
Identification of Runs of Homozygosity Islands and Functional Variants in Wenchang Chicken

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Genotypes
2.2. Genetic Diversity and Linkage Disequilibrium (LD) Analysis
2.3. Identification of ROH
2.4. Assessment of Inbreeding Coefficients
2.5. Identification of Candidate Genes within ROH Islands
3. Results
3.1. SNP Identification
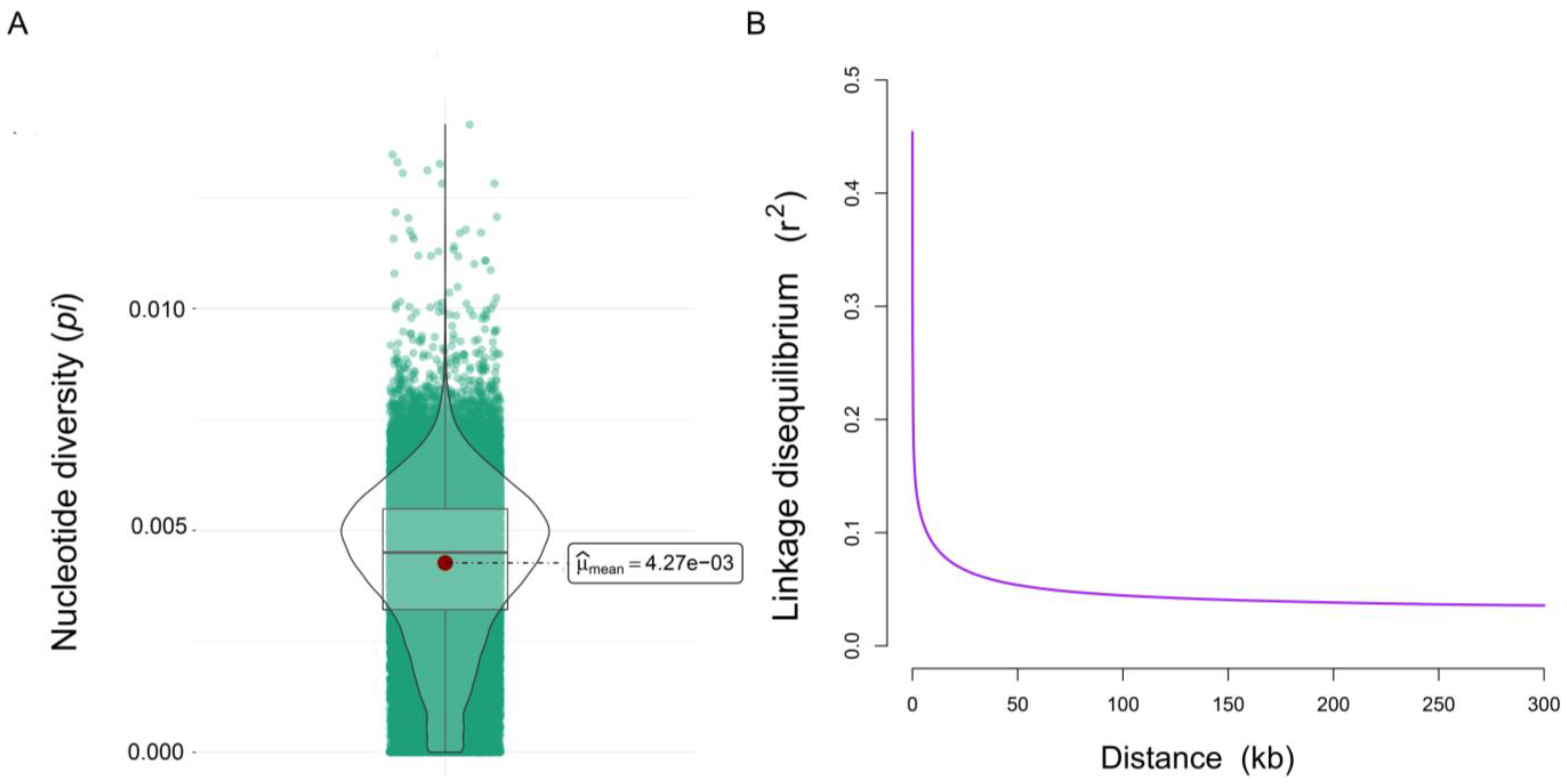
3.2. Genetic Diversity and LD Analysis
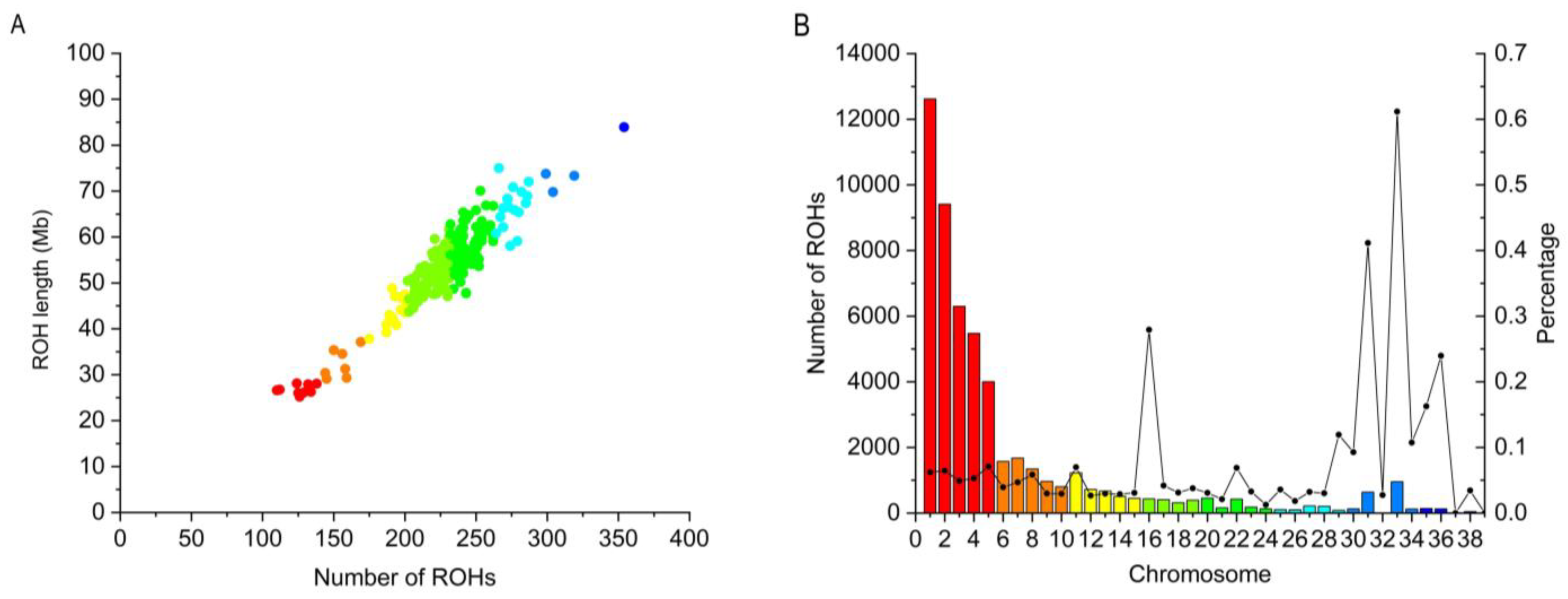
3.3. Genomic Distribution of ROH
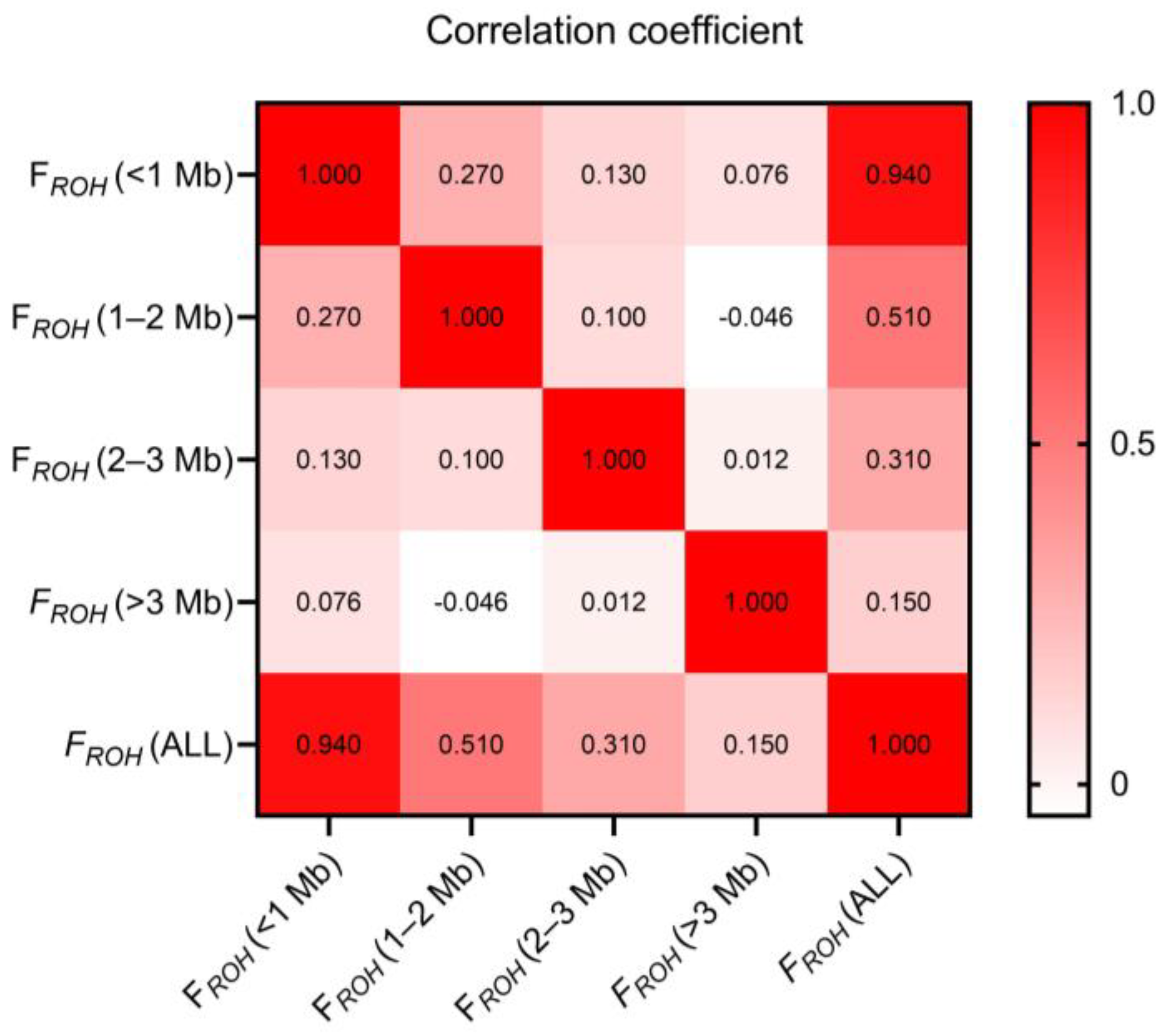
3.4. Inbreeding Coefficients
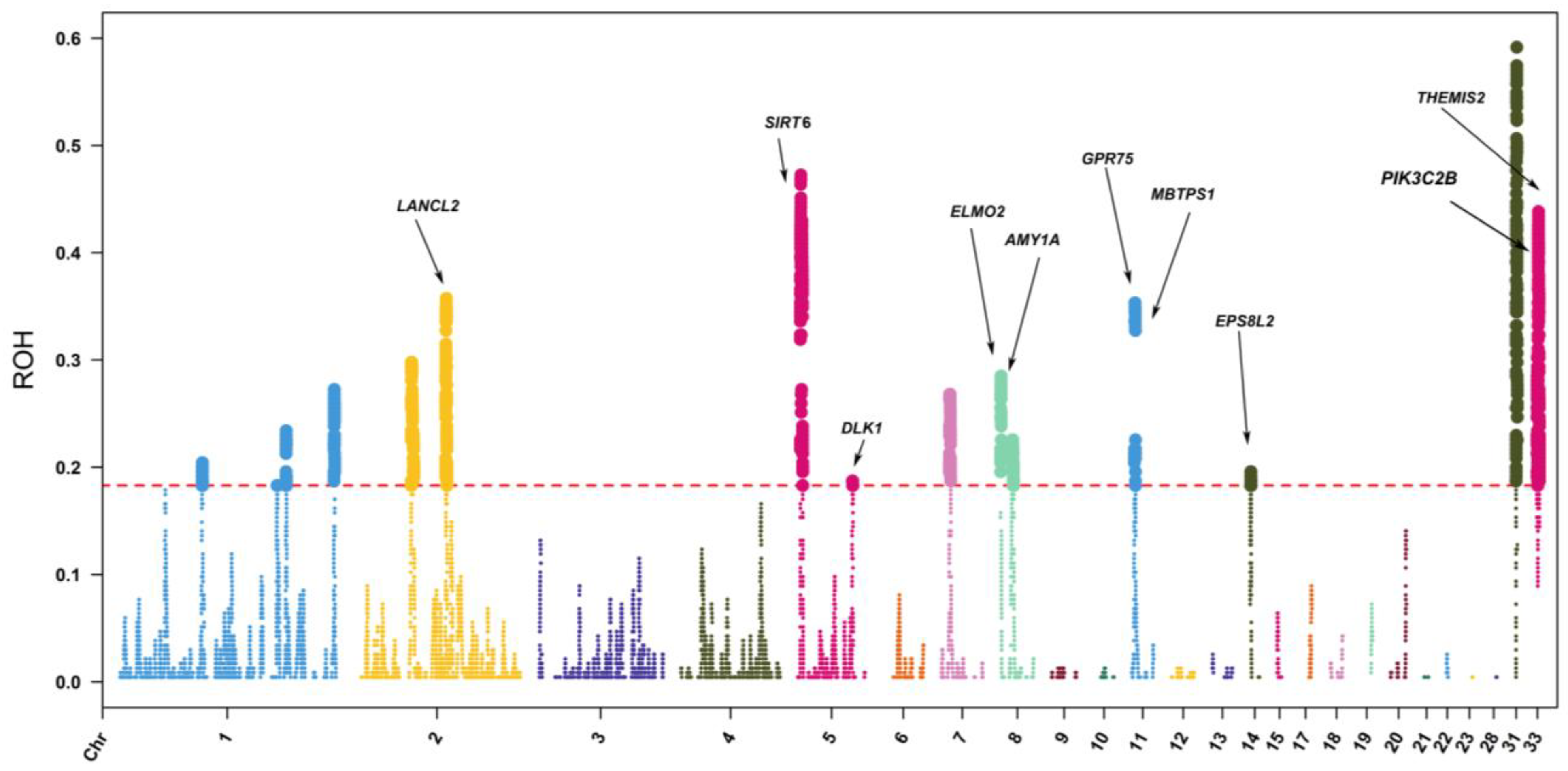
3.5. ROH-Based Selective Signal Analysis
4. Discussion
4.1. Genetic Diversity of the Wenchang Chicken Sample Population
4.2. Characteristics of the Identified ROH
4.3. Inbreeding Coefficients
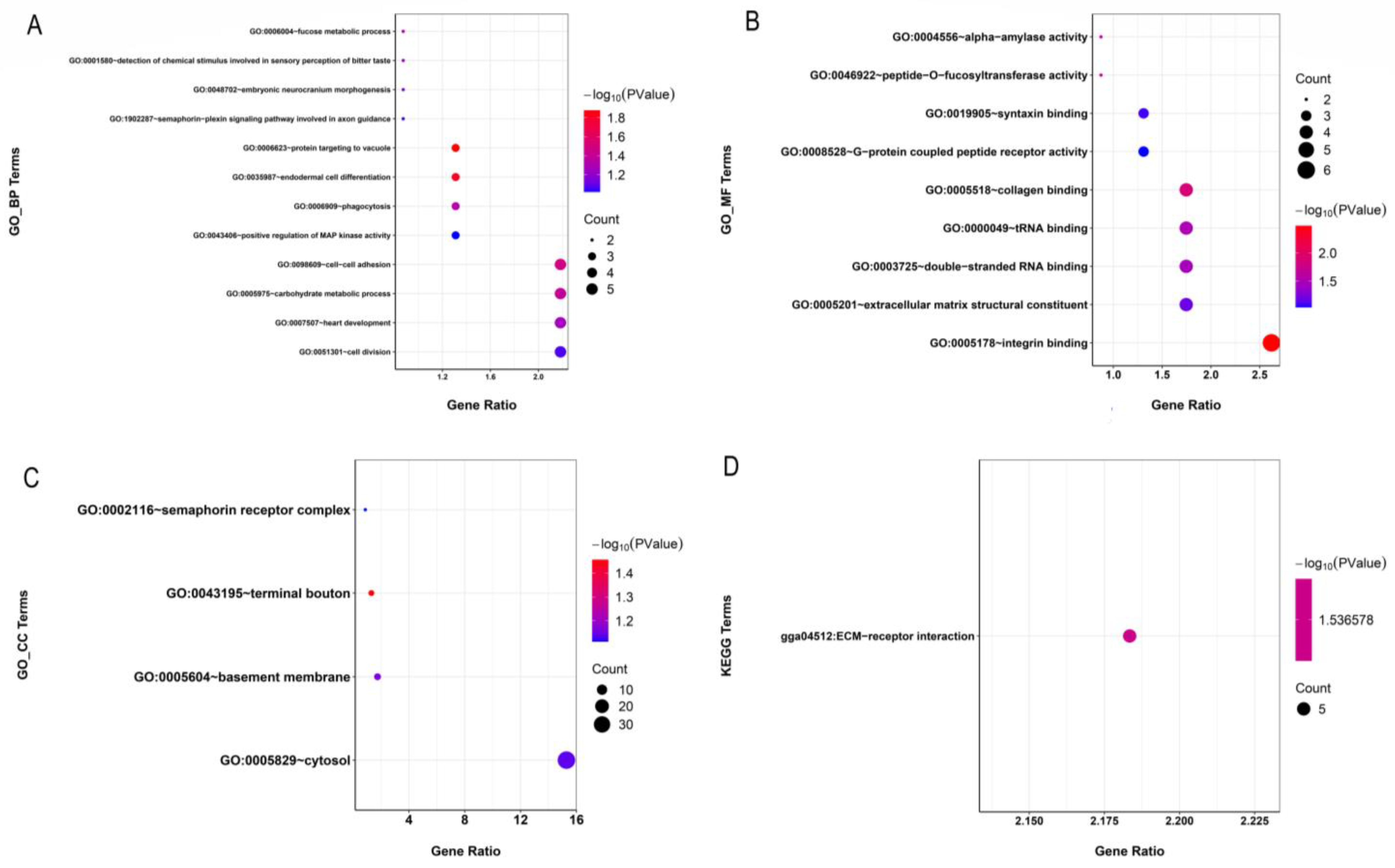
4.4. Candidate Genes within ROH Islands
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, C.; Lin, D.; Wang, Y.; Peng, D.; Li, H.; Fei, J.; Chen, K.; Yang, N.; Hu, X.; Zhao, Y.; et al. Widespread introgression in Chinese indigenous chicken breeds from commercial broiler. Evol. Appl. 2019, 12, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Makanjuola, B.O.; Maltecca, C.; Miglior, F.; Marras, G.; Abdalla, E.A.; Schenkel, F.S.; Baes, C.F. Identification of unique ROH regions with unfavorable effects on production and fertility traits in Canadian Holsteins. Genet. Sel. Evol. 2021, 53, 68. [Google Scholar] [CrossRef] [PubMed]
- Rostamzadeh Mahdabi, E.; Esmailizadeh, A.; Ayatollahi Mehrgardi, A.; Asadi Fozi, M. A genome-wide scan to identify signatures of selection in two Iranian indigenous chicken ecotypes. Genet. Sel. Evol. 2021, 53, 72. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of artificial selection on runs of homozygosity in u.s. Holstein cattle. PLoS ONE 2013, 8, e80813. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.; Ventura, R.V.; Baldi, F.; da Silva, M. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef]
- Schachler, K.; Distl, O.; Metzger, J. Tracing selection signatures in the pig genome gives evidence for selective pressures on a unique curly hair phenotype in Mangalitza. Sci. Rep. 2020, 10, 22142. [Google Scholar] [CrossRef]
- Tao, L.; He, X.; Wang, F.; Zhong, Y.; Pan, L.; Wang, X.; Gan, S.; Di, R.; Chu, M. Luzhong mutton sheep: Inbreeding and selection signatures. J. Anim. Sci. Technol. 2020, 62, 777–789. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z.; Fang, Y.; Cao, C.; Zhang, Z.; Pan, Y.; Wang, Q. Runs of Homozygosity Revealed Reproductive Traits of Hu Sheep. Genes 2022, 13, 1848. [Google Scholar] [CrossRef]
- Tian, S.; Li, W.; Zhong, Z.; Wang, F.; Xiao, Q. Genome-wide re-sequencing data reveals the genetic diversity and population structure of Wenchang chicken in China. Anim. Genet. 2023, 54, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Davies, R.W.; Flint, J.; Myers, S.; Mott, R. Rapid genotype imputation from sequence without reference panels. Nat. Genet. 2016, 48, 965–969. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Cendron, F.; Mastrangelo, S.; Tolone, M.; Perini, F.; Lasagna, E.; Cassandro, M. Genome-wide analysis reveals the patterns of genetic diversity and population structure of 8 Italian local chicken breeds. Poult. Sci. 2021, 100, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Han, W.; Tang, H.; Li, G.; Zhang, M.; Xu, R.; Liu, Y.; Yang, T.; Li, W.; Zou, J.; et al. Genomic diversity dynamics in conserved chicken populations are revealed by genome-wide SNPs. BMC Genom. 2018, 19, 598. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Adams, S.M.; Derks, M.F.L.; Makanjuola, B.O.; Marras, G.; Wood, B.J.; Baes, C.F. Investigating inbreeding in the turkey (Meleagris gallopavo) genome. Poult. Sci. 2021, 100, 101366. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Bizarria Dos Santos, W.; Pimenta Schettini, G.; Fonseca, M.G.; Pereira, G.L.; Loyola Chardulo, L.A.; Rodrigues Machado Neto, O.; Baldassini, W.A.; Nunes de Oliveira, H.; Abdallah Curi, R. Fine-scale estimation of inbreeding rates, runs of homozygosity and genome-wide heterozygosity levels in the Mangalarga Marchador horse breed. J. Anim. Breed. Genet. 2021, 138, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Abebe, A.S.; Mikko, S.; Johansson, A.M. Genetic diversity of five local Swedish chicken breeds detected by microsatellite markers. PLoS ONE 2015, 10, e0120580. [Google Scholar] [CrossRef]
- Luo, W.; Luo, C.; Wang, M.; Guo, L.; Chen, X.; Li, Z.; Zheng, M.; Folaniyi, B.S.; Luo, W.; Shu, D.; et al. Genome diversity of Chinese indigenous chicken and the selective signatures in Chinese gamecock chicken. Sci. Rep. 2020, 10, 14532. [Google Scholar] [CrossRef]
- Yuan, J.; Li, S.; Sheng, Z.; Zhang, M.; Liu, X.; Yuan, Z.; Yang, N.; Chen, J. Genome-wide run of homozygosity analysis reveals candidate genomic regions associated with environmental adaptations of Tibetan native chickens. BMC Genom. 2022, 23, 91. [Google Scholar] [CrossRef]
- Sun, J.; Chen, T.; Zhu, M.; Wang, R.; Huang, Y.; Wei, Q.; Yang, M.; Liao, Y. Whole-genome sequencing revealed genetic diversity and selection of Guangxi indigenous chickens. PLoS ONE 2022, 17, e0250392. [Google Scholar] [CrossRef]
- Li, D.; Sun, G.; Zhang, M.; Cao, Y.; Zhang, C.; Fu, Y.; Li, F.; Li, G.; Jiang, R.; Han, R.; et al. Breeding history and candidate genes responsible for black skin of Xichuan black-bone chicken. BMC Genom. 2020, 21, 511. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Ribani, A.; Moscatelli, G.; Bonacini, M.; Prandi, M.; Mancin, E.; Mantovani, R.; Dall’Olio, S.; Fontanesi, L. Comparative analysis of inbreeding parameters and runs of homozygosity islands in 2 Italian autochthonous cattle breeds mainly raised in the Parmigiano-Reggiano cheese production region. J. Dairy Sci. 2022, 105, 2408–2425. [Google Scholar] [CrossRef]
- Pengelly, R.J.; Gheyas, A.A.; Kuo, R.; Mossotto, E.; Seaby, E.G.; Burt, D.W.; Ennis, S.; Collins, A. Commercial chicken breeds exhibit highly divergent patterns of linkage disequilibrium. Heredity 2016, 117, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, F.C.; Hazelhurst, S.; Ramsay, M. Assessing runs of Homozygosity: A comparison of SNP Array and whole genome sequence low coverage data. BMC Genom. 2018, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.S.; Weigend, S.; Simianer, H.; Weigend, A.; Rothschild, M.; Schmidt, C.; Ashwell, C.; Persia, M.; Reecy, J.; Lamont, S.J. Genomic Comparison of Indigenous African and Northern European Chickens Reveals Putative Mechanisms of Stress Tolerance Related to Environmental Selection Pressure. G3 Genes Genomes Genet. 2017, 7, 1525–1537. [Google Scholar] [CrossRef]
- Zhang, J.; Nie, C.; Li, X.; Ning, Z.; Chen, Y.; Jia, Y.; Han, J.; Wang, L.; Lv, X.; Yang, W.; et al. Genome-Wide Population Genetic Analysis of Commercial, Indigenous, Game, and Wild Chickens Using 600K SNP Microarray Data. Front. Genet. 2020, 11, 543294. [Google Scholar] [CrossRef]
- Hill, W.G.; Weir, B.S. Variation in actual relationship as a consequence of Mendelian sampling and linkage. Genet. Res. 2011, 93, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Almeida, O.A.C.; Moreira, G.C.M.; Rezende, F.M.; Boschiero, C.; de Oliveira Peixoto, J.; Ibelli, A.M.G.; Ledur, M.C.; de Novais, F.J.; Coutinho, L.L. Identification of selection signatures involved in performance traits in a paternal broiler line. BMC Genom. 2019, 20, 449. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Mesarič, M.; Cotman, M.; Brem, G. Analysis of ROH patterns in the Noriker horse breed reveals signatures of selection for coat color and body size. Anim. Genet. 2019, 50, 334–346. [Google Scholar] [CrossRef]
- Talebi, R.; Szmatoła, T.; Mészáros, G.; Qanbari, S. Runs of Homozygosity in Modern Chicken Revealed by Sequence Data. G3 Genes Genomes Genet. 2020, 10, 4615–4623. [Google Scholar] [CrossRef]
- Aderibigbe, A.; Cowieson, A.J.; Sorbara, J.O.; Adeola, O. Growth phase and dietary α-amylase supplementation effects on nutrient digestibility and feedback enzyme secretion in broiler chickens. Poult. Sci. 2020, 99, 6867–6876. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, Y.; Sun, X.; Yin, D.; Wang, Y.; Mahmood, T.; Yuan, J. Effects of exogenous α-(1,4)-amylase on the utilisation of corn starch and glucose metabolism in broiler chickens. Animal 2021, 15, 100396. [Google Scholar] [CrossRef]
- Mir, N.A.; Rafiq, A.; Kumar, F.; Singh, V.; Shukla, V. Determinants of broiler chicken meat quality and factors affecting them: A review. J. Food Sci. Technol. 2017, 54, 2997–3009. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Li, X.; Wang, J.; Sun, G.; Mu, X.; Ji, R. Ovarian transcriptome profile from pre-laying period to broody period of Xupu goose. Poult. Sci. 2021, 100, 101403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhong, H.; Lin, S.; Liang, L.; Ye, S.; Xu, Z.; Ji, C.; Zhang, Z.; Zhang, D.; Zhang, X. Polymorphisms of AMY1A gene and their association with growth, carcass traits and feed intake efficiency in chickens. Genomics 2021, 113, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Banos, G.; Lindsay, V.; Desta, T.T.; Bettridge, J.; Sanchez-Molano, E.; Vallejo-Trujillo, A.; Matika, O.; Dessie, T.; Wigley, P.; Christley, R.M.; et al. Integrating Genetic and Genomic Analyses of Combined Health Data Across Ecotypes to Improve Disease Resistance in Indigenous African Chickens. Front. Genet. 2020, 11, 543890. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Lim, K.S.; Byun, M.; Lee, K.T.; Yang, Y.R.; Park, M.; Lim, D.; Chai, H.H.; Bang, H.T.; Hwangbo, J.; et al. Identification of the acclimation genes in transcriptomic responses to heat stress of White Pekin duck. Cell. Stress. Chaperones 2017, 22, 787–797. [Google Scholar] [CrossRef]
- Renaville, B.; Piasentier, E.; Fan, B.; Vitale, M.; Prandi, A.; Rothschild, M.F. Candidate gene markers involved in San Daniele ham quality. Meat Sci. 2010, 85, 441–445. [Google Scholar] [CrossRef]
- Chen, F.Y.; Niu, H.; Wang, J.Q.; Lei, C.Z.; Lan, X.Y.; Zhang, C.L.; Li, M.J.; Hua, L.S.; Wang, J.; Chen, H. Polymorphism of DLK1 and CLPG gene and their association with phenotypic traits in Chinese cattle. Mol. Biol. Rep. 2011, 38, 243–248. [Google Scholar] [CrossRef]
- Offenhäuser, N.; Borgonovo, A.; Disanza, A.; Romano, P.; Ponzanelli, I.; Iannolo, G.; Di Fiore, P.P.; Scita, G. The eps8 family of proteins links growth factor stimulation to actin reorganization generating functional redundancy in the Ras/Rac pathway. Mol. Biol. Cell. 2004, 15, 91–98. [Google Scholar] [CrossRef]
- Dutta, D.; Lai, K.Y.; Reyes-Ordoñez, A.; Chen, J.; van der Donk, W.A. Lanthionine synthetase C-like protein 2 (LanCL2) is important for adipogenic differentiation. J. Lipid Res. 2018, 59, 1433–1445. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HE | HO | PN | MAF | pi |
|---|---|---|---|---|
| 0.241613 ± 0.162092 | 0.228047 ± 0.154646 | 0.8264 | 0.170253 ± 0.143041 | 0.004268 ± 0.001726 |
| ROH Length (Mb) | ROH Number | Percent (%) | Mean Length (Mb) | Genome Coverage (%) |
|---|---|---|---|---|
| 0–1 | 52,526 | 98.17% | 0.212 ± 0.147 | 88.53% |
| 1–2 | 853 | 1.59% | 1.323 ± 0.264 | 8.97% |
| 2–3 | 111 | 0.21% | 2.302 ± 0.259 | 2.03% |
| >3 | 16 | 0.03% | 3.64 ± 0.600 | 0.46% |
| Total | 53,506 | 100.00% | 0.235 ± 0.232 | 100.00% |
| FROH (<1 MB) | FROH (1–2 MB) | FROH (2–3 MB) | FROH (>3 MB) | FROH (ALL) | FHOM | FGRM |
|---|---|---|---|---|---|---|
| 0.0501 ± 0.0084 | 0.0051 ± 0.0027 | 0.0012 ± 0.0017 | 0.0003 ± 0.001 | 0.0566 ± 0.0100 | 0.0561 ± 0.0205 | 0.0600 ± 0.0144 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, S.; Tang, W.; Zhong, Z.; Wang, Z.; Xie, X.; Liu, H.; Chen, F.; Liu, J.; Han, Y.; Qin, Y.; et al. Identification of Runs of Homozygosity Islands and Functional Variants in Wenchang Chicken. Animals 2023, 13, 1645. https://doi.org/10.3390/ani13101645
Tian S, Tang W, Zhong Z, Wang Z, Xie X, Liu H, Chen F, Liu J, Han Y, Qin Y, et al. Identification of Runs of Homozygosity Islands and Functional Variants in Wenchang Chicken. Animals. 2023; 13(10):1645. https://doi.org/10.3390/ani13101645
Chicago/Turabian StyleTian, Shuaishuai, Wendan Tang, Ziqi Zhong, Ziyi Wang, Xinfeng Xie, Hong Liu, Fuwen Chen, Jiaxin Liu, Yuxin Han, Yao Qin, and et al. 2023. "Identification of Runs of Homozygosity Islands and Functional Variants in Wenchang Chicken" Animals 13, no. 10: 1645. https://doi.org/10.3390/ani13101645
APA StyleTian, S., Tang, W., Zhong, Z., Wang, Z., Xie, X., Liu, H., Chen, F., Liu, J., Han, Y., Qin, Y., Tan, Z., & Xiao, Q. (2023). Identification of Runs of Homozygosity Islands and Functional Variants in Wenchang Chicken. Animals, 13(10), 1645. https://doi.org/10.3390/ani13101645