
Exploration of Mediators Associated with Myocardial Remodelling in Feline Hypertrophic Cardiomyopathy


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Echocardiography
2.3. Heart Collection and Histopathological Examination
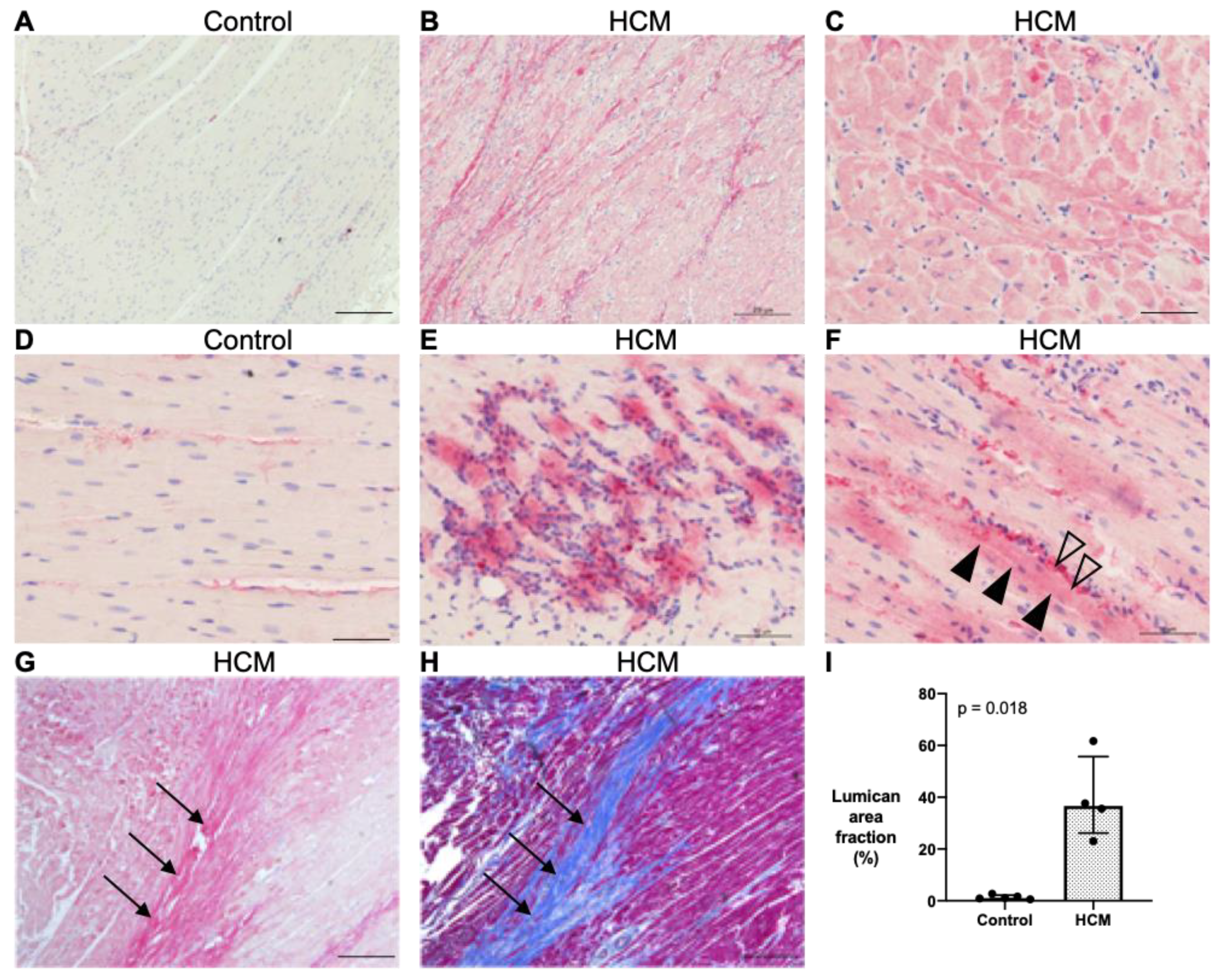
2.4. Collagen and Non-Collagen Quantification—Sirius Red and Fast Green
2.5. Measurement of Cardiomyocyte Width
2.6. RNA Extraction and Preparation of Complementary DNA
2.7. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.8. Western Blotting
2.9. Histological Immunostaining
2.10. Image Analysis
2.11. Soluble and Insoluble Collagen Quantification
2.12. Statistical Analysis
3. Results
3.1. Study Population
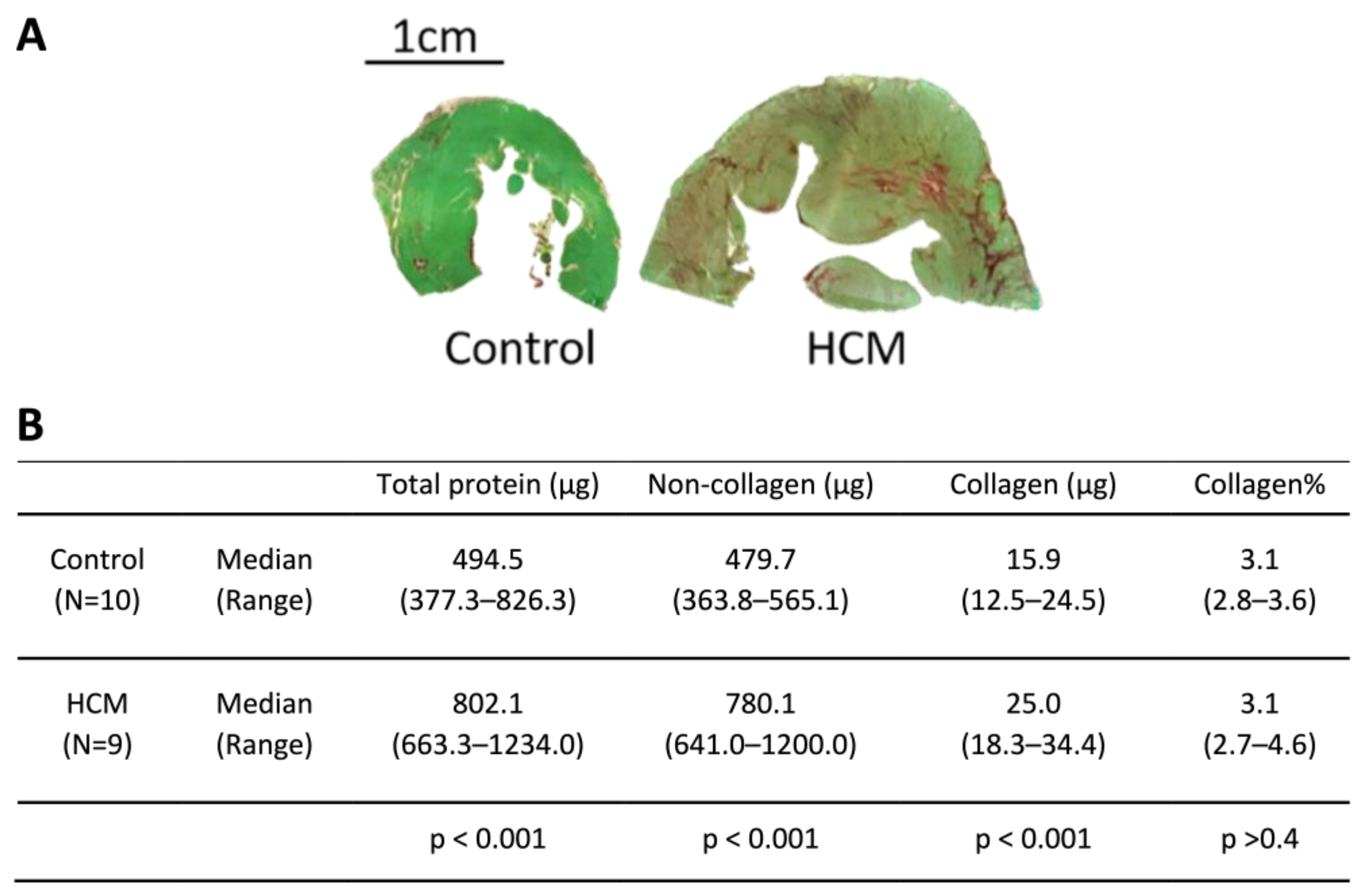
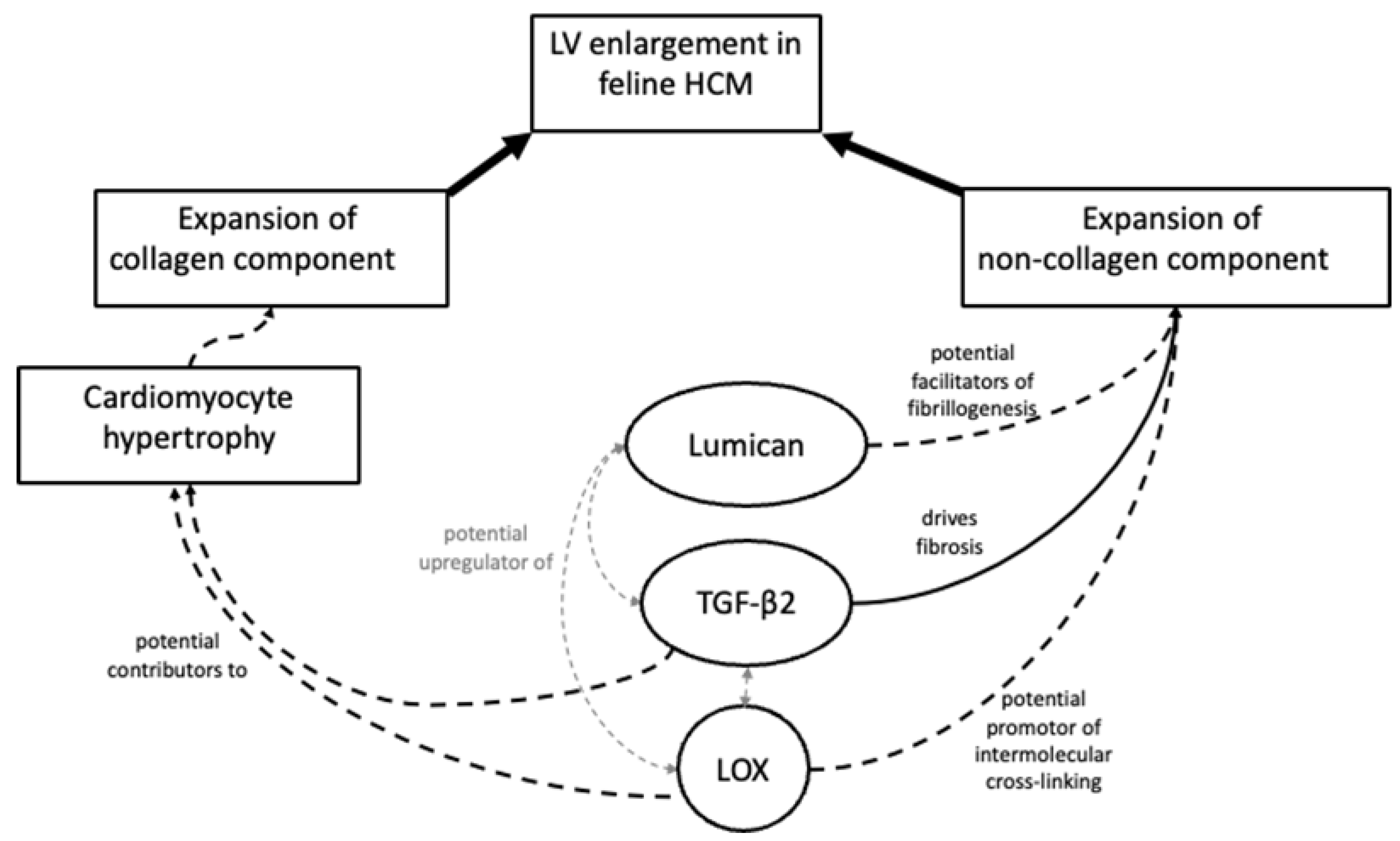
3.2. Exploration of Myocardial Remodelling in HCM
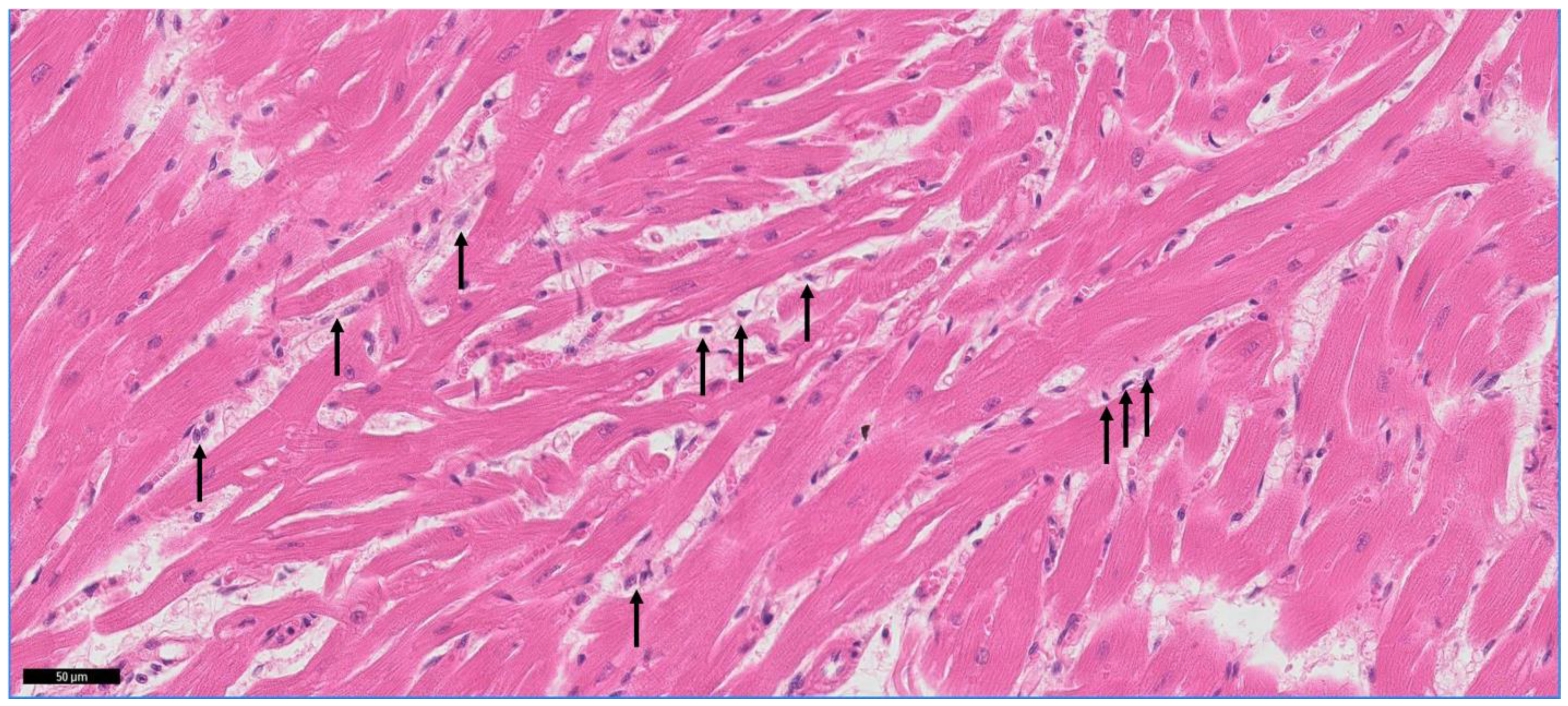
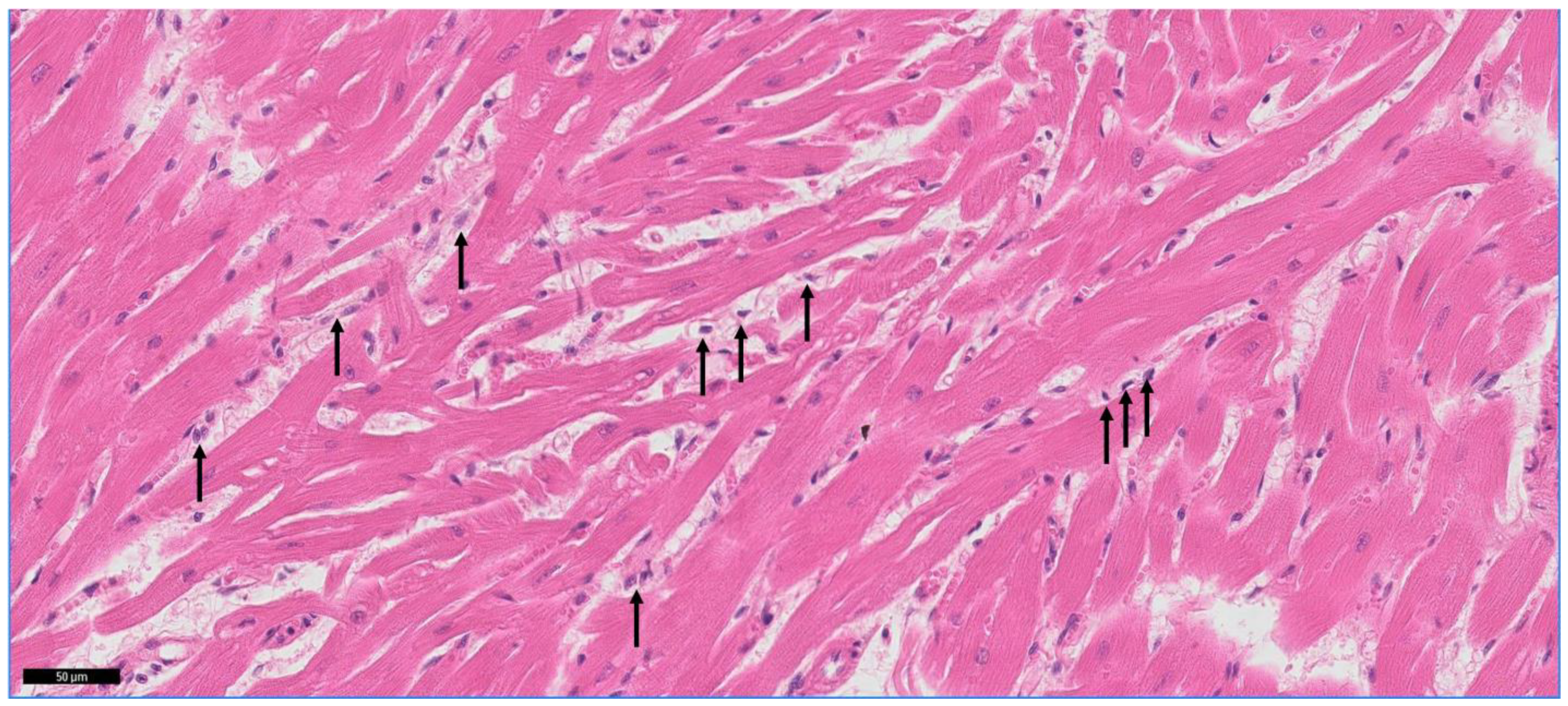
3.3. Mononuclear Cell Infiltration in the Myocardium
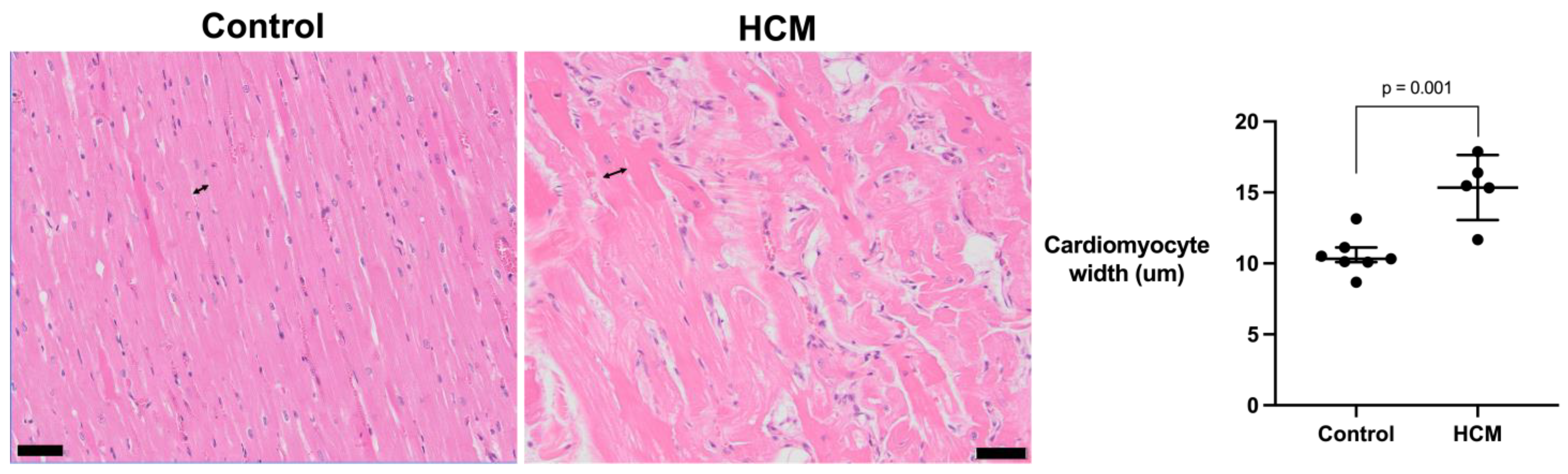
3.4. Assessment of the Cardiomyocyte Width
3.5. Expression and Localisation of Proteins Associated with Myocardial Remodelling Based on Other Species
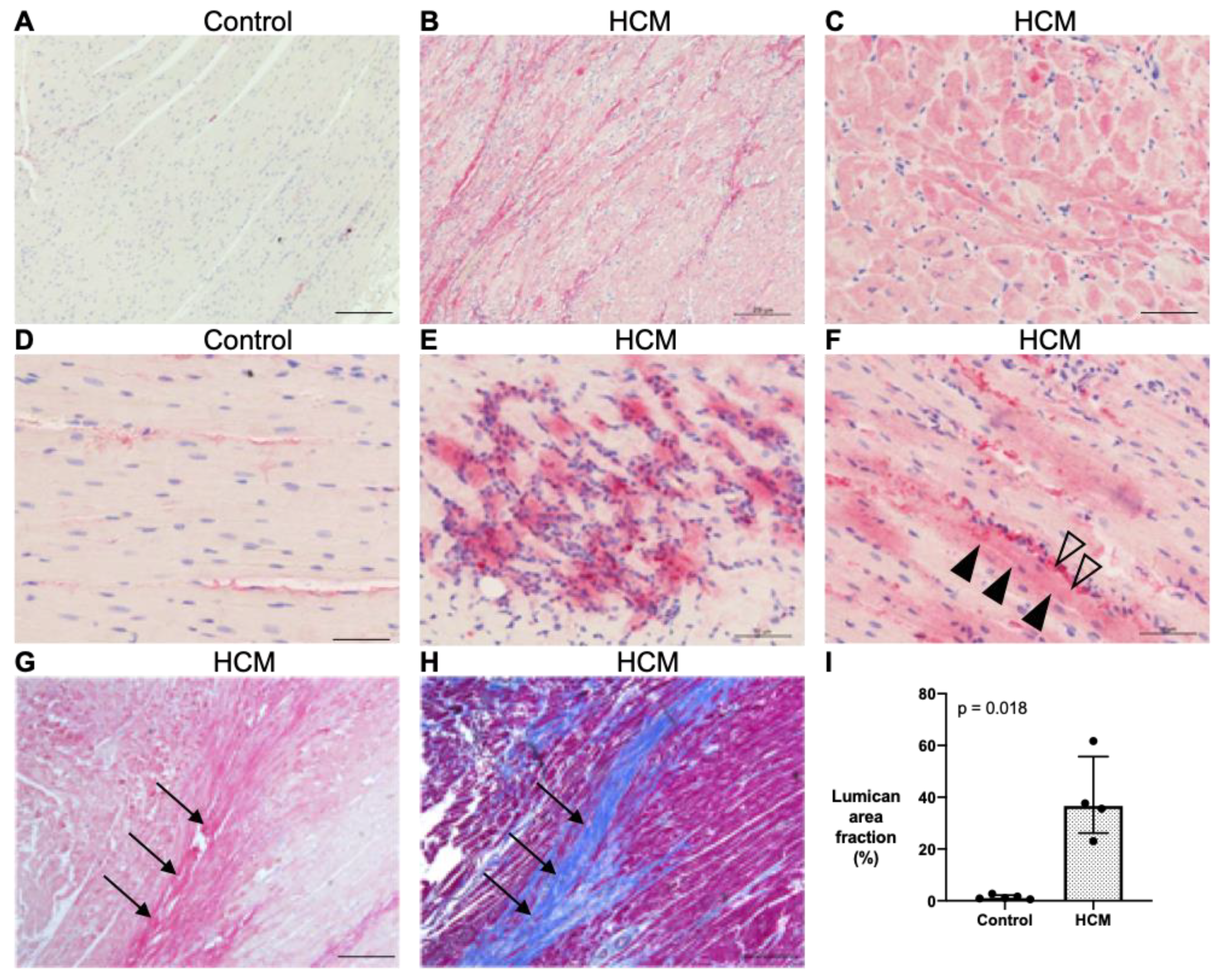
3.5.1. Localisation of Lumican Protein in Feline Myocardium
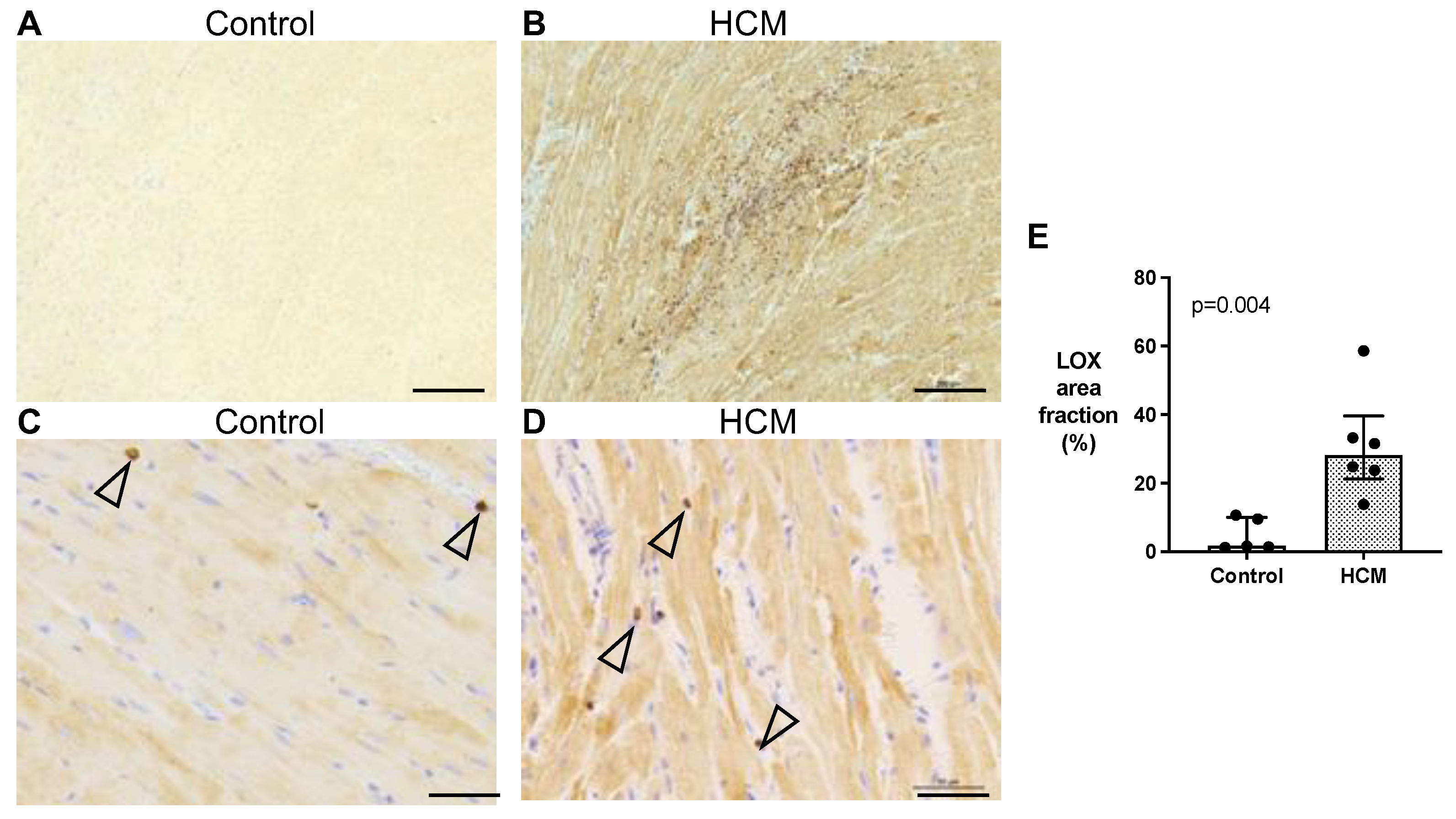
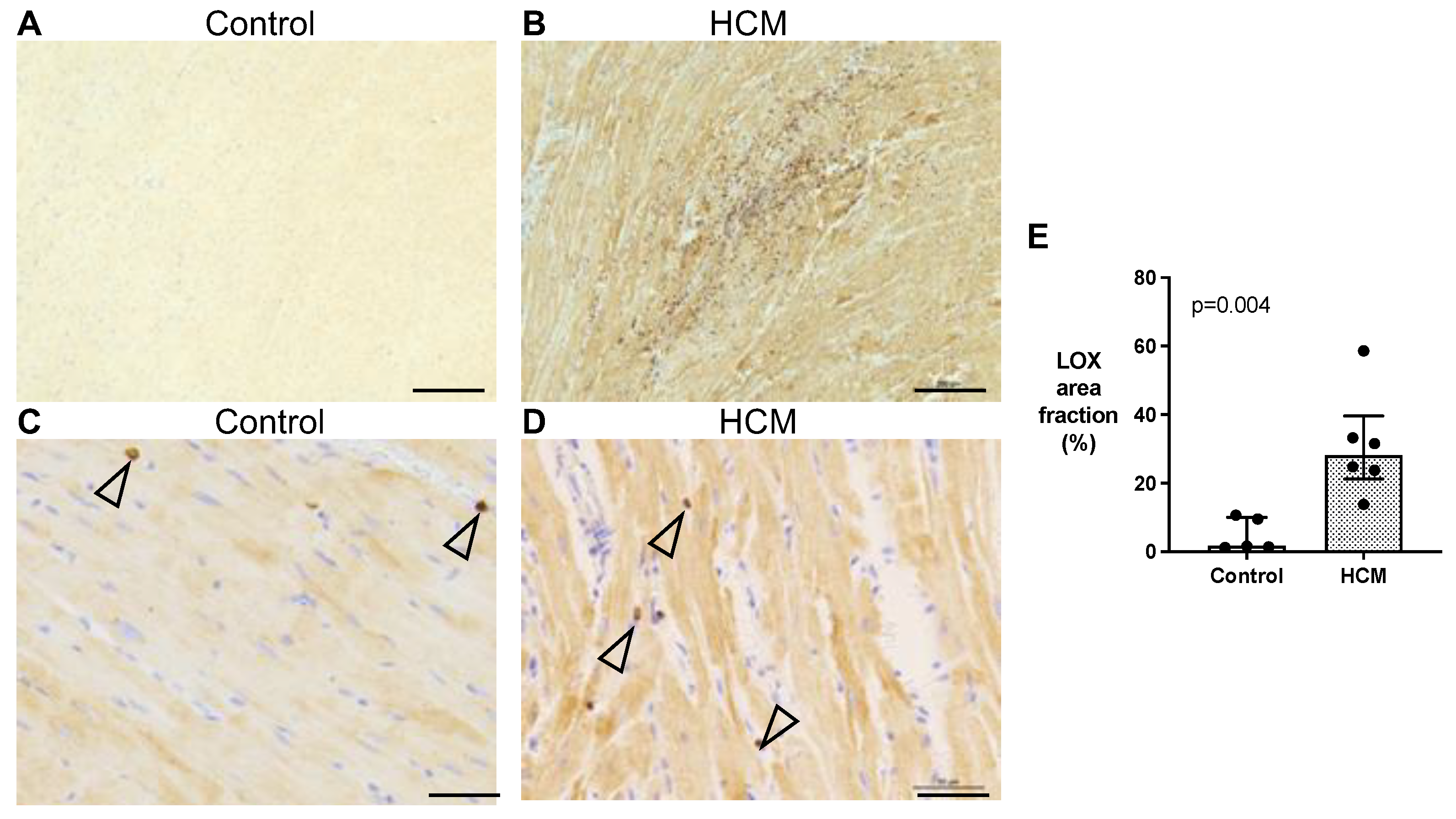
3.5.2. Localisation of LOX Protein in Feline Myocardium
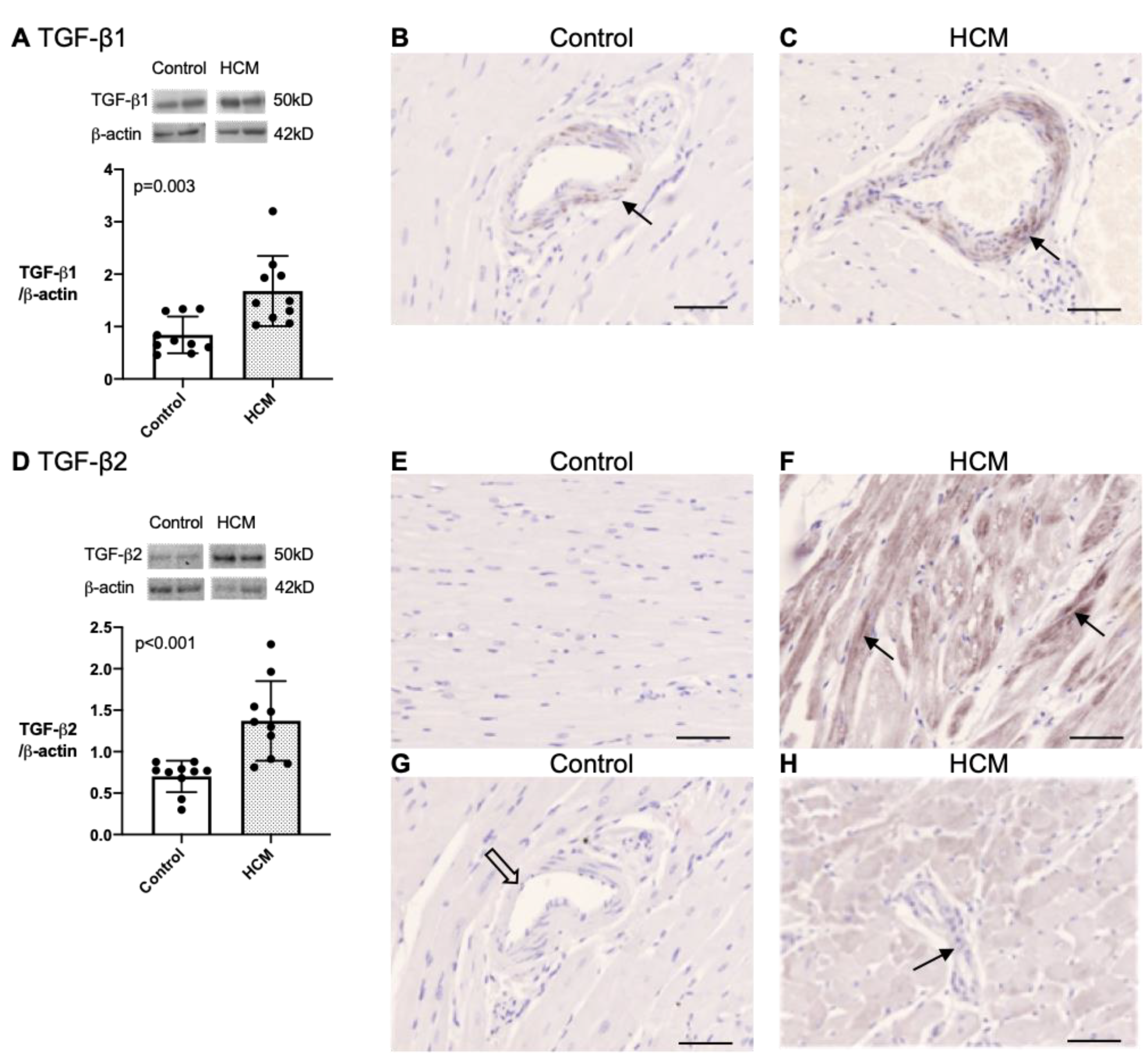
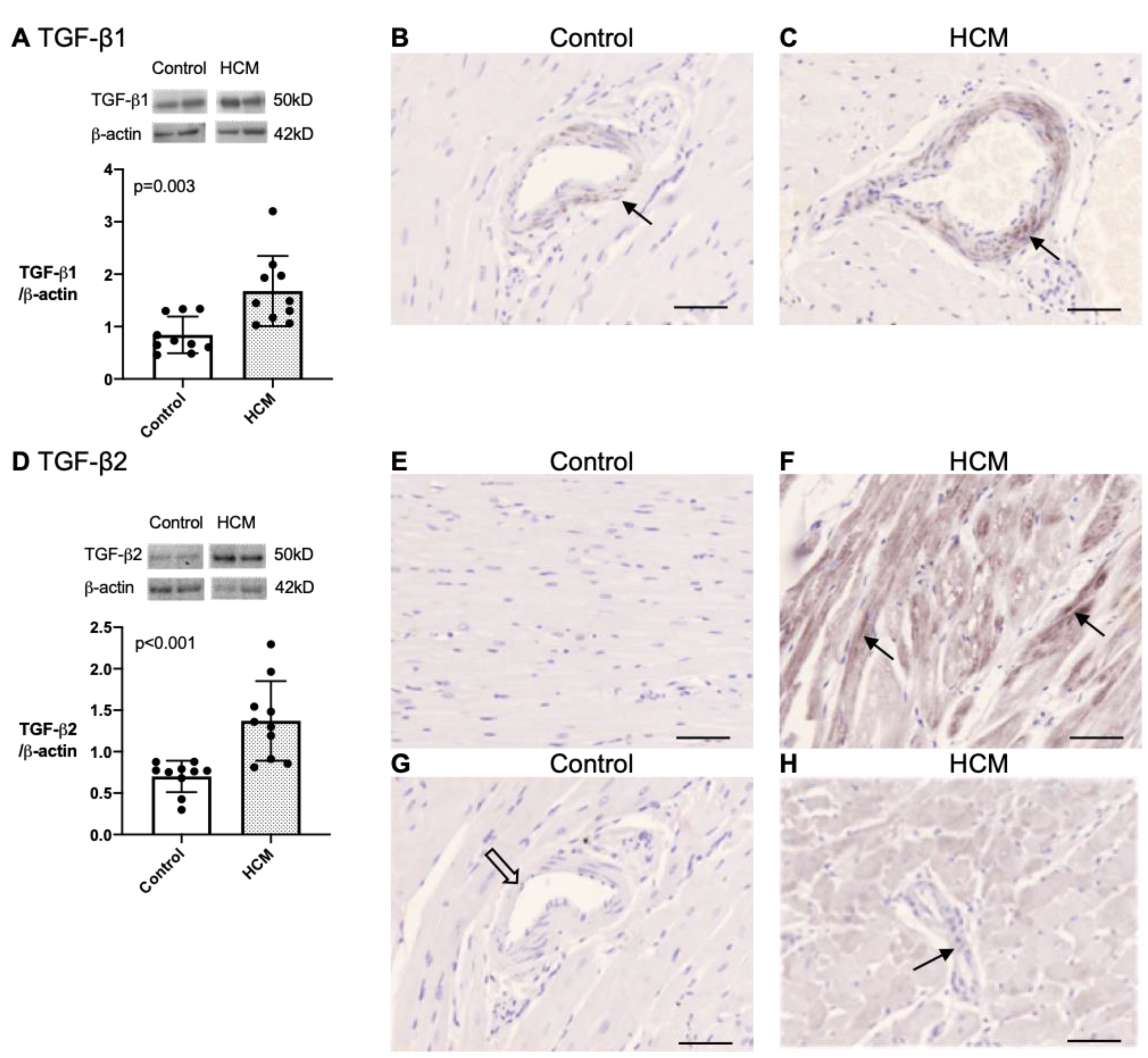
3.5.3. Quantification and Localisation of TGF-β1 and TGF-β2 Protein in Feline Myocardium
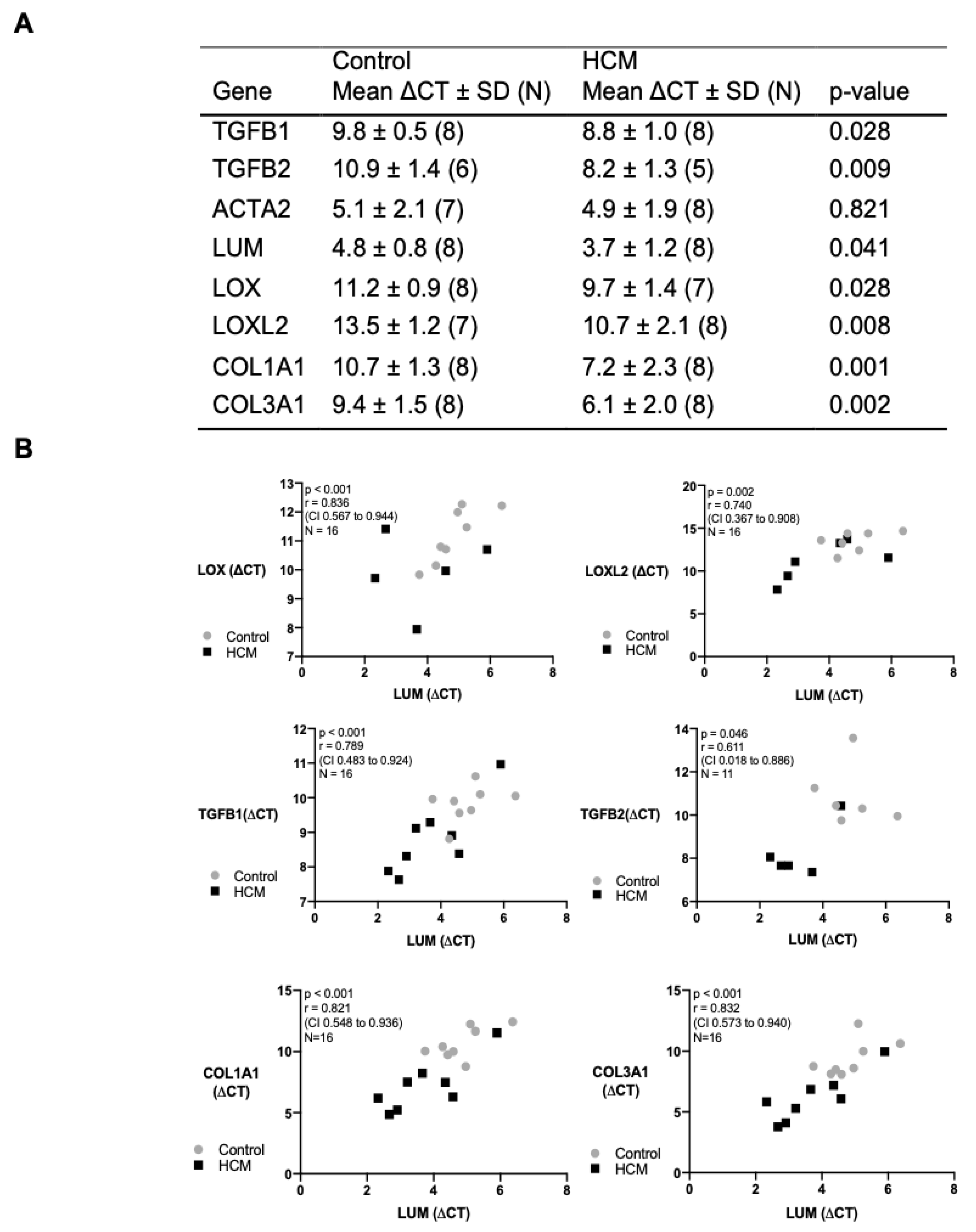
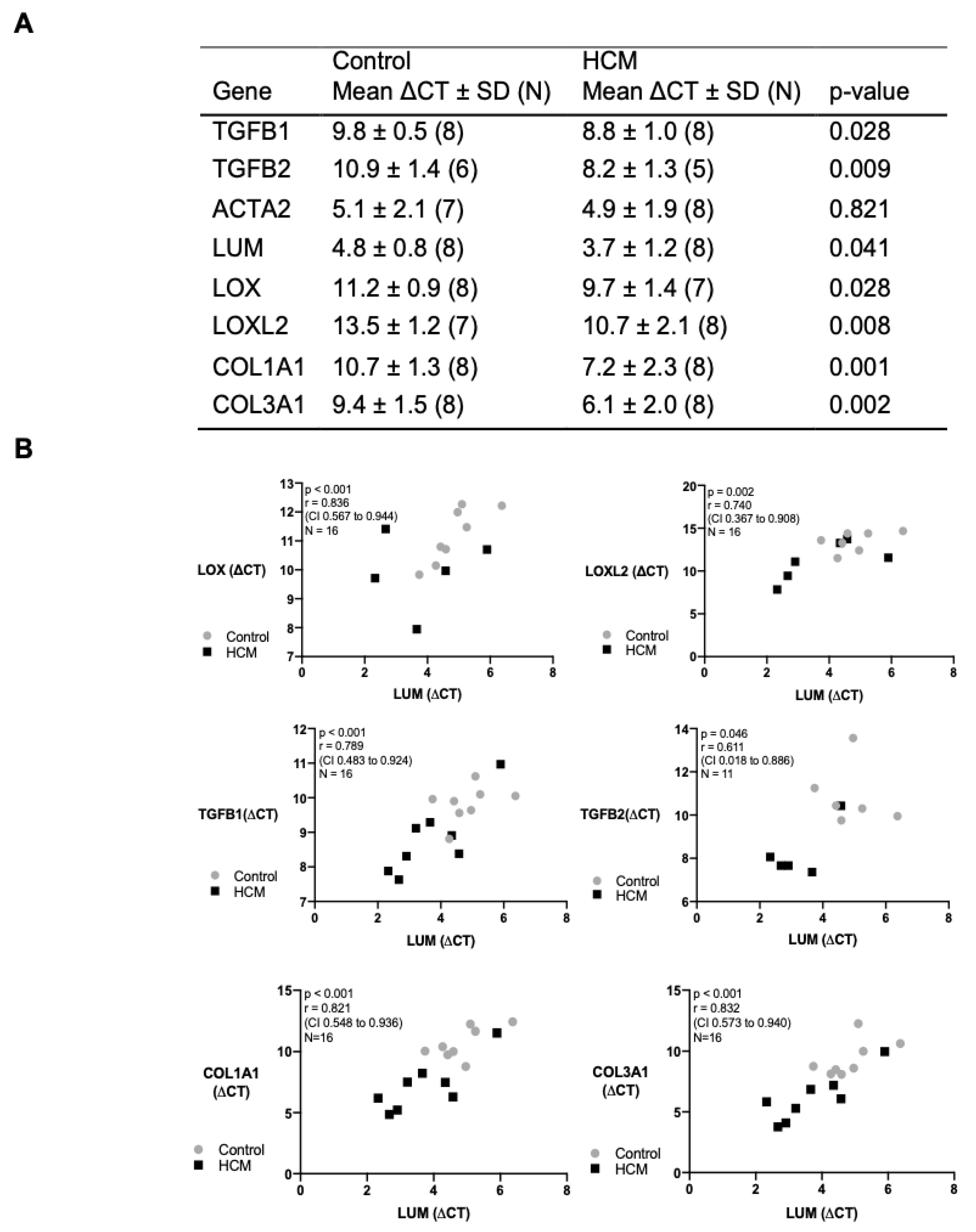
3.6. Transcription and Correlation of Genes Associated with Myocardial Remodelling
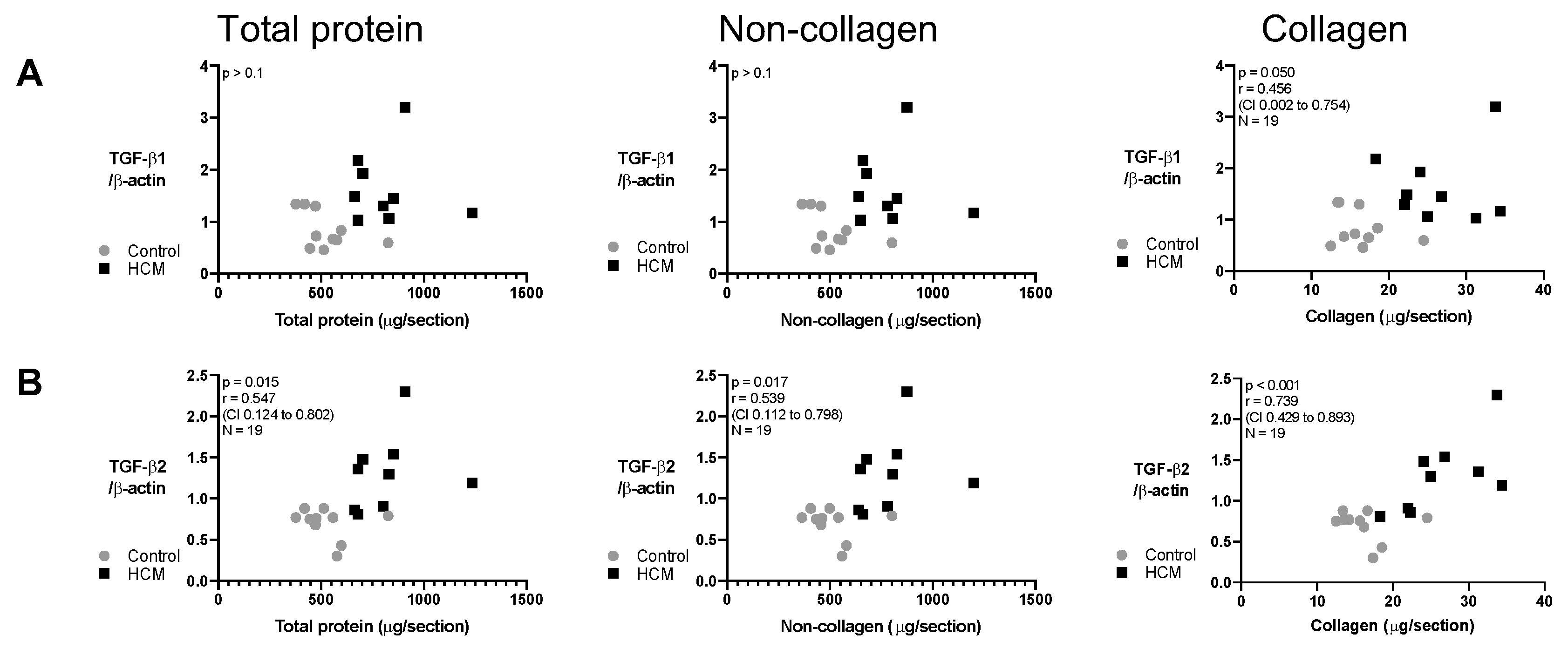
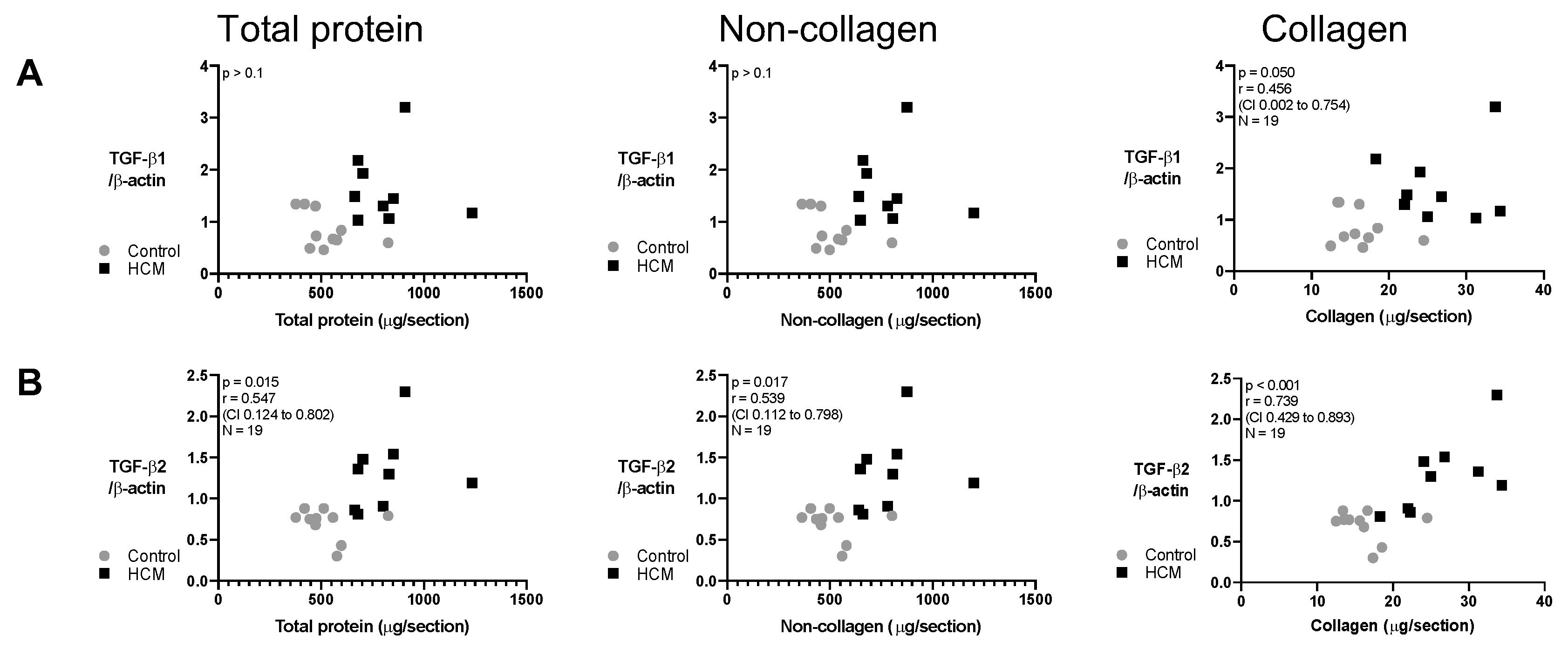
3.7. Relationship between Collagen and Non-Collagen Myocardial Components and TGF-β Isoforms
3.8. Exploration of Soluble and Insoluble Collagen
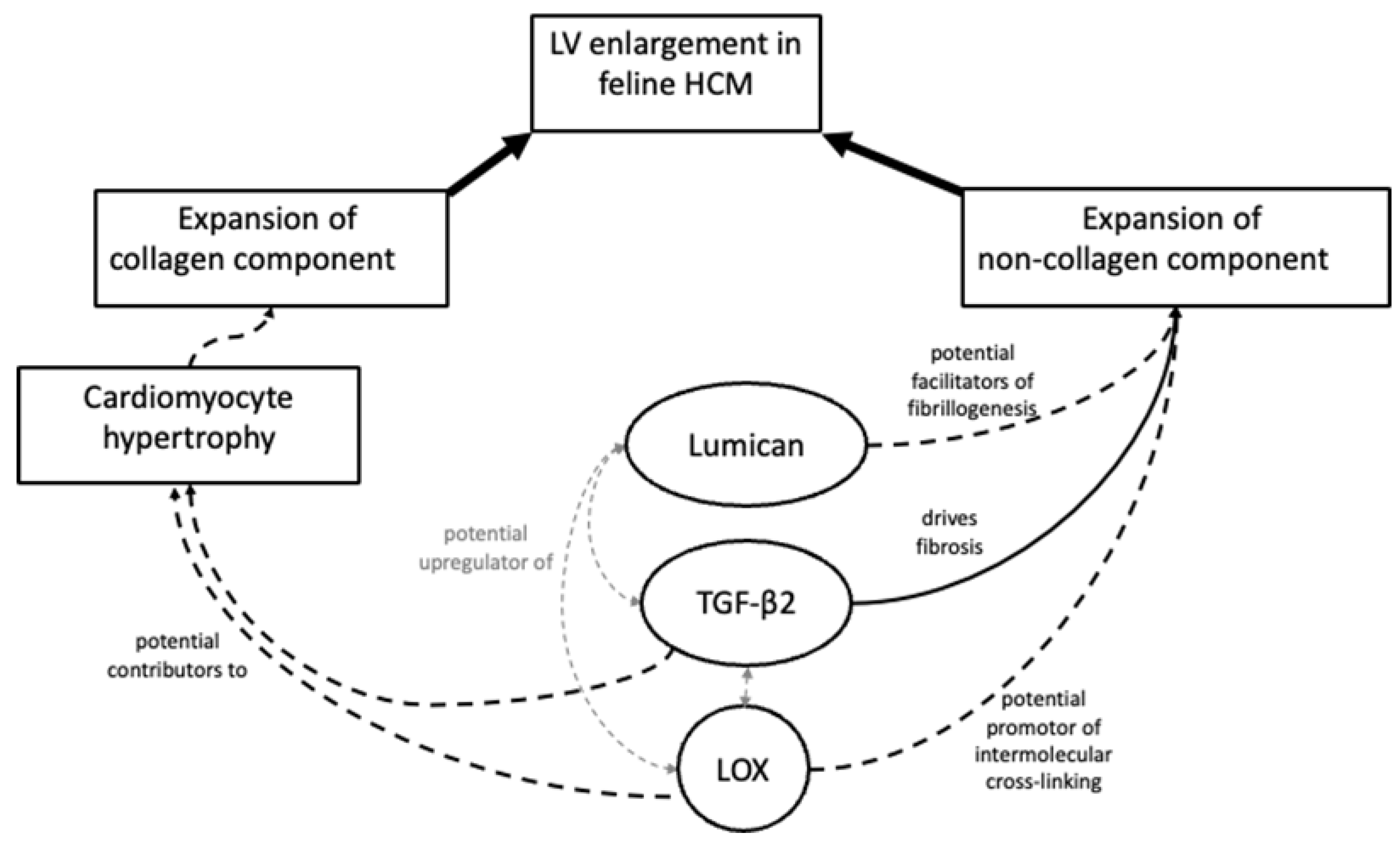
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Messer, A.E.; Chan, J.; Daley, A.; Copeland, O.; Marston, S.B.; Connolly, D.J. Investigations into the Sarcomeric Protein and Ca2+-Regulation Abnormalities Underlying Hypertrophic Cardiomyopathy in Cats (Felix catus). Front. Physiol. 2017, 8, 348. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, L.; Prats, C.; Hyttel, P.; Koch, J. Ultrastructural myocardial changes in seven cats with spontaneous hypertrophic cardiomyopathy. J. Vet. Cardiol. 2015, 17, S220–S232. [Google Scholar] [CrossRef] [Green Version]
- Khor, K.; Campbell, F.; Owen, H.; Shiels, I.; Mills, P. Myocardial collagen deposition and inflammatory cell infiltration in cats with pre-clinical hypertrophic cardiomyopathy. Vet. J. 2015, 203, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Maron, B.J.; Fox, P.R. Hypertrophic cardiomyopathy in man and cats. J. Vet. Cardiol. 2015, 17, S6–S9. [Google Scholar] [CrossRef]
- Aupperle, H.; Baldauf, K.; März, I. An Immunohistochemical Study of Feline Myocardial Fibrosis. J. Comp. Pathol. 2011, 145, 158–173. [Google Scholar] [CrossRef]
- Kitz, S.; Fonfara, S.; Hahn, S.; Hetzel, U.; Kipar, A. Feline Hypertrophic Cardiomyopathy: The Consequence of Cardiomyocyte-Initiated and Macrophage-Driven Remodeling Processes? Vet. Pathol. 2019, 56, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Kittleson, M.D.; Meurs, K.; Harris, S.P. The genetic basis of hypertrophic cardiomyopathy in cats and humans. J. Vet. Cardiol. 2015, 17, S53–S73. [Google Scholar] [CrossRef] [Green Version]
- Meurs, K.M.; Norgard, M.M.; Ederer, M.M.; Hendrix, K.P.; Kittleson, M.D. A substitution mutation in the myosin binding protein C gene in ragdoll hypertrophic cardiomyopathy. Genomics 2007, 90, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.M.; Rush, J.E.; Stern, J.; Huggins, G.S.; Maron, M.S. Feline Hypertrophic Cardiomyopathy: A Spontaneous Large Animal Model of Human HCM. Cardiol. Res. 2017, 8, 139–142. [Google Scholar] [CrossRef]
- Fox, P.R. Hypertrophic Cardiomyopathy. Clinical and Pathologic Correlates. J. Vet. Cardiol. 2003, 5, 39–45. [Google Scholar] [CrossRef]
- O’Hanlon, R.; Grasso, A.; Roughton, M.; Moon, J.C.; Clark, S.; Wage, R.; Webb, J.; Kulkarni, M.; Dawson, D.; Sulaibeekh, L.; et al. Prognostic Significance of Myocardial Fibrosis in Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Raman, B.; Ariga, R.; Spartera, M.; Sivalokanathan, S.; Chan, K.; Dass, S.; Petersen, S.E.; Daniels, M.J.; Francis, J.; Smillie, R.; et al. Progression of myocardial fibrosis in hypertrophic cardiomyopathy: Mechanisms and clinical implications. Eur. Heart J.-Cardiovasc. Imaging 2019, 20, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Fries, R.C.; Kadotani, S.; Keating, S.C.J.; Stack, J.P. Cardiac extracellular volume fraction in cats with preclinical hypertrophic cardiomyopathy. J. Vet. Intern. Med. 2021, 35, 812–822. [Google Scholar] [CrossRef]
- Borgeat, K.; Casamian-Sorrosal, D.; Helps, C.; Fuentes, V.L.; Connolly, D.J. Association of the myosin binding protein C3 mutation (MYBPC3 R820W) with cardiac death in a survey of 236 Ragdoll cats. J. Vet. Cardiol. 2014, 16, 73–80. [Google Scholar] [CrossRef]
- White, A.J.M. End-Stage Hypertrophic Cardiomyopathy in a Cat. Can. Vet. J. 2015, 56, 509–511. [Google Scholar]
- Rienks, M.; Papageorgiou, A.-P.; Frangogiannis, N.G.; Heymans, S. Myocardial Extracellular Matrix: An Ever-Changing and Diverse Entity. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef] [Green Version]
- Kalamajski, S.; Oldberg, A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010, 29, 248–253. [Google Scholar] [CrossRef]
- Mohammadzadeh, N.; Lunde, I.G.; Andenæs, K.; Strand, M.E.; Aronsen, J.M.; Skrbic, B.; Marstein, H.S.; Bandlien, C.; Nygård, S.; Gorham, J.; et al. The extracellular matrix proteoglycan lumican improves survival and counteracts cardiac dilatation and failure in mice subjected to pressure overload. Sci. Rep. 2019, 9, 9206. [Google Scholar] [CrossRef] [Green Version]
- Engebretsen, K.V.T.; Lunde, I.G.; Strand, M.E.; Waehre, A.; Sjaastad, I.; Marstein, H.S.; Skrbic, B.; Dahl, C.P.; Askevold, E.T.; Christensen, G.; et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J. 2013, 280, 2382–2398. [Google Scholar] [CrossRef]
- Voloshenyuk, T.G.; Landesman, E.S.; Khoutorova, E.; Hart, A.D.; Gardner, J.D. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine 2011, 55, 90–97. [Google Scholar] [CrossRef]
- Neff, L.S.; Bradshaw, A.D. Cross your heart? Collagen cross-links in cardiac health and disease. Cell. Signal. 2021, 79, 109889. [Google Scholar] [CrossRef]
- López, B.; Querejeta, R.; González, A.; Beaumont, J.; Larman, M.; Díez, J. Impact of Treatment on Myocardial Lysyl Oxidase Expression and Collagen Cross-Linking in Patients With Heart Failure. Hypertension 2009, 53, 236–242. [Google Scholar] [CrossRef] [Green Version]
- López, B.; Querejeta, R.; González, A.; Larman, M.; Díez, J. Collagen Cross-Linking But Not Collagen Amount Associates With Elevated Filling Pressures in Hypertensive Patients With Stage C Heart Failure. Hypertension 2012, 60, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, C.; Martínez-González, J. The Role of Lysyl Oxidase Enzymes in Cardiac Function and Remodeling. Cells 2019, 8, 1483. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 2016, 7, 13710. [Google Scholar] [CrossRef]
- Bi, X.; Song, Y.; Song, Y.; Yuan, J.; Cui, J.; Zhao, S.; Qiao, S. Collagen Cross-Linking Is Associated With Cardiac Remodeling in Hypertrophic Obstructive Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e017752. [Google Scholar] [CrossRef]
- Coats, C.J.; Heywood, W.E.; Virasami, A.; Ashrafi, N.; Syrris, P.; dos Remedios, C.; Treibel, T.A.; Moon, J.C.; Lopes, L.R.; McGregor, C.G.; et al. Proteomic Analysis of the Myocardium in Hypertrophic Obstructive Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001974. [Google Scholar] [CrossRef] [Green Version]
- Kuusisto, J.; Kärjä, V.; Sipola, P.; Kholová, I.; Peuhkurinen, K.; Jääskeläinen, P.; Naukkarinen, A.; Ylä-Herttuala, S.; Punnonen, K.; Laakso, M. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart 2012, 98, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Ellims, A.H.; Beale, A.L.; Taylor, A.J.; Murphy, A.; Dart, A.M. Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Am. J. Transl. Res. 2017, 9, 5063–5073. [Google Scholar]
- Van Hoek, I.; Hodgkiss-Geere, H.; Bode, E.F.; Hamilton-Elliott, J.; Mõtsküla, P.; Palermo, V.; Pereira, Y.M.; Culshaw, G.J.; Ivanova, A.; Dukes-McEwan, J. Associations among echocardiography, cardiac biomarkers, insulin metabolism, morphology, and inflammation in cats with asymptomatic hypertrophic cardiomyopathy. J. Vet. Intern. Med. 2020, 34, 591–599. [Google Scholar] [CrossRef]
- Becker, R.C.; Owens, A.P.; Sadayappan, S. Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. J. Thromb. Thrombolysis 2020, 49, 177–183. [Google Scholar] [CrossRef]
- Fonfara, S.; Kitz, S.; Monteith, G.; Hahn, S.; Kipar, A. Myocardial transcription of inflammatory and remodeling markers in cats with hypertrophic cardiomyopathy and systemic diseases associated with an inflammatory phenotype. Res. Vet. Sci. 2021, 136, 484–494. [Google Scholar] [CrossRef]
- Melacini, P.; Basso, C.; Angelini, A.; Calore, C.; Bobbo, F.; Tokajuk, B.; Bellini, N.; Smaniotto, G.; Zucchetto, M.; Iliceto, S.; et al. Clinicopathological profiles of progressive heart failure in hypertrophic cardiomyopathy. Eur. Heart J. 2010, 31, 2111–2123. [Google Scholar] [CrossRef] [Green Version]
- Linney, C.J.; Dukes-McEwan, J.; Stephenson, H.M.; López-Alvarez, J.; Fonfara, S. Left atrial size, atrial function and left ventricular diastolic function in cats with hypertrophic cardiomyopathy. J. Small Anim. Pract. 2014, 55, 198–206. [Google Scholar] [CrossRef]
- Finney, J.; Moon, H.-J.; Ronnebaum, T.; Lantz, M.; Mure, M. Human copper-dependent amine oxidases. Arch. Biochem. Biophys. 2014, 546, 19–32. [Google Scholar] [CrossRef]
- Galán, M.; Varona, S.; Guadall, A.; Orriols, M.; Navas, M.; Aguiló, S.; de Diego, A.; Navarro, M.A.; García-Dorado, D.; Rodríguez-Sinovas, A.; et al. Lysyl oxidase overexpression accelerates cardiac remodeling and aggravates angiotensin II–induced hypertrophy. FASEB J. 2017, 31, 3787–3799. [Google Scholar] [CrossRef]
- Ohmura, H.; Yasukawa, H.; Minami, T.; Sugi, Y.; Oba, T.; Nagata, T.; Kyogoku, S.; Ohshima, H.; Aoki, H.; Imaizumi, T. Cardiomyocyte-specific transgenic expression of lysyl oxidase-like protein-1 induces cardiac hypertrophy in mice. Hypertens. Res. 2012, 35, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Borger, M.A.; Williams, W.G.; Weisel, R.D.; Mickle, D.A.; Wigle, E.; Li, R.-K. Regional overexpression of insulin-like growth factor-I and transforming growth factor-β1 in the myocardium of patients with hypertrophic obstructive cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2002, 123, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Cartledge, J.E.; Kane, C.; Dias, P.; Tesfom, M.; Clarke, L.; McKee, B.; Al Ayoubi, S.; Chester, A.; Yacoub, M.H.; Camelliti, P.; et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Cardiovasc. Res. 2015, 105, 260–270. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Q.; Meng, L.; Fang, W.; Zhu, N.; Na, R.; Liu, B.; Chen, L.; Tu, W.; Yu, Q. Association between Collagen Cross-Linking and Cardiac Function and Remodeling in Rats with Furazolidone-Induced Dilated Cardiomyopathy. Int. J. Clin. Exp. Pathol. 2016, 9, 1626–1634. [Google Scholar]
- Ceccato, T.L.; Starbuck, R.B.; Hall, J.K.; Walker, C.J.; Brown, T.E.; Killgore, J.P.; Anseth, K.S.; Leinwand, L.A. Defining the Cardiac Fibroblast Secretome in a Fibrotic Microenvironment. J. Am. Heart Assoc. 2020, 9, e017025. [Google Scholar] [CrossRef]
- Seo, J.; Payne, J.R.; Matos, J.N.; Fong, W.W.; Connolly, D.J.; Fuentes, V.L. Biomarker changes with systolic anterior motion of the mitral valve in cats with hypertrophic cardiomyopathy. J. Vet. Intern. Med. 2020, 34, 1718–1727. [Google Scholar] [CrossRef]
- Fox, P.R.; Liu, S.-K.; Maron, B.J. Echocardiographic Assessment of Spontaneously Occurring Feline Hypertrophic Cardiomyopathy: An Animal Model of Human Disease. Circulation 1995, 92, 2645–2651. [Google Scholar] [CrossRef]
- Wilkie, L.; Smith, K.; Fuentes, V.L. Cardiac pathology findings in 252 cats presented for necropsy; a comparison of cats with unexpected death versus other deaths. J. Vet. Cardiol. 2015, 17, S329–S340. [Google Scholar] [CrossRef]
- Kittleson, M.D.; Côté, E. The Feline Cardiomyopathies: 2. Hypertrophic cardiomyopathy. J. Feline Med. Surg. 2021, 23, 1028–1051. [Google Scholar] [CrossRef]
- Biasato, I.; Francescone, L.; La Rosa, G.; Tursi, M. Anatomopathological staging of feline hypertrophic cardiomyopathy through quantitative evaluation based on morphometric and histopathological data. Res. Vet. Sci. 2015, 102, 136–141. [Google Scholar] [CrossRef]
- Penning, L.C.; Vrieling, H.E.; Brinkhof, B.; Riemers, F.M.; Rothuizen, J.; Rutteman, G.R.; Hazewinkel, H.A. A validation of 10 feline reference genes for gene expression measurements in snap-frozen tissues. Vet. Immunol. Immunopathol. 2007, 120, 212–222. [Google Scholar] [CrossRef]
- Cheng, W.-C.; Wilkie, L.; Kurosawa, T.A.; Dobromylskyj, M.; Priestnall, S.L.; Fuentes, V.L.; Connolly, D.J. Immunohistological Evaluation of Von Willebrand Factor in the Left Atrial Endocardium and Atrial Thrombi from Cats with Cardiomyopathy. Animals 2021, 11, 1240. [Google Scholar] [CrossRef]
- Naqvi, R.U.; MacLeod, K.T. Effect of hypertrophy on mechanisms of relaxation in isolated cardiac myocytes from guinea pig. Am. J. Physiol.-Heart Circ. Physiol. 1994, 267, H1851–H1861. [Google Scholar] [CrossRef]
- Payne, J.; Borgeat, K.; Brodbelt, D.; Connolly, D.; Fuentes, V.L. Risk factors associated with sudden death vs. congestive heart failure or arterial thromboembolism in cats with hypertrophic cardiomyopathy. J. Vet. Cardiol. 2015, 17, S318–S328. [Google Scholar] [CrossRef] [Green Version]
- Li, R.-K.; Li, G.; Mickle, D.A.G.; Weisel, R.D.; Merante, F.; Luss, H.; Rao, V.; Christakis, G.T.; Williams, W.G. Overexpression of Transforming Growth Factor-β1 and Insulin-Like Growth Factor-I in Patients With Idiopathic Hypertrophic Cardiomyopathy. Circulation 1997, 96, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, S.; Magnuson, T.; Lass, J.H.; Jepsen, K.J.; LaMantia, C.; Carroll, H. Lumican Regulates Collagen Fibril Assembly: Skin Fragility and Corneal Opacity in the Absence of Lumican. J. Cell Biol. 1998, 141, 1277–1286. [Google Scholar] [CrossRef]
- Dupuis, L.E.; Berger, M.G.; Feldman, S.; Doucette, L.; Fowlkes, V.; Chakravarti, S.; Thibaudeau, S.; Alcala, N.E.; Bradshaw, A.D.; Kern, C.B. Lumican deficiency results in cardiomyocyte hypertrophy with altered collagen assembly. J. Mol. Cell. Cardiol. 2015, 84, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Rixon, C.; Andreassen, K.; Shen, X.; Erusappan, P.M.; Almaas, V.M.; Palmero, S.; Dahl, C.P.; Ueland, T.; Sjaastad, I.; Louch, W.E.; et al. Lumican accumulates with fibrillar collagen in fibrosis in hypertrophic cardiomyopathy. ESC Heart Fail. 2023, 10, 858–871. [Google Scholar] [CrossRef]
- Heras-Bautista, C.O.; Mikhael, N.; Lam, J.; Shinde, V.; Katsen-Globa, A.; Dieluweit, S.; Molcanyi, M.; Uvarov, V.; Jütten, P.; Sahito, R.G.; et al. Cardiomyocytes facing fibrotic conditions re-express extracellular matrix transcripts. Acta Biomater. 2019, 89, 180–192. [Google Scholar] [CrossRef]
- Ying, S.; Shiraishi, A.; Kao, C.W.-C.; Converse, R.L.; Funderburgh, J.L.; Swiergiel, J.; Roth, M.R.; Conrad, G.W.; Kao, W.W.-Y. Characterization and Expression of the Mouse Lumican Gene. J. Biol. Chem. 1997, 272, 30306–30313. [Google Scholar] [CrossRef] [Green Version]
- Baba, H.; Ishiwata, T.; Takashi, E.; Xu, G.; Asano, G. Expression and Localization of Lumican in the Ischemic and Reperfused Rat Heart. Jpn. Circ. J. 2001, 65, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Waehre, A.; Vistnes, M.; Sjaastad, I.; Nygård, S.; Husberg, C.; Lunde, I.G.; Aukrust, P.; Yndestad, A.; Vinge, L.E.; Behmen, D.; et al. Chemokines regulate small leucine-rich proteoglycans in the extracellular matrix of the pressure-overloaded right ventricle. J. Appl. Physiol. 2012, 112, 1372–1382. [Google Scholar] [CrossRef] [Green Version]
- Carlson, E.C.; Lin, M.; Liu, C.-Y.; Kao, W.W.-Y.; Perez, V.L.; Pearlman, E. Keratocan and Lumican Regulate Neutrophil Infiltration and Corneal Clarity in Lipopolysaccharide-induced Keratitis by Direct Interaction with CXCL1. J. Biol. Chem. 2007, 282, 35502–35509. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Liang, T.; Zhuang, Z.; He, R.; Ren, J.; Jiang, S.; Zhu, L.; Wang, K.; Shi, D. Lumican promotes joint fibrosis through TGF-β signaling. FEBS Open Bio 2020, 10, 2478–2488. [Google Scholar] [CrossRef]
- Meng, Q.; Bhandary, B.; Bhuiyan, S.; James, J.; Osinska, H.; Valiente-Alandi, I.; Shay-Winkler, K.; Gulick, J.; Molkentin, J.D.; Blaxall, B.C.; et al. Myofibroblast-Specific TGFβ Receptor II Signaling in the Fibrotic Response to Cardiac Myosin Binding Protein C-Induced Cardiomyopathy. Circ. Res. 2018, 123, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- López, B.; González, A.; Hermida, N.; Valencia, F.; de Teresa, E.; Díez, J. Role of lysyl oxidase in myocardial fibrosis: From basic science to clinical aspects. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H1–H9. [Google Scholar] [CrossRef] [Green Version]
- El Hajj, E.C.; El Hajj, M.C.; Ninh, V.K.; Bradley, J.M.; Claudino, M.A.; Gardner, J.D. Detrimental role of lysyl oxidase in cardiac remodeling. J. Mol. Cell. Cardiol. 2017, 109, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Martínez-González, J.; Varona, S.; Cañes, L.; Galán, M.; Briones, A.; Cachofeiro, V.; Rodríguez, C. Emerging Roles of Lysyl Oxidases in the Cardiovascular System: New Concepts and Therapeutic Challenges. Biomolecules 2019, 9, 610. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Qin, Q.; Yao, J.; Sun, L.; Qin, X. Induction of LOX by TGF-β1/Smad/AP-1 signaling aggravates rat myocardial fibrosis and heart failure. IUBMB Life 2019, 71, 1729–1739. [Google Scholar] [CrossRef]
- Erasmus, M.; Samodien, E.; Lecour, S.; Cour, M.; Lorenzo, O.; Dludla, P.; Pheiffer, C.; Johnson, R. Linking LOXL2 to Cardiac Interstitial Fibrosis. Int. J. Mol. Sci. 2020, 21, 5913. [Google Scholar] [CrossRef]
- Wu, J.; Jackson-Weaver, O.; Xu, J. The TGFβ superfamily in cardiac dysfunction. Acta Biochim. Biophys. Sin. 2018, 50, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Vakrou, S.; Liu, Y.; Zhu, L.; Greenland, G.V.; Simsek, B.; Hebl, V.B.; Guan, Y.; Woldemichael, K.; Talbot, C.C.; Aon, M.A.; et al. Differences in molecular phenotype in mouse and human hypertrophic cardiomyopathy. Sci. Rep. 2021, 11, 13163. [Google Scholar] [CrossRef]
- Sethi, A.; Mao, W.; Wordinger, R.J.; Clark, A.F. Transforming Growth Factor–β Induces Extracellular Matrix Protein Cross-Linking Lysyl Oxidase (LOX) Genes in Human Trabecular Meshwork Cells. Investig. Opthalmology Vis. Sci. 2011, 52, 5240–5250. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Wang, C.; Huang, D.-Y.; Zhang, Y.; Xu, J.; Kolesnikov, S.S.; Sung, K.; Zhao, H. TGF-beta1 induces the different expressions of lysyl oxidases and matrix metalloproteinases in anterior cruciate ligament and medial collateral ligament fibroblasts after mechanical injury. J. Biomech. 2013, 46, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Tovar-Vidales, T.; Fitzgerald, A.M.; Clark, A.F.; Wordinger, R.J. Transforming Growth Factor-β2 Induces Expression of Biologically Active Bone Morphogenetic Protein-1 in Human Trabecular Meshwork Cells. Investig. Opthalmology Vis. Sci. 2013, 54, 4741–4748. [Google Scholar] [CrossRef] [Green Version]
- Foà, A.; Agostini, V.; Rapezzi, C.; Olivotto, I.; Corti, B.; Potena, L.; Biagini, E.; Suarez, S.M.; Rotellini, M.; Cecchi, F.; et al. Histopathological comparison of intramural coronary artery remodeling and myocardial fibrosis in obstructive versus end-stage hypertrophic cardiomyopathy. Int. J. Cardiol. 2019, 291, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Nabel, E.G.; Shum, L.; Pompili, V.J.; Yang, Z.Y.; San, H.; Shu, H.B.; Liptay, S.; Gold, L.; Gordon, D.; Derynck, R. Direct transfer of transforming growth factor beta 1 gene into arteries stimulates fibrocellular hyperplasia. Proc. Natl. Acad. Sci. USA 1993, 90, 10759–10763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majesky, M.W.; Lindner, V.; Twardzik, D.R.; Schwartz, S.M.; Reidy, M.A. Production of transforming growth factor beta 1 during repair of arterial injury. J. Clin. Investig. 1991, 88, 904–910. [Google Scholar] [CrossRef]
- Smith, J.D.; Bryant, S.R.; Couper, L.L.; Vary, C.P.H.; Gotwals, P.J.; Koteliansky, V.E.; Lindner, V. Soluble Transforming Growth Factor-β Type II Receptor Inhibits Negative Remodeling, Fibroblast Transdifferentiation, and Intimal Lesion Formation But Not Endothelial Growth. Circ. Res. 1999, 84, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Baillie, R.; Coombes, R.; Smith, J. Multiple forms of TGF-β1 in breast tissues: A biologically active form of the small latent complex of TGF-β1. Eur. J. Cancer A 1996, 32, 1566–1573. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Orban, J.M.; Wilson, L.B.; Kofroth, J.A.; El-Kurdi, M.S.; Maul, T.M.; Vorp, D.A. Crosslinking of collagen gels by transglutaminase. J. Biomed. Mater. Res. 2004, 68A, 756–762. [Google Scholar] [CrossRef]
- Bakris, G.L.; Bank, A.J.; Kass, D.A.; Neutel, J.M.; Preston, R.A.; Oparil, S. Advanced glycation end-product cross-link breakersA novel approach to cardiovascular pathologies related to the aging process. Am. J. Hypertens. 2004, 17, S23–S30. [Google Scholar] [CrossRef] [Green Version]
- Zieman, S.J.; Kass, D.A. Advanced Glycation End Product Cross-Linking: Pathophysiologic Role and Therapeutic Target in Cardiovascular Disease. Congest. Heart Fail. 2004, 10, 144–151. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.-C.; Lawson, C.; Liu, H.-H.; Wilkie, L.; Dobromylskyj, M.; Luis Fuentes, V.; Dudhia, J.; Connolly, D.J. Exploration of Mediators Associated with Myocardial Remodelling in Feline Hypertrophic Cardiomyopathy. Animals 2023, 13, 2112. https://doi.org/10.3390/ani13132112
Cheng W-C, Lawson C, Liu H-H, Wilkie L, Dobromylskyj M, Luis Fuentes V, Dudhia J, Connolly DJ. Exploration of Mediators Associated with Myocardial Remodelling in Feline Hypertrophic Cardiomyopathy. Animals. 2023; 13(13):2112. https://doi.org/10.3390/ani13132112
Chicago/Turabian StyleCheng, Wan-Ching, Charlotte Lawson, Hui-Hsuan Liu, Lois Wilkie, Melanie Dobromylskyj, Virginia Luis Fuentes, Jayesh Dudhia, and David J. Connolly. 2023. "Exploration of Mediators Associated with Myocardial Remodelling in Feline Hypertrophic Cardiomyopathy" Animals 13, no. 13: 2112. https://doi.org/10.3390/ani13132112
APA StyleCheng, W.-C., Lawson, C., Liu, H.-H., Wilkie, L., Dobromylskyj, M., Luis Fuentes, V., Dudhia, J., & Connolly, D. J. (2023). Exploration of Mediators Associated with Myocardial Remodelling in Feline Hypertrophic Cardiomyopathy. Animals, 13(13), 2112. https://doi.org/10.3390/ani13132112