

Genetic and Haplotype Diversity of Manila Clam Ruditapes philippinarum in Different Regions of China Based on Three Molecular Markers


 and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Amplification and Sequencing
2.3. Data Analysis
3. Results
3.1. Gene Sequence Base Composition of the R. philippinarum Population
3.2. Genetic Diversity Analysis of R. philippinarum Population
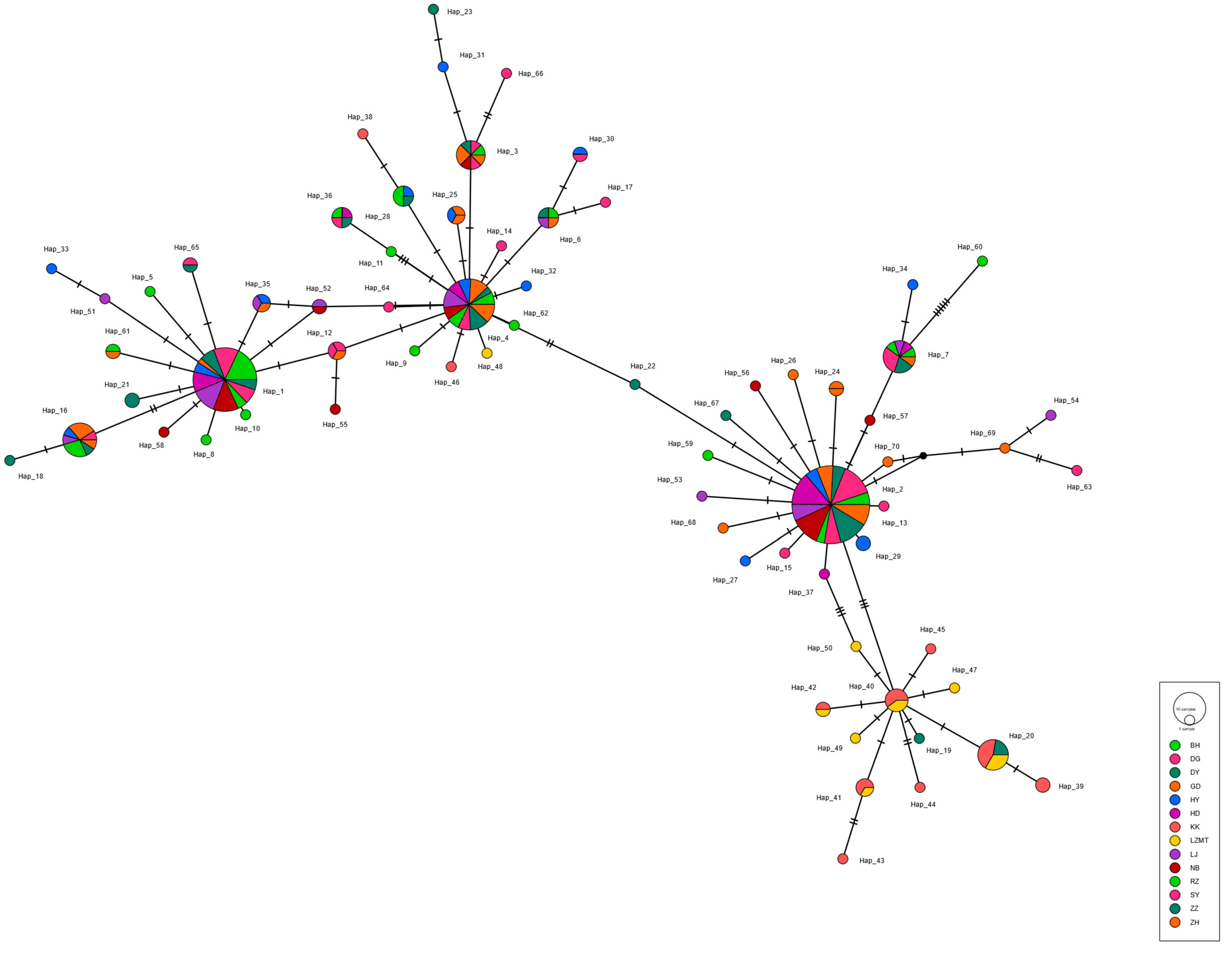
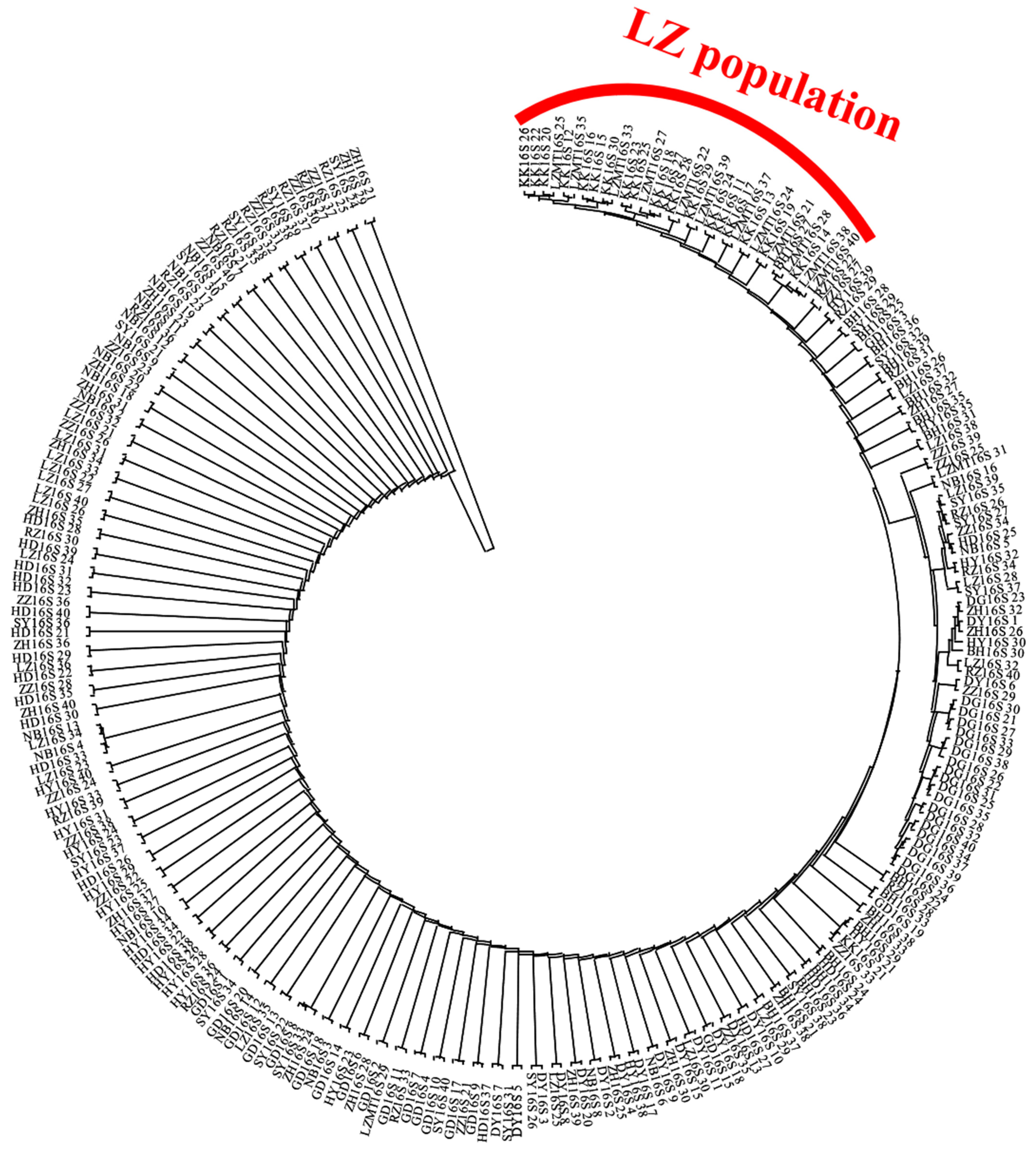
3.3. Genetic Structure of the R. philippinarum Population
4. Discussion
4.1. Haplotype and Genetic Diversity of R. philippinarum
4.2. Genetic Differentiation among Populations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A



References
- Sekine, Y.; Yamakawa, H.; Takazawa, S.; Lin, Y.P.; Toba, M. Geographic variation of the COX1 gene of the short-neck clam Ruditapes philippinarum in coastal regions of Japan and China. Venus 2006, 65, 229–240. [Google Scholar]
- Avolio, M.L.; Beaulieu, J.M.; Lo, E.Y.; Smith, M.D. Measuring genetic diversity in ecological studies. Plant Ecol. 2012, 213, 1105–1115. [Google Scholar]
- Ellegren, H.; Galtier, N. Determinants of genetic diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [PubMed]
- Montero-Pau, J.; Gómez, A.; Serra, M. Founder effects drive the genetic structure of passively dispersed aquatic invertebrates. PeerJ 2018, 6, e6094. [Google Scholar] [PubMed]
- Launey, S.; Hedgecock, D. High genetic load in the pacific oyster Crassostrea gigas. Genetics 2001, 159, 255–265. [Google Scholar] [CrossRef]
- Li, Q. Development of microsatellite DNA markers for marine shellfish and their application in genetic studies. Chin. Fish. Sci. 2006, 13, 502–509. [Google Scholar]
- Lallias, D.; Boudry, P.; Lapègue, S.; King, J.W.; Beaumont, A.R. Strategies for the retention of high genetic variability in European flat oyster (Ostrea edulis) restoration programmes. Conserv. Genet. 2010, 11, 1899–1910. [Google Scholar]
- Ma, L.; Ji, Y.J.; Zhang, D.X. Statistical measures of genetic differentiation of populations: Rationales, history and current states. Curr. Zool. 2016, 61, 886–897. [Google Scholar] [CrossRef]
- Sun, S.E.; Li, Q.I.; Kong, L.; Yu, H.; Zheng, X.; Yu, R.; Dai, L.; Sun, Y.; Chen, J.; Liu, J. DNA barcoding reveal patterns of species diversity among northwestern Pacific molluscs. Sci. Rep. 2016, 6, 33367. [Google Scholar] [CrossRef][Green Version]
- Cordero, D.; Delgado, M.; Liu, B.; Ruesink, J.; Saavedra, C. Population genetics of the Manila clam (Ruditapes philippinarum) introduced in North America and Europe. Sci. Rep. 2017, 7, 39745. [Google Scholar]
- Duran, S.; Palacín, C.; Becerro, M.A.; Turon, X.; Giribet, G. Genetic diversity and population structure of the commercially harvested sea urchin Paracentrotus lividus (Echinodermata, Echinoidea). Mol. Ecol. 2004, 13, 3317–3328. [Google Scholar]
- Pertoldi, C.; Bijlsma, R.; Loeschcke, V. Conservation genetics in a globally changing environment: Present problems, paradoxes and future challenges. Biodivers. Conserv. 2007, 16, 4147–4163. [Google Scholar]
- Guo, C.; Xu, S. Advances in the study of shellfish of the family Clam. J. Biol. 2016, 33, 7. [Google Scholar]
- Thorpe, J.P.; Solé-Cava, A.M.; Watts, P.C. Exploited marine invertebrates: Genetics and fisheries. Mar. Genet. 2000, 144, 165–184. [Google Scholar]
- Wong, Y.T.; Meier, R.; Tan, K.S. High haplotype variability in established Asian populations of the invasive Caribbean bivalve Mytilopsis sallei (Dreissenidae). Biol. Invasions 2011, 13, 341–348. [Google Scholar] [CrossRef]
- Yuan, T.; He, M.; Huang, L. Intraspecific genetic variation in mitochondrial 16S rRNA and COI genes in domestic and wild populations of Huaguizhikong scallop Chlamys nobilis Reeve. Aquaculture 2009, 289, 19–25. [Google Scholar] [CrossRef]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [PubMed]
- Kim, W.J.; Dammannagoda, S.T.; Jung, H.; Baek, I.S.; Yoon, H.S.; Choi, S.D. Mitochondrial DNA sequence analysis from multiple gene fragments reveals genetic heterogeneity of Crassostrea ariakensis in East Asia. Genes Genom. 2014, 36, 611–624. [Google Scholar] [CrossRef]
- Ni, G.; Li, Q.; Kong, L.; Yu, H. Mitochondrial phylogeography of a surf clam Mactra veneriformis in the East China Sea: Genetic homogeneity across two biogeographic boundaries. Biochem. Syst. Ecol. 2015, 61, 493–500. [Google Scholar]
- Gosling, E. Bivalve Molluscs: Biology, Ecology and Culture. In Fishing News Books; Blackwell Publishing: Oxford, UK, 2003. [Google Scholar]
- Chiesa, S.; Lucentini, L.; Freitas, R.; Marzano, F.N.; Argese, E. Genetic diversity of introduced manila clam Ruditapes philippinarum populations inferred by 16SrDNA. Biochem. Syst. Ecol. 2014, 57, 52–59. [Google Scholar] [CrossRef]
- DOF. China Fisheries Statistic Yearbook; China Agriculture Press: Beijing, China, 2021. [Google Scholar]
- An, H.S.; Park, K.J.; Cho, K.C.; Han, H.S.; Myeong, J.I. Genetic structure of Korean populations of the clam Ruditapes philippinarum inferred from microsatellite marker analysis. Biochem. Syst. Ecol. 2012, 44, 186–195. [Google Scholar] [CrossRef]
- Tan, Y.; Fang, L.; Qiu, M.; Huo, Z.; Yan, X. Population genetics of the Manila clam (Ruditapes philippinarum) in East Asia. Sci. Rep. 2020, 10, 21890. [Google Scholar] [CrossRef]
- Zheng, S.; Zhang, T.; Tu, K.; Li, L.; Liu, Z.; Wu, B.; Zhou, L.; Sun, X. Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers. Biology 2023, 12, 557. [Google Scholar] [CrossRef]
- Chen, J.; Li, Q.; Kong, L.; Zheng, X.; Yu, R. Analysis of DNA barcoding based on COI sequences in the shellfish of the subfamily Phyllostomidae from coastal China. Zool. Res. 2010, 4, 345–352. [Google Scholar] [CrossRef]
- Mao, Y.; Gao, T.; Yanagimoto, T.; Xiao, Y. Molecular phylogeography of Ruditapes philippinarum in the Northwestern Pacific Ocean based on COI gene. J. Exp. Mar. Biol. Ecol. 2011, 407, 171–181. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, Z.; Ma, P.; Wang, H. Genetic diversity study of 10 wild populations of R. philippinarum. Ocean. Lakes 2016, 47, 549–556. [Google Scholar]
- Tan, Y.; Qiu, M.; Li, J.; Zuo, L.; Wu, W.; Huo, Z.; Yan, X. Genetic diversity analysis of wild populations of Manila clam Ruditapes philippinarum from China, Japan and Democratic People’s Republic of Korea based on mitochondrial 16S rRNA gene sequence. J. Dalian Ocean. Univ. 2021, 36, 127–134. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial Cytochrome C oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Anderson, F.E. Phylogeny and historical biogeography of the loliginid squids (Mollusca: Cephalopoda) based on mitochondrial DNA sequence data. Mol. Phylogenetics Evol. 2000, 15, 191–214. [Google Scholar] [CrossRef]
- Heath, D.D.; Rawson, P.D.; Hilbish, T.J. Pcr-based nuclear markers identify alien blue mussel (mytilus spp.) genotypes on the west coast of Canada. Can. J. Fish. Aquat. Sci. 1995, 52, 2621–2627. [Google Scholar] [CrossRef]
- Swindell, S.R.; Plasterer, T.N. SEQMAN: Contig assembly. Methods Mol. Biol. 1997, 70, 75. [Google Scholar] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11 Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6 molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the genetics of populations. a treatise in four volumes. volume 4. variability within and among natural populations. J. Biosoc. Sci. 1972, 4, 253–256. [Google Scholar]
- Nie, H.; Niu, H.; Zhao, L.; Yang, F.; Yan, X.; Zhang, G. Genetic diversity and structure of Manila clam (Ruditapes philippinarum) populations from Liaodong peninsula revealed by SSR markers. Biochem. Syst. Ecol. 2015, 59, 116–125. [Google Scholar] [CrossRef]
- Luttikhuizen, P.C.; Drent, J.; Baker, A.J. Disjunct distribution of highly diverged mitochondrial lineage clade and population subdivision in a marine bivalve with pelagic larval dispersal. Mol. Ecol. 2003, 12, 2215–2229. [Google Scholar] [CrossRef]
- Asif, J.H.; Krug, P.J. Lineage distribution and barriers to gene flow among populations of the globally invasive marine mussel musculista senhousia. Biol. Invasions 2012, 14, 1431–1444. [Google Scholar] [CrossRef]
- Hedgecock, D.; Li, G.; Hubert, S.; Bucklin, K.; Ribes, V. Widespread null alleles and poor cross-species amplification of microsatellite dna loci cloned from the pacific oyster, Crassostrea gigas. J. Shellfish. Res. 2004, 23, 379–385. [Google Scholar]
- Grant, W.S.; Clark, A.M.; Bowen, B.W. Why restriction fragment length polymorphism analysis of mitochondrial DNA failed to resolve sardine (Sardinops) biogeography: Insights from mitochondrial DNA cytochrome b sequences. Can. J. Fish. Aquat. Sci. 1998, 55, 2539–2547. [Google Scholar] [CrossRef]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Vacquier, C.V.D. Exploring the phylogenetic utility of ITS sequences for animals: A test case for abalone (Haliotis). J. Mol. Evol. 2002, 54, 246–257. [Google Scholar]
- Nie, H.; Li, J.; Huo, Z.; Guo, W.; Yan, X. Analysis of genetic variability in selected lines and a wild population of Ruditapes philippinarum using microsatellite markers. J. Fish. Sci. China 2016, 3, 538–546. [Google Scholar]
- Yu, Z.; Yan, X.; Yang, F.; Wang, J.; Zhang, Y.; Yang, F.; Zhang, G. Genetic diversity of different generations of the Dalian population of Manila clam Ruditapes philippinarum through selective breeding. Acta Ecol. Sin. 2011, 31, 4199–4206. [Google Scholar]
- Evans, F.; Matson, S.; Brake, J.; Langdon, C. The effects of inbreeding on performance traITS of adult Pacific oysters (Crassostrea gigas). Aquaculture 2003, 230, 89–98. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Name | Sample Size | Number of Sequences | Number of Haplotypes | Haplotype Diversity Index | Nucleotide Diversity Index | Average Nucleotide Differences |
|---|---|---|---|---|---|---|---|
| BH | Beihai | 20 | 20 | 11 | 0.86842 | 0.00514 | 2.86316 |
| DG | Donggang | 20 | 20 | 9 | 0.80000 | 0.00592 | 3.30000 |
| DY | Dongying | 20 | 17 | 11 | 0.94118 | 0.00840 | 4.67647 |
| GD | Guangdong | 20 | 16 | 8 | 0.89167 | 0.00639 | 3.55833 |
| HY | Haiyang | 20 | 19 | 14 | 0.96491 | 0.00707 | 3.93567 |
| HD | Hongdao | 20 | 17 | 6 | 0.74265 | 0.00520 | 2.89706 |
| KK | Laizhou shell-wide | 20 | 17 | 10 | 0.91912 | 0.00610 | 3.39706 |
| LZMT | Laizhou dock | 20 | 11 | 8 | 0.92727 | 0.00470 | 2.61818 |
| LJ | Lianjiang | 20 | 20 | 11 | 0.90000 | 0.00610 | 3.40000 |
| NB | Ningbo | 20 | 20 | 9 | 0.83158 | 0.00542 | 3.02105 |
| RZ | Rizhao | 20 | 17 | 11 | 0.94853 | 0.00850 | 4.75529 |
| SY | Sanya | 20 | 20 | 12 | 0.93158 | 0.00719 | 4.00526 |
| ZZ | Zhangzhou | 20 | 19 | 9 | 0.84795 | 0.00596 | 3.32164 |
| ZH | Zhuanghe | 20 | 20 | 14 | 0.93158 | 0.00609 | 3.39474 |
| Population | Number of Sequences | Number of Haplotypes | Haplotype Diversity Index | Nucleotide Diversity Index | Average Nucleotide Differences |
|---|---|---|---|---|---|
| BH | 19 | 3 | 0.20468 | 0.0049 | 0.21053 |
| DG | 20 | 2 | 0.10000 | 0.0047 | 0.20000 |
| DY | 16 | 3 | 0.24167 | 0.0058 | 0.25000 |
| GD | 18 | 1 | 0.00000 | 0.0000 | 0.0000 |
| HY | 20 | 3 | 0.19474 | 0.0070 | 0.30000 |
| HD | 20 | 2 | 0.10000 | 0.00023 | 10.000 |
| KK | 20 | 5 | 0.36842 | 0.00158 | 0.67895 |
| LZMT | 14 | 5 | 0.59341 | 0.00251 | 1.07692 |
| LJ | 20 | 4 | 0.28421 | 0.0070 | 0.30000 |
| NB | 19 | 3 | 0.20468 | 0.0049 | 0.21053 |
| RZ | 20 | 4 | 0.36316 | 0.00091 | 0.38947 |
| SY | 19 | 3 | 0.29240 | 0.0016 | 1.35673 |
| ZZ | 20 | 6 | 0.44737 | 0.00117 | 0.50000 |
| ZH | 20 | 2 | 0.18947 | 0.00044 | 0.18947 |
| Population | Number of Sequences | Number of Haplotypes | Haplotype Diversity Index | Nucleotide Diversity Index | Average Nucleotide Differences |
|---|---|---|---|---|---|
| BH | 18 | 11 | 0.89542 | 0.00994 | 2.80392 |
| DG | 19 | 9 | 0.86550 | 0.01974 | 5.56725 |
| DY | 15 | 11 | 0.95238 | 0.02948 | 8.31429 |
| GD | 16 | 15 | 0.99167 | 0.03183 | 8.97500 |
| HY | 15 | 12 | 0.96190 | 0.07693 | 21.69524 |
| HD | 18 | 12 | 0.93464 | 0.01808 | 5.09804 |
| KK | 20 | 13 | 0.91053 | 0.02714 | 7.65263 |
| LZMT | 13 | 8 | 0.88462 | 0.02741 | 7.73077 |
| LJ | 18 | 12 | 0.93464 | 0.04334 | 12.22222 |
| NB | 18 | 13 | 0.94771 | 0.02603 | 7.33987 |
| RZ | 18 | 13 | 0.95425 | 0.01539 | 4.33987 |
| SY | 19 | 14 | 0.96491 | 0.12986 | 36.61988 |
| ZZ | 16 | 10 | 0.93333 | 0.02813 | 7.93333 |
| ZH | 19 | 12 | 0.92398 | 0.03142 | 8.85965 |
| BH | DG | DY | GD | HY | HD | KK | LZMT | LJ | NB | RZ | SY | ZZ | ZH | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BH | 0.00591 | 0.00704 | 0.00601 | 0.00622 | 0.00601 | 0.01331 | 0.01289 | 0.00556 | 0.00548 | 0.00684 | 0.00645 | 0.00622 | 0.00630 | |
| DG | 0.06335 | 0.00722 | 0.00604 | 0.00635 | 0.00540 | 0.01143 | 0.01093 | 0.00593 | 0.00546 | 0.00728 | 0.00645 | 0.00577 | 0.00587 | |
| DY | 0.04202 | 0.00973 | 0.00745 | 0.00771 | 0.00705 | 0.01121 | 0.01070 | 0.00724 | 0.00695 | 0.00853 | 0.00786 | 0.00736 | 0.00749 | |
| GD | 0.04338 | −0.01941 | 0.00749 | 0.00650 | 0.00589 | 0.01220 | 0.01174 | 0.00615 | 0.00581 | 0.00727 | 0.00667 | 0.00611 | 0.00613 | |
| HY | 0.01890 | −0.02271 | −0.00152 | −0.03498 | 0.00617 | 0.01238 | 0.01192 | 0.00640 | 0.00608 | 0.00763 | 0.00692 | 0.00642 | 0.00646 | |
| HD | 0.13961 * | −0.03027 | 0.03579 | 0.01730 | 0.00487 | 0.01038 | 0.00983 | 0.00583 | 0.00523 | 0.00719 | 0.00614 | 0.00533 | 0.00549 | |
| KK | 0.57961 ** | 0.47448 ** | 0.35331 ** | 0.48834 ** | 0.46703 ** | 0.45572 ** | 0.00517 | 0.01234 | 0.01146 | 0.01377 | 0.01222 | 0.01097 | 0.01123 | |
| LZMT | 0.61486 ** | 0.50388 ** | 0.36994 ** | 0.51929 ** | 0.49066 ** | 0.49308 ** | −0.04672 | 0.01189 | 0.01098 | 0.01336 | 0.01176 | 0.01047 | 0.01076 | |
| LJ | −0.01182 | −0.01438 | 0.00072 | −0.01556 | −0.02847 | 0.03034 | 0.50555 ** | 0.53468 ** | 0.00561 | 0.00715 | 0.00658 | 0.00611 | 0.00619 | |
| NB | 0.03695 | −0.03965 | 0.00771 | −0.01519 | −0.02675 | −0.01641 | 0.49859 ** | 0.53304 ** | −0.02655 | 0.00699 | 0.00618 | 0.00558 | 0.00569 | |
| RZ | 0.00562 | 0.01118 | 0.00947 | −0.02491 | −0.02017 | 0.04760 | 0.46995 ** | 0.48764 ** | −0.01939 | 0.00620 | 0.00780 | 0.00733 | 0.00746 | |
| SY | 0.04340 | −0.01745 | 0.00955 | −0.01932 | −0.03060 | −0.01028 | 0.45446 ** | 0.47639 ** | −0.01096 | −0.01978 | −0.00529 | 0.00641 | 0.00648 | |
| ZZ | 0.10741 * | −0.03039 | 0.02568 | −0.01007 | −0.01467 | −0.04753 | 0.45050 ** | 0.48127 ** | 0.01225 | −0.01944 | 0.01474 | −0.02607 | 0.00581 | |
| ZH | 0.10856 * | −0.02444 | 0.03416 | −0.01850 | −0.01869 | −0.03001 | 0.45718 ** | 0.48734 ** | 0.01454 | −0.01275 | 0.02362 | −0.02493 | −0.03835 |
| BH | DG | DY | GD | HY | HD | KK | LZMT | LJ | NB | RZ | SY | ZZ | ZH | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BH | 0.002687 | 0.000656 | 0.000656 | 0.000714 | 0.000598 | 0.004891 | 0.004989 | 0.000830 | 0.000611 | 0.000830 | 0.002079 | 0.000946 | 0.000592 | |
| DG | 0.81439 ** | 0.002596 | 0.002449 | 0.002657 | 0.002552 | 0.006845 | 0.006961 | 0.002784 | 0.002564 | 0.002784 | 0.004033 | 0.002900 | 0.002529 | |
| DY | 0.00088 | 0.80105 ** | 0.000419 | 0.000624 | 0.000522 | 0.004814 | 0.004930 | 0.000754 | 0.000534 | 0.000754 | 0.002003 | 0.000870 | 0.000493 | |
| GD | −0.00293 | 0.89497 ** | 0.00758 | 0.000470 | 0.000342 | 0.004653 | 0.004769 | 0.000580 | 0.000373 | 0.000593 | 0.001821 | 0.000709 | 0.000361 | |
| HY | −0.00047 | 0.78166 ** | −0.02422 | −0.00544 | 0.000563 | 0.004873 | 0.004989 | 0.000795 | 0.000580 | 0.000789 | 0.002030 | 0.000917 | 0.000557 | |
| HD | 0.00097 | 0.85714 ** | 0.00577 | −0.00544 | −0.02564 | 0.004757 | 0.004873 | 0.000667 | 0.000464 | 0.000673 | 0.001896 | 0.000801 | 0.000464 | |
| KK | 0.78114 ** | 0.85103 ** | 0.76653 ** | 0.81782 ** | 0.76692 ** | 0.80526 ** | 0.001972 | 0.004988 | 0.004769 | 0.004884 | 0.006238 | 0.005000 | 0.004757 | |
| LZMT | 0.71819 ** | 0.80999 ** | 0.69803 ** | 0.75908 ** | 0.70484 ** | 0.74768 ** | −0.02881 | 0.005104 | 0.004885 | 0.005005 | 0.006354 | 0.005121 | 0.004873 | |
| LJ | −0.00047 | 0.78261 ** | −0.00116 | −0.00544 | −0.01695 | −0.02564 | 0.76692 ** | 0.70484 ** | 0.000696 | 0.000905 | 0.002128 | 0.001032 | 0.000696 | |
| NB | 0.00000 | 0.81439 ** | 0.00088 | −0.00293 | −0.02144 | −0.03440 | 0.78114 ** | 0.71922 ** | −0.02144 | 0.000684 | 0.001931 | 0.000812 | 0.000476 | |
| RZ | 0.01633 | 0.75439 ** | 0.01314 | 0.01980 | −0.01393 | −0.01974 | 0.74622 ** | 0.68159 ** | −0.01393 | −0.01854 | 0.002128 | 0.001009 | 0.000696 | |
| SY | 0.00741 | 0.54331** | 0.00026 | 0.00543 | −0.00401 | −0.00421 | 0.61588 ** | 0.54166 ** | −0.00728 | −0.00672 | −0.01014 | 0.002268 | 0.001945 | |
| ZZ | −0.00109 | 0.72000 ** | −0.00414 | −0.00544 | −0.01266 | −0.01695 | 0.72646 ** | 0.65992 ** | −0.01266 | −0.01599 | −0.02238 | −0.00385 | 0.000812 | |
| ZH | 0.02581 | 0.82134 ** | −0.03271 | 0.04509 | −0.01974 | 0.03509 | 0.78819 ** | 0.72861 ** | 0.02105 | 0.02581 | 0.03509 | 0.01648 | 0.01504 |
| BH | DG | DY | GD | HY | HD | LZ | LZMT | LJ | NB | RZ | SY | ZZ | ZH | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BH | 0.0177 | 0.0224 | 0.5036 | 0.0477 | 0.0188 | 0.0228 | 0.0232 | 0.0305 | 0.0210 | 0.0156 | 0.0810 | 0.0223 | 0.0247 | |
| DG | 0.00077 | 0.0258 | 0.5062 | 0.0511 | 0.0222 | 0.0266 | 0.0275 | 0.0342 | 0.0249 | 0.0192 | 0.0849 | 0.0265 | 0.0286 | |
| DY | 0.00389 | −0.00231 | 0.5078 | 0.0545 | 0.0265 | 0.0309 | 0.0315 | 0.0377 | 0.0289 | 0.0239 | 0.0892 | 0.0304 | 0.0328 | |
| GD | 0.96041 ** | 0.95073 ** | 0.94023 ** | 0.5134 | 0.5054 | 0.5082 | 0.5088 | 0.5097 | 0.5063 | 0.5068 | 0.5255 | 0.5064 | 0.5089 | |
| HY | 0.05375 ** | 0.03393 * | −0.00240 | 0.89671 ** | 0.0513 | 0.0550 | 0.0560 | 0.0620 | 0.0534 | 0.0487 | 0.1107 | 0.0556 | 0.0566 | |
| HD | 0.01454 | −0.01671 | −0.00551 | 0.95207 ** | 0.01638 | 0.0274 | 0.0279 | 0.0347 | 0.0256 | 0.0202 | 0.0856 | 0.0271 | 0.0293 | |
| LZ | 0.03103 * | 0.01026 | 0.00238 | 0.94325 ** | 0.01662 | 0.00992 | 0.0316 | 0.0388 | 0.0296 | 0.0245 | 0.0890 | 0.0315 | 0.0331 | |
| LZMT | 0.07220 ** | 0.04760 * | 0.01877 | 0.94196 ** | 0.01538 | 0.05842 ** | 0.02937 * | 0.0394 | 0.0307 | 0.0250 | 0.0903 | 0.0316 | 0.0335 | |
| LJ | 0.00400 | 0.00707 | −0.02152 | 0.92652 ** | −0.00184 | −0.00031 | 0.00625 | 0.01056 | 0.0371 | 0.0319 | 0.0960 | 0.0384 | 0.0405 | |
| NB | 0.00164 | −0.00466 | −0.01783 | 0.94399 ** | 0.00440 | −0.01355 | −0.00632 | 0.03024 | −0.01248 | 0.0229 | 0.0875 | 0.0297 | 0.0316 | |
| RZ | 0.01507 | 0.00191 | 0.01419 | 0.95490 ** | 0.03070 * | −0.00127 | 0.01878 | 0.05320 ** | 0.00554 | 0.00676 | 0.0828 | 0.0242 | 0.0259 | |
| SY | 0.08917 | 0.08211 | 0.06136 | 0.84080 ** | 0.03720 | 0.08015 | 0.07718 | 0.06848 | 0.05694 | 0.06890 | 0.08312 | 0.0893 | 0.0906 | |
| ZZ | −0.00058 | −0.00320 | −0.02070 | 0.94163 ** | 0.01622 | −0.00576 | 0.01419 | 0.02137 | −0.02041 | −0.00815 | 0.00194 | 0.06673 | 0.0333 | |
| ZH | 0.01815 | 0.01137 | −0.00262 | 0.93873 ** | 0.00082 | 0.00220 | 0.00581 | 0.01076 | −0.00737 | −0.00905 | −0.00905 | 0.06779 | 0.00196 |
| Source of Variation | Degree of Freedom | Sum of Squares | Variance Component | Percentage of Variation (%) |
|---|---|---|---|---|
| Among populations | 13 | 103.073 | 0.34184 Va | 16.27 |
| Within populations | 239 | 420.591 | 1.75980 Vb | 83.73 |
| Total | 252 | 523.664 | 2.10163 | 100 |
| Total genetic differentiation index (FST): 0.16265 | ||||
| Source of Variation | Degree of Freedom | Sum of Squares | Variance Component | Percentage of Variation (%) |
|---|---|---|---|---|
| Among populations | 13 | 64.935 | 0.25350 Va | 55.94 |
| Within populations | 251 | 50.125 | 0.19970 Vb | 44.06 |
| Total | 264 | 115.060 | 0.45321 | 100 |
| Total genetic differentiation index (FST): 0.55936 | ||||
| Source of Variation | Degree of Freedom | Sum of Squares | Variance Component | Percentage of Variation (%) |
|---|---|---|---|---|
| Among populations | 13 | 2127.224 | 9.17407 Va | 63.82 |
| Within populations | 228 | 1185.718 | 5.20052 Vb | 36.18 |
| Total | 241 | 3312.942 | 14.37459 | 100 |
| Total genetic differentiation index (FST): 0.63821 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, D.; Zheng, S.; Wang, S.; Yan, J.; Liu, Z.; Zhou, L.; Wu, B.; Sun, X. Genetic and Haplotype Diversity of Manila Clam Ruditapes philippinarum in Different Regions of China Based on Three Molecular Markers. Animals 2023, 13, 2886. https://doi.org/10.3390/ani13182886
Wei D, Zheng S, Wang S, Yan J, Liu Z, Zhou L, Wu B, Sun X. Genetic and Haplotype Diversity of Manila Clam Ruditapes philippinarum in Different Regions of China Based on Three Molecular Markers. Animals. 2023; 13(18):2886. https://doi.org/10.3390/ani13182886
Chicago/Turabian StyleWei, Di, Sichen Zheng, Songlin Wang, Jingkai Yan, Zhihong Liu, Liqing Zhou, Biao Wu, and Xiujun Sun. 2023. "Genetic and Haplotype Diversity of Manila Clam Ruditapes philippinarum in Different Regions of China Based on Three Molecular Markers" Animals 13, no. 18: 2886. https://doi.org/10.3390/ani13182886
APA StyleWei, D., Zheng, S., Wang, S., Yan, J., Liu, Z., Zhou, L., Wu, B., & Sun, X. (2023). Genetic and Haplotype Diversity of Manila Clam Ruditapes philippinarum in Different Regions of China Based on Three Molecular Markers. Animals, 13(18), 2886. https://doi.org/10.3390/ani13182886