
Preliminary Analysis of Placental DNA Methylation Profiles in Piglets with Extreme Birth Weight Variations
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. H&E Staining and Immunohistochemistry
2.3. Real-Time Quantitative PCR
2.4. RNA-Seq and Data Analysis
2.5. Genome Bisulfite Sequencing and Data Analysis
2.6. Cell Culture
2.7. Methylation-Specific PCR
- (1)
- Genomic DNA from weak and normal placentas was extracted and bisulfite-treated using the EZ DNA Methylation-Direct Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions.
- (2)
- Specific MS-PCR primers were designed using MethPrimer [24] and are listed in Supplementary Table S6.
- (3)
- PCR products were analyzed by 3% agarose gel electrophoresis (150 V) with 6× loading buffer (1:5 ratio). Samples were loaded with a DNA marker and visualized after electrophoresis.
- (4)
- Methylation status was determined as follows: Unmethylated: Only unmethylated bands visible; Fully methylated: Only methylated bands visible; Partially methylated: Both methylated and unmethylated bands present.
- (5)
- Methylation level was quantified using grayscale analysis: Methylation level (%) = [Methylated band intensity/(Methylated + Unmethylated band intensities)] × 100.
2.8. Statistical Analysis
3. Results
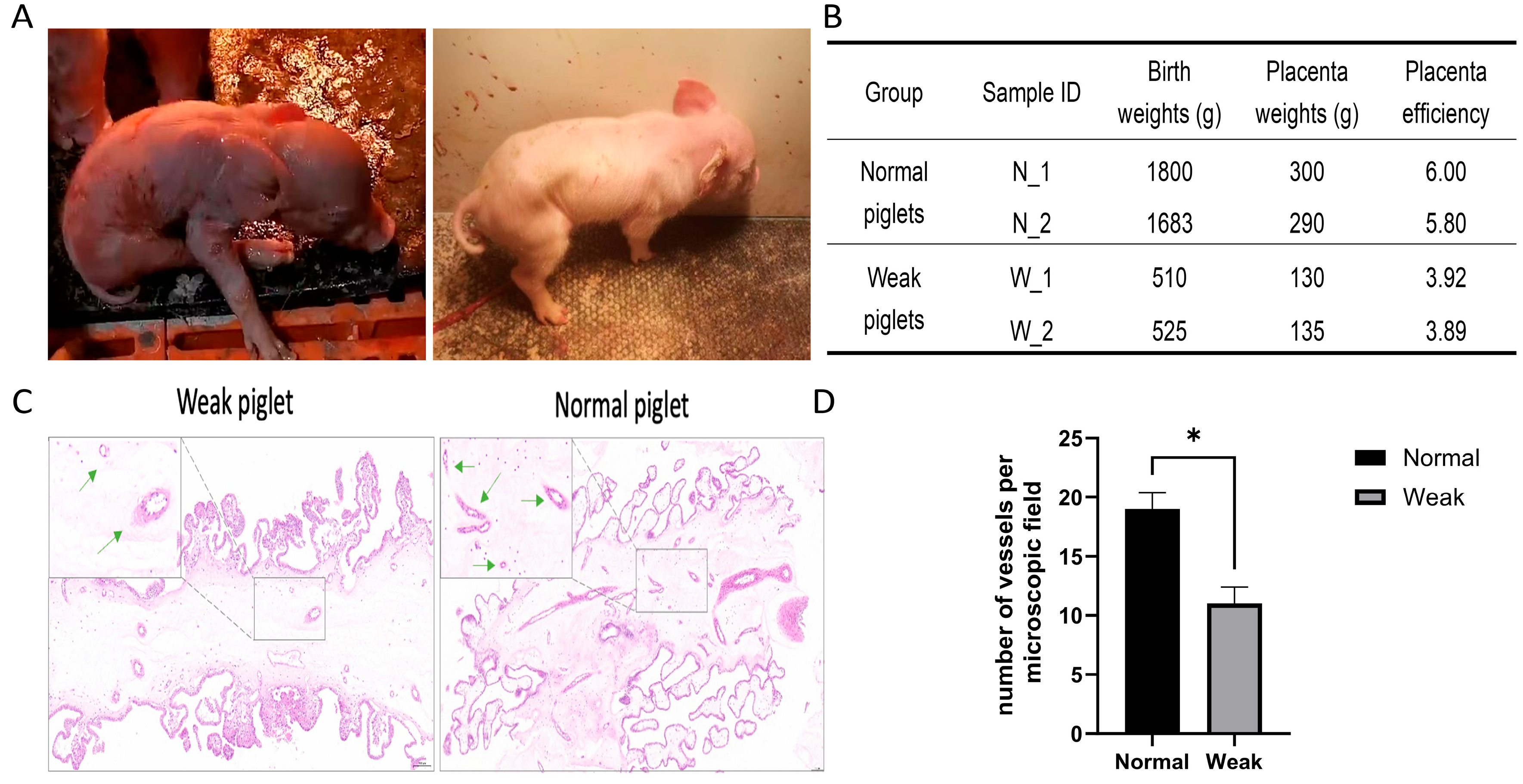
3.1. Morphology and Gene Expression Changes Between Placentas from Weak and Normal Piglets
3.2. Characteristics of DNA Methylome Between Placentas from Weak and Normal Piglets
3.3. Differences in DNA Methylation Between Placentas from Weak and Normal Piglets
3.4. Regulation of DNA Methylation in Gene Transcription in Placentas from Weak and Normal Piglets
3.5. Validation of DNA Methylation-Regulated Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vilanova, C.S.; Hirakata, V.N.; de Souza Buriol, V.C.; Nunes, M.; Goldani, M.Z.; da Silva, C.H. The relationship between the different low birth weight strata of newborns with infant mortality and the influence of the main health determinants in the extreme south of Brazil. Popul. Health Metr. 2019, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Dean, W. DNA methylation and mammalian epigenetics. Electrophoresis 2001, 22, 2838–2843. [Google Scholar] [CrossRef] [PubMed]
- Koukoura, O.; Sifakis, S.; Spandidos, D.A. DNA methylation in the human placenta and fetal growth (review). Mol. Med. Rep. 2012, 5, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Apicella, C.; Ruano, C.S.M.; Méhats, C.; Miralles, F.; Vaiman, D. The Role of Epigenetics in Placental Development and the Etiology of Preeclampsia. Int. J. Mol. Sci. 2019, 20, 2837. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Ren, R.; Cao, J.; Zhao, S.; Yu, M. Genome-Wide Identification of Histone Modifications Involved in Placental Development in Pigs. Front. Genet. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Sferruzzi-Perri, A.N.; Coan, P.M.; Constancia, M.; Burton, G.J. Placental efficiency and adaptation: Endocrine regulation. J. Physiol. 2009, 587 Pt 14, 3459–3472. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.R.; King, M.; Perez-Garcia, V.; Bogutz, A.B.; Caley, M.; Fineberg, E.; Lefebvre, L.; Cook, S.J.; Dean, W.; Hemberger, M.; et al. Maternal DNA Methylation Regulates Early Trophoblast Development. Dev. Cell 2016, 36, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, Y.N.; Im, D.; Cho, S.H.; Kim, J.; Kim, J.H.; Kim, K. DNA Methylation and gene expression patterns are widely altered in fetal growth restriction and associated with FGR development. Anim. Cells Syst. 2021, 25, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Fantone, S.; Giannubilo, S.R.; Marzioni, D.; Tossetta, G. HTRA family proteins in pregnancy outcome. Tissue Cell 2021, 10, 101549. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Xiao, L.; Wang, Y.; Zhou, C.; Huang, H.; Li, Z.; Hong, L.; Cai, G.; Wu, Z.; Gu, T. Comprehensive Analysis of Placental DNA Methylation Changes and Fetal Birth Weight in Pigs. Int. J. Mol. Sci. 2024, 25, 7702. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Zhou, C.; Zang, X.; Zhao, X.; Xiao, L.; Zeng, J.; Hong, L.; Wu, Z.; Gu, T. Integrated Analysis of DNA Methylation and Gene Expression in Porcine Placental Development. Int. J. Mol. Sci. 2023, 24, 5169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Analysis of the reasons for the formation and prevention of weak piglets in pig farms. Chin. Livest. Poult. Breed. 2021, 17, 129–130. [Google Scholar]
- Luo, N.; Cheng, W.; Zhou, Y.; Gu, B.; Zhao, Z.; Zhao, Y. Screening Candidate Genes Regulating Placental Development from Trophoblast Transcriptome at Early Pregnancy in Dazu Black Goats (Capra hircus). Animals 2021, 11, 2132. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gao, L.; Zhang, S. Comparative pan-cancer DNA methylation analysis reveals cancer common and specific patterns. Brief. Bioinform. 2017, 18, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, S.; Song, T.; Yin, Y.; Tan, C. Placental Angiogenesis in Mammals: A Review of the Regulatory Effects of Signaling Pathways and Functional Nutrients. Adv. Nutr. 2021, 12, 2415–2434. [Google Scholar] [CrossRef] [PubMed]
- Svensson-Arvelund, J.; Mehta, R.B.; Lindau, R.; Mirrasekhian, E.; Rodriguez-Martinez, H.; Berg, G.; Lash, G.E.; Jenmalm, M.C.; Ernerudh, J. The human fetal placenta promotes tolerance against the semiallogeneic fetus by inducing regulatory T cells and homeostatic M2 macrophages. J. Immunol. 2015, 194, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Tilburgs, T.; Roelen, D.L.; van der Mast, B.J.; Kanhai, H.H.; Scherjon, S.; Claas, F.H. Comparison of macrophage phenotype between decidua basalis and decidua parietalis by flow cytometry. Placenta 2008, 29, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Ruano, C.S.M.; Apicella, C.; Jacques, S.; Gascoin, G.; Gaspar, C.; Miralles, F.; Méhats, C.; Vaiman, D. Alternative splicing in normal and pathological human placentas is correlated to genetic variants. Hum. Genet. 2021, 140, 827–848. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Gaccioli, F.; Dopierala, J.; Sovio, U.; Cook, E.; Volders, P.-J.; Martens, L.; Kirk, P.D.W.; Richardson, S.; Smith, G.C.S.; et al. The RNA landscape of the human placenta in health and disease. Nat. Commun. 2021, 12, 2639. [Google Scholar] [CrossRef] [PubMed]
- Simner, C.; Novakovic, B.; Lillycrop, K.A.; Bell, C.G.; Harvey, N.C.; Cooper, C.; Saffery, R.; Lewis, R.M.; Cleal, J.K. DNA methylation of amino acid transporter genes in the human placenta. Placenta 2017, 60, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Cleal, J.K.; Glazier, J.D.; Ntani, G.; Crozier, S.R.; Day, P.E.; Harvey, N.C.; Robinson, S.M.; Cooper, C.; Godfrey, K.M.; Hanson, M.A.; et al. Facilitated transporters mediate net efflux of amino acids to the fetus across the basal membrane of the placental syncytiotrophoblast. J. Physiol. 2011, 589 Pt 4, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Loubière, L.S.; Vasilopoulou, E.; Bulmer, J.N.; Taylor, P.M.; Stieger, B.; Verrey, F.; McCabe, C.J.; Franklyn, J.A.; Kilby, M.D.; Chan, S.Y. Expression of thyroid hormone transporters in the human placenta and changes associated with intrauterine growth restriction. Placenta 2010, 31, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Powell, T.L.; Jansson, T. Regulation of placental amino acid transport in health and disease. Acta Physiol. 2024, 240, e14157. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Tan, B.; Su, J.; Xue, J.; Xiao, L.; Li, Z.; Hong, L.; Cai, G.; Gu, T. Preliminary Analysis of Placental DNA Methylation Profiles in Piglets with Extreme Birth Weight Variations. Animals 2025, 15, 2168. https://doi.org/10.3390/ani15152168
Zhang Z, Tan B, Su J, Xue J, Xiao L, Li Z, Hong L, Cai G, Gu T. Preliminary Analysis of Placental DNA Methylation Profiles in Piglets with Extreme Birth Weight Variations. Animals. 2025; 15(15):2168. https://doi.org/10.3390/ani15152168
Chicago/Turabian StyleZhang, Zhiyuan, Baohua Tan, Jiawei Su, Jiaming Xue, Liyao Xiao, Zicong Li, Linjun Hong, Gengyuan Cai, and Ting Gu. 2025. "Preliminary Analysis of Placental DNA Methylation Profiles in Piglets with Extreme Birth Weight Variations" Animals 15, no. 15: 2168. https://doi.org/10.3390/ani15152168
APA StyleZhang, Z., Tan, B., Su, J., Xue, J., Xiao, L., Li, Z., Hong, L., Cai, G., & Gu, T. (2025). Preliminary Analysis of Placental DNA Methylation Profiles in Piglets with Extreme Birth Weight Variations. Animals, 15(15), 2168. https://doi.org/10.3390/ani15152168