023, 12, 11294. [Google Scholar] [CrossRef] [PubMed]
- Woting, A.; Pfeiffer, N.; Loh, G.; Klaus, S.; Blaut, M. Clostridium ramosum promotes High-Fat diet-induced obesity in Gnotobiotic Mouse Models. MBio 2014, 5, e01530-14. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Hatasa, M.; Ohsugi, Y.; Tsuchiya, Y.; Liu, A.; Niimi, H.; Morita, K.; Shimohira, T.; Sasaki, N.; Maekawa, S. Porphyromonas gingivalis administration induces gestational obesity, alters gene expression in the liver and brown adipose tissue in pregnant mice, and causes underweight in fetuses. Front. Cell Infect. Microbiol. 2022, 11, 745117. [Google Scholar] [CrossRef] [PubMed]
- Hatasa, M.; Ohsugi, Y.; Katagiri, S.; Yoshida, S.; Niimi, H.; Morita, K.; Tsuchiya, Y.; Shimohira, T.; Sasaki, N.; Maekawa, S. Endotoxemia by Porphyromonas gingivalis alters endocrine functions in brown adipose tissue. Front. Cell Infect. Microbiol. 2021, 10, 580577. [Google Scholar] [CrossRef] [PubMed]
- Just, S.; Mondot, S.; Ecker, J.; Wegner, K.; Rath, E.; Gau, L.; Streidl, T.; Hery-Arnaud, G.; Schmidt, S.; Lesker, T.R. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 2018, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2020, 19, 77–94. [Google Scholar] [CrossRef]
- Yu, Y.; Raka, F.; Adeli, K. The Role of the Gut Microbiota in Lipid and Lipoprotein Metabolism. J. Clin. Med. 2019, 8, 2227. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Prawitt, J.; Abdelkarim, M.; Stroeve, J.H.M.; Popescu, I.; Duez, H.; Velagapudi, V.R.; Dumont, J.; Bouchaert, E.; Van Dijk, T.H.; Lucas, A. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes 2011, 60, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Nishina, P.M.; Freedland, R.A. Effects of propionate on lipid biosynthesis in isolated rat hepatocytes. J. Nutr. 1990, 120, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Madrid, S.; Martínez-Álvaro, M.; Paucar, Y.; Hernández, P.; Blasco, A. Efecto de la selección divergente por grasa intramuscular en caracteres de eficiencia alimentaria. In XVII Jornadas sobre Producción Animal; AIDA, ITEA: Zaragoza, Spain, 2017; pp. 480–482. [Google Scholar]
- Serena, C.; Ceperuelo-Mallafré, V.; Keiran, N.; Queipo-Ortuño, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Pérez-Brocal, V.; Andrés-Lacueva, C. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-Mcdermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Diaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yeisoo, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittman, B.E. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Venema, K. Role of gut microbiota in the control of energy and carbohydrate metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 432–438. [Google Scholar] [CrossRef]
- Mi, L.; Yang, B.; Hu, X.; Luo, Y.; Liu, J.; Yu, Z.; Wang, J. Comparative analysis of the microbiota between sheep rumen and rabbit cecum provides new insight into their differential methane production. Front. Microbiol. 2018, 9, 575. [Google Scholar] [CrossRef]
- Smith, N.W.; Shorten, P.R.; Altermann, E.H.; Roy, N.C.; McNabb, W.C. Hydrogen cross-feeders of the human gastrointestinal tract. Gut Microbes 2019, 10, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.H.; Su, Y.; Wright, A.D.G.; Zhang, L.L.; Smidt, H.; Zhu, W.Y. Lean breed landrace pigs harbor fecal Methanogens at higher diversity and density than obese breed Erhualian pigs. Archaea 2012, 2012, 605289. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.H. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Maraninchi, M.; Henry, M.; Armougom, F.; Richet, H.; Carrieri, P.; Valero, R.; Raccah, D.; Vialettes, B.; Raoult, D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. 2012, 36, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, S.H.; Ren, F.; Li, J.T.; Chen, J.J.; Yao, C.B.; Yang, H.B.; Jiang, S.X.; Yan, G.Q.; Wang, D. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Marty, J.F.; Vernay, M.Y.; Abravanel, G.M. Acetate absorption and metabolism in the rabbit hindgut. Gut 1985, 26, 562–569. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Álvaro, M.; Zubiri-Gaitán, A.; Hernández, P.; Casto-Rebollo, C.; Ibáñez-Escriche, N.; Santacreu, M.A.; Artacho, A.; Pérez-Brocal, V.; Blasco, A. Correlated Responses to Selection for Intramuscular Fat on the Gut Microbiome in Rabbits. Animals 2024, 14, 2078. https://doi.org/10.3390/ani14142078
Martínez-Álvaro M, Zubiri-Gaitán A, Hernández P, Casto-Rebollo C, Ibáñez-Escriche N, Santacreu MA, Artacho A, Pérez-Brocal V, Blasco A. Correlated Responses to Selection for Intramuscular Fat on the Gut Microbiome in Rabbits. Animals. 2024; 14(14):2078. https://doi.org/10.3390/ani14142078
Chicago/Turabian StyleMartínez-Álvaro, Marina, Agostina Zubiri-Gaitán, Pilar Hernández, Cristina Casto-Rebollo, Noelia Ibáñez-Escriche, Maria Antonia Santacreu, Alejandro Artacho, Vicente Pérez-Brocal, and Agustín Blasco. 2024. "Correlated Responses to Selection for Intramuscular Fat on the Gut Microbiome in Rabbits" Animals 14, no. 14: 2078. https://doi.org/10.3390/ani14142078
APA StyleMartínez-Álvaro, M., Zubiri-Gaitán, A., Hernández, P., Casto-Rebollo, C., Ibáñez-Escriche, N., Santacreu, M. A., Artacho, A., Pérez-Brocal, V., & Blasco, A. (2024). Correlated Responses to Selection for Intramuscular Fat on the Gut Microbiome in Rabbits. Animals, 14(14), 2078. https://doi.org/10.3390/ani14142078