
Assessing Genetic Variation in Wild and Domesticated Pikeperch Populations: Implications for Conservation and Fish Farming

 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
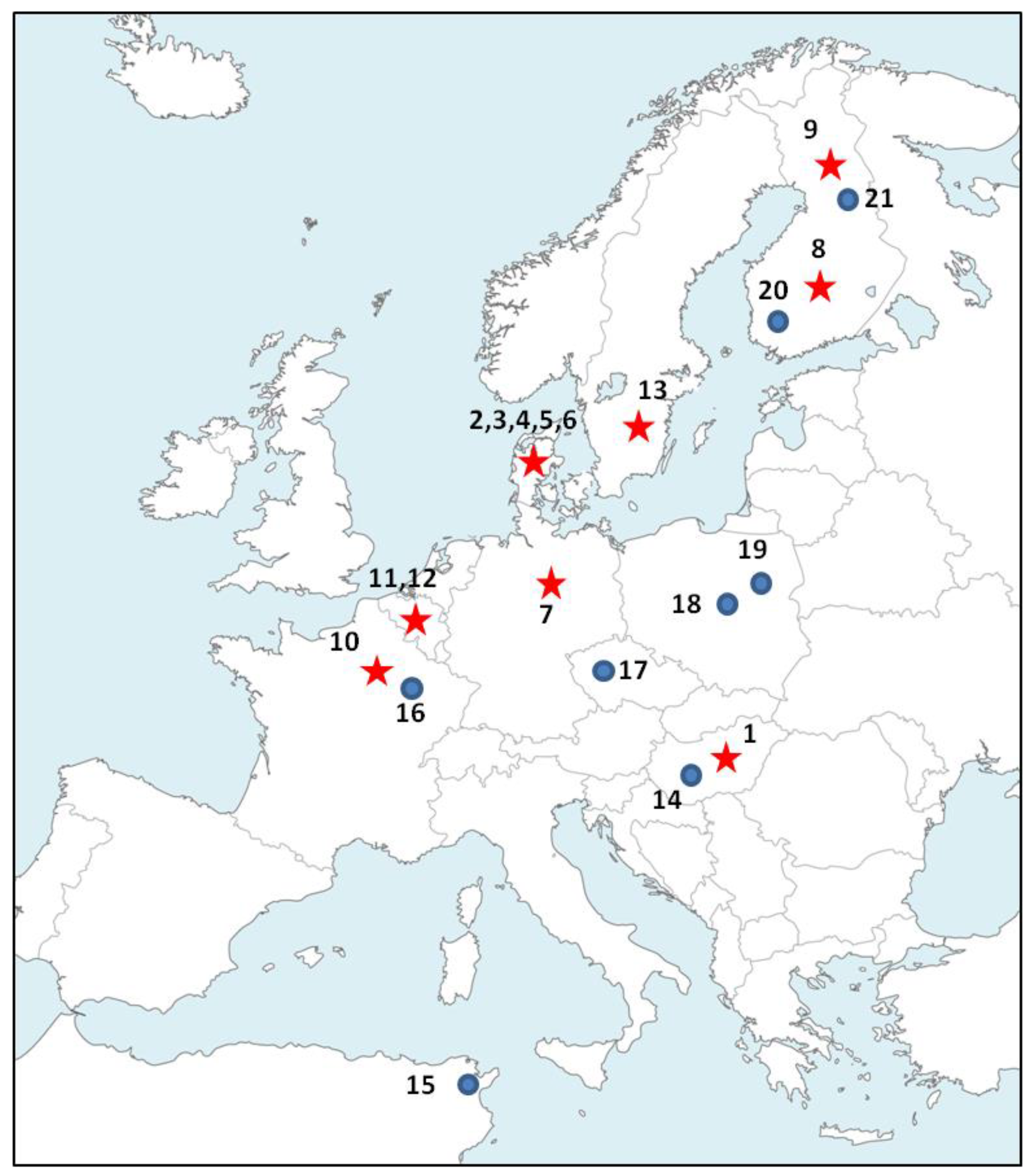
2.1. Biological Material and DNA Extractions
2.2. Microsatellite Genetic Markers and Population Genetics Analyses
2.3. Mitochondrial DNA Sequencing, Sequence Diversity and Phylogeographic Analyses
3. Results
3.1. Polymorphism of Microsatellites
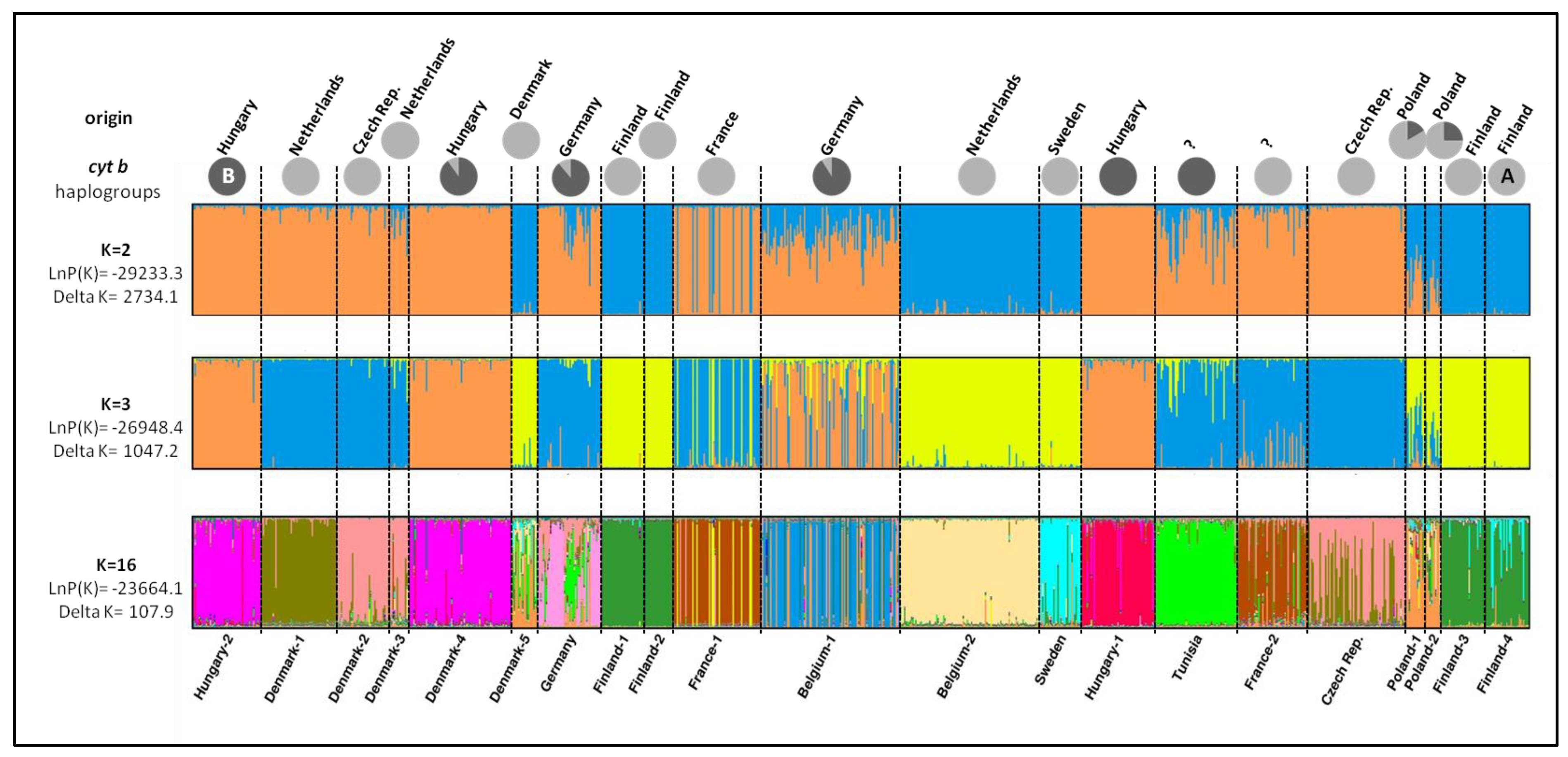
3.2. Population Genetics Analyses
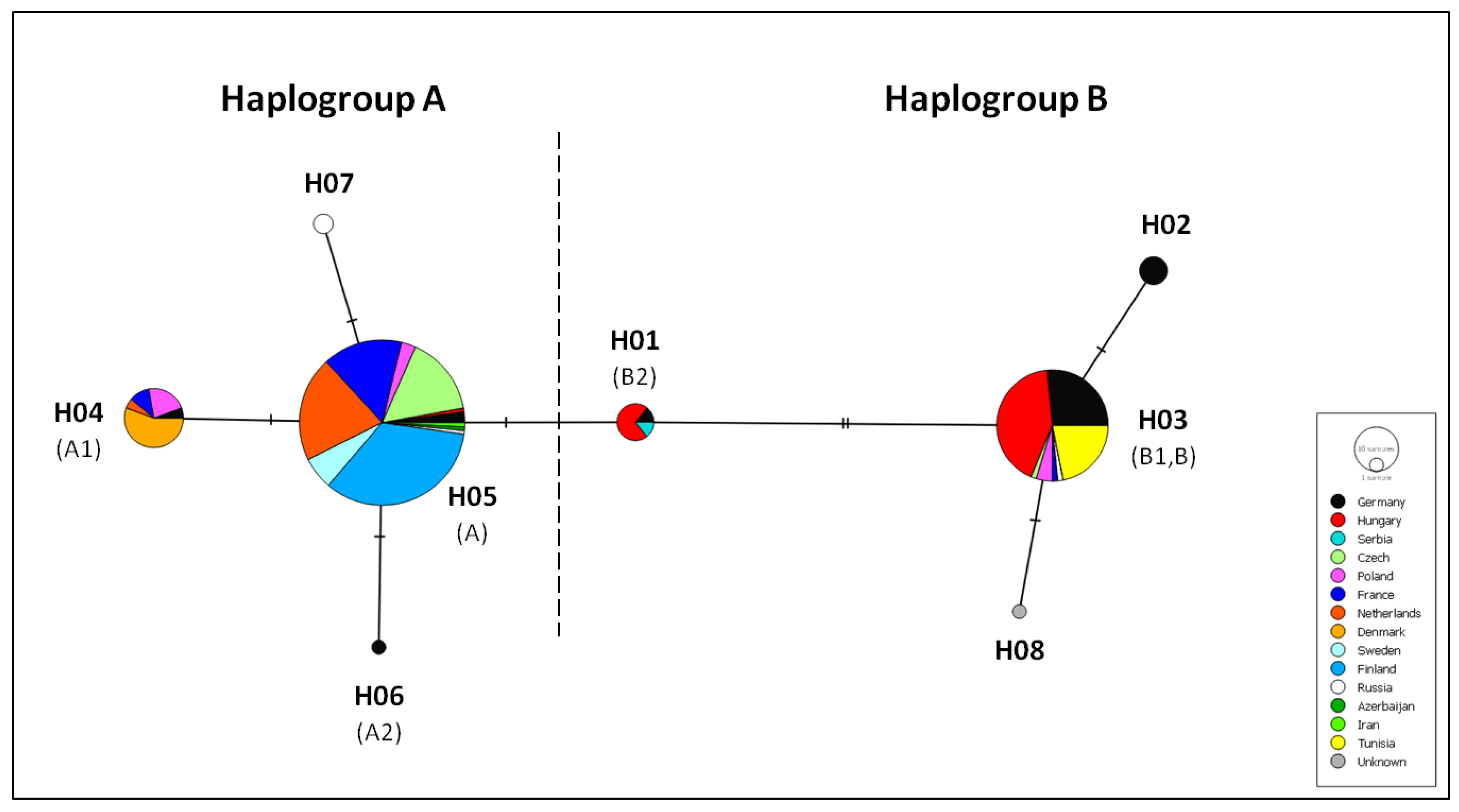
3.3. Cytochrome b Diversity and Phylogeographic Analysis
4. Discussion
4.1. Genetic Diversity of Wild and Domesticated Populations
4.2. Differentiation and Genetic Structure
4.3. Guideline for Future Pikeperch Aquaculture Development
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kestemont, P.; Dabrowski, K.; Summerfelt, R.C. Biology and Culture of Percid Fishes: Principles and Practices; Kestemont, P., Summerfelt, R.C., Dabrowski, K., Eds.; Springer: Dordrecht, The Netherlands, 2015; ISBN 9789401772273. [Google Scholar]
- Haponski, A.E.; Stepien, C.A. Phylogenetic and biogeographical relationships of the sander pikeperches (percidae: Perciformes): Patterns across North America and Eurasia. Biol. J. Linn. Soc. 2013, 110, 156–179. [Google Scholar] [CrossRef]
- Eschbach, E.; Nolte, A.W.; Kohlmann, K.; Kersten, P.; Kail, J.; Arlinghaus, R. Population differentiation of zander (Sander lucioperca) across native and newly colonized ranges suggests increasing admixture in the course of an invasion. Evol. Appl. 2014, 7, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Mustamäki, N.; Bergström, U.; Ådjers, K.; Sevastik, A.; Mattila, J. Pikeperch (Sander lucioperca (L.)) in Decline: High Mortality of Three Populations in the Northern Baltic Sea. Ambio 2014, 43, 325–336. [Google Scholar] [CrossRef] [PubMed]
- FAO. Global Capture Production; FAO FishStat, Fisheries and Aquaculture Department: Electronic Database, 2019. [Google Scholar]
- Colchen, T.; Gisbert, E.; Krauss, D.; Ledoré, Y.; Pasquet, A.; Fontaine, P. Improving pikeperch larviculture by combining environmental, feeding and populational factors. Aquac. Rep. 2020, 17, 100337. [Google Scholar] [CrossRef]
- Poulet, N.; Balaresque, P.; Aho, T.; Björklund, M. Genetic structure and dynamics of a small introduced population: The pikeperch, Sander lucioperca, in the Rhône delta. Genetica 2009, 135, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Salminen, M.; Koljonen, M.L.; Säisä, M.; Ruuhijärvi, J. Genetic effects of supportive stockings on native pikeperch populations in boreal lakes—Three cases, three different outcomes. Hereditas 2012, 149, 1–15. [Google Scholar] [CrossRef]
- Björklund, M.; Aho, T.; Larsson, L.C. Genetic differentiation in pikeperch (Sander lucioperca): The relative importance of gene flow, drift and common history. J. Fish Biol. 2007, 71, 264–278. [Google Scholar] [CrossRef]
- Khurshut, E.; Kohlmann, K. Application of nine species-specific microsatellite loci to characterize three pike-perch (Sander lucioperca) populations from the Aral Sea basin in Uzbekistan. Environ. Biotechnol. 2009, 5, 3–10. [Google Scholar]
- Säisä, M.; Salminen, M.; Koljonen, M.L.; Ruuhijärvi, J. Coastal and freshwater pikeperch (Sander lucioperca) populations differ genetically in the Baltic Sea basin. Hereditas 2010, 147, 205–214. [Google Scholar] [CrossRef][Green Version]
- Mirza Ahmadi, F.; Norouzi, M.; Samiei, M.H.; Behrouz, M. Study of Genetic structure and variation of pikeperch (Sander lucioperca) in Lake of Aras dame and Anzali Wetland. Wetl. Ecobiol. 2015, 6, 83–92. [Google Scholar]
- Kohlmann, K.; Poulet, N.; Louati, M.; Kersten, P.; Ben Hassine, O.; Bahri-Sfar, L. Detection of two major cytochrome b lineages in pike-perch, Sander lucioperca, and first data on their distribution in European populations. Environ. Biotechnol. 2013, 9, 1–5. [Google Scholar]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics, 2nd ed.; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Kánainé Sipos, D.; Kovács, G.; Buza, E.; Csenki-Bakos, K.; Ősz, Á.; Ljubobratović, U.; Cserveni-Szücs, R.; Bercsényi, M.; Lehoczky, I.; Urbányi, B.; et al. Comparative genetic analysis of natural and farmed populations of pike-perch (Sander lucioperca). Aquac. Int. 2019, 27, 991–1007. [Google Scholar] [CrossRef]
- Teletchea, F.; Fontaine, P. Levels of domestication in fish: Implications for the sustainable future of aquaculture. Fish Fish. 2014, 15, 181–195. [Google Scholar] [CrossRef]
- Larson, G.; Burger, J. A population genetics view of animal domestication. Trends Genet. 2013, 29, 197–205. [Google Scholar] [CrossRef]
- Mignon-Grasteau, S.; Boissy, A.; Bouix, J.; Faure, J.-M.; Fisher, A.D.; Hinch, G.N.; Jensen, P.; Le Neindre, P.; Mormède, P.; Prunet, P.; et al. Genetics of adaptation and domestication in livestock. Livest. Prod. Sci. 2005, 93, 3–14. [Google Scholar] [CrossRef]
- Rhymer, J.M.; Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 1996, 27, 83–109. [Google Scholar] [CrossRef]
- Danancher, D.; Garcia-Vazquez, E. Genetic population structure in flatfishes and potential impact of aquaculture and stock enhancement on wild populations in Europe. Rev. Fish Biol. Fish. 2011, 21, 441–462. [Google Scholar] [CrossRef]
- Champagnon, J.; Elmberg, J.; Guillemain, M.; Gauthier-Clerc, M.; Lebreton, J.-D.D. Conspecifics can be aliens too: A review of effects of restocking practices in vertebrates. J. Nat. Conserv. 2012, 20, 231–241. [Google Scholar] [CrossRef]
- Lecocq, T.; Coppée, A.; Michez, D.; Brasero, N.; Rasplus, J.-Y.; Valterová, I.; Rasmont, P. The alien’s identity: Consequences of taxonomic status for the international bumblebee trade regulations. Biol. Conserv. 2016, 195, 169–176. [Google Scholar] [CrossRef]
- Cross, T.F. Genetic implications of translocation and stocking of fish species, with particular reference to Western Australia. Aquac. Res. 2000, 31, 83–94. [Google Scholar] [CrossRef]
- Vehanen, T.; Huusko, A.; Hokki, R. Competition between hatchery-raised and wild brown trout Salmo trutta in enclosures—Do hatchery releases have negative effects on wild populations? Ecol. Freshw. Fish 2009, 18, 261–268. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, D.; Wirth, T.; Bernatchez, L. Isolation and characterization of microsatellite loci in the yellow perch (Perca flavescens), and cross- species amplification within the family Percidae. Mol. Ecol. 2000, 9, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Borer, S.O.; Miller, L.M.; Kapuscinski, A.R. Microsatellites in walleye Stizostedion vitreum. Mol. Ecol. 1999, 8, 336–338. [Google Scholar] [CrossRef]
- Dubut, V.; Grenier, R.; Meglécz, E.; Chappaz, R.; Costedoat, C.; Danancher, D.; Descloux, S.; Malausa, T.; Martin, J.F.; Pech, N.; et al. Development of 55 novel polymorphic microsatellite loci for the critically endangered Zingel asper L. (Actinopterygii: Perciformes: Percidae) and cross-species amplification in five other percids. Eur. J. Wildl. Res. 2010, 56, 931–938. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 1.2): A Computer Program to Calculate F-Statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Lagnel, J. ParaStructure. 2015. Available online: https://github.com/jacqueslagnel/parastructure (accessed on 2 December 2019).
- Rosenberg, N.A. distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Meyer, A.; Bouvetv, J. Unusual mitochondrial DNA polymorphism in two local populations of blue tit Parus caeruleus. Mol. Ecol. 1992, 1, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.B.; Vincent, A.; Ahnesjö, I.; Meyer, A. Male Pregnancy in Seahorses and Pipefishes (Family Syngnathidae): Rapid Diversification of Paternal Brood Pouch Morphology Inferred From a Molecular Phylogeny. J. Hered. 2001, 92, 159–166. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Kalous, L.; Šlechtová, V.; Petrtýl, M.; Kohout, J.; Čech, M. Do small fish mean no voucher? Using a flatbed desktop scanner to document larval and small specimens before destructive analyses. J. Appl. Ichthyol. 2010, 26, 614–617. [Google Scholar] [CrossRef]
- Kahilainen, K.K.; Teacher, A.G.F.; Kähkönen, K.; Vinni, M.; Lehtonen, H.; Merilä, J. First Record of Natural Hybridization and Introgression between Pikeperch (Sander lucioperca) and Perch (Perca fluviatilis). Ann. Zool. Fennici 2011, 48, 39–44. [Google Scholar] [CrossRef]
- Ma, Z.; Bercsenyi, M.; Yang, X.; Wei, K.; Yang, R. The complete mitochondrial genome of pike-perch, Sander lucioperca (Perciformes: Percidae). Mitochondrial DNA Part A 2016, 27, 3135–3136. [Google Scholar] [CrossRef]
- Louati, M.; Kohlmann, K.; Ben Hassine, O.K.; Kersten, P.; Poulet, N.; Bahri-Sfar, L. Genetic characterization of introduced Tunisian and French populations of pike-perch (Sander lucioperca) by species-specific microsatellites and mitochondrial haplotypes. Czech J. Anim. Sci. 2016, 61, 159–171. [Google Scholar] [CrossRef]
- Norris, A.T.; Bradley, D.G.; Cunningham, E.P. Microsatellite genetic variation between and within farmed and wild Atlantic salmon (Salmo salar) populations. Aquaculture 1999, 180, 247–264. [Google Scholar] [CrossRef]
- Robertson, A. The interpretation of genotypic ratios in domestic animal populations. Anim. Sci. 1965, 7, 319–324. [Google Scholar] [CrossRef]
- Seifertová, M.; Bryja, J.; Vyskočilová, M.; Martínková, N.; Šimková, A. Multiple Pleistocene refugia and post-glacial colonization in the European chub (Squalius cephalus) revealed by combined use of nuclear and mitochondrial markers. J. Biogeogr. 2012, 39, 1024–1040. [Google Scholar] [CrossRef]
- Li, D.; Wang, S.; Shen, Y.; Meng, X.; Xu, X.; Wang, R.; Li, J. A multiplex microsatellite PCR method for evaluating genetic diversity in grass carp (Ctenopharyngodon idellus). Aquac. Fish. 2018, 3, 238–245. [Google Scholar] [CrossRef]
- Borrell, Y.J.; Arias-Pérez, A.; Freire, R.; Valdés, A.; Sánchez, J.A.; Méndez, J.; Martínez, D.; López, J.; Carleos, C.; Blanco, G.; et al. Microsatellites and multiplex PCRs for assessing aquaculture practices of the grooved carpet shell Ruditapes decussatus in Spain. Aquaculture 2014, 426–427, 49–59. [Google Scholar] [CrossRef]
- Xu, J.; Feng, J.; Peng, W.; Liu, X.; Feng, J.; Xu, P. Development and evaluation of a high-throughput single nucleotide polymorphism multiplex assay for assigning pedigrees in common carp. Aquac. Res. 2017, 48, 1866–1876. [Google Scholar] [CrossRef]
- Toomey, L.; Fontaine, P.; Lecocq, T. Unlocking the intraspecific aquaculture potential from the wild biodiversity to facilitate aquaculture development. Rev. Aquac. 2020, 12, 2212–2227. [Google Scholar] [CrossRef]
- Orbán, L.; Shen, X.; Phua, N.; Varga, L. Toward Genome-Based Selection in Asian Seabass: What Can We Learn From Other Food Fishes and Farm Animals? Front. Genet. 2021, 12, 506754. [Google Scholar] [CrossRef] [PubMed]
- Granier, S.; Audet, C.; Bernatchez, L. Heterosis and outbreeding depression between strains of young-of-the-year brook trout (Salvelinus fontinalis). Can. J. Zool. 2011, 89, 190–198. [Google Scholar] [CrossRef]
- Fleming, I.A.; Hindar, K.; Mjolnerod, I.B.; Jonsson, B.; Balstad, T.; Lamberg, A. Lifetime success and interactions of farm salmon invading a native population. Proc. R. Soc. B Biol. Sci. 2000, 267, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, P.; Prodöhl, P.; Ferguson, A.; Hynes, R.; Maoiléidigh, N.Ó.; Baker, N.; Cotter, D.; O’Hea, B.; Cooke, D.; Rogan, G.; et al. Fitness reduction and potential extinction of wild populations of Atlantic salmon, Salmo salar, as a result of interactions with escaped farm salmon. Proc. R. Soc. B Biol. Sci. 2003, 270, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Summerfelt, S.T.; Vinci, B.J. Better Management Practices for Recirculating Aquaculture Systems. In Environmental Best Management Practices for Aquaculture; Blackwell Publishing: Ames, IA, USA, 2008; pp. 389–426. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microsatellite Analyses | Cyt b Analysis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Population | Origin | N | Na | HO | uHE | Ar | HWE | FIS | N | nh | h | π% | |
| 1 | Hungary-2 | Hungary | 49 | 7.8 | 0.676 | 0.726 | 5.4 | ns | 0.070 | 10 | 2 | 0.356 | 0.125 |
| 2 | Denmark-1 | Netherlands | 54 | 2.6 | 0.680 | 0.472 | 2.4 | * | −0.446 | 10 | 1 | 0 | 0 |
| 3 | Denmark-2 | Czech Rep. | 38 | 3.3 | 0.488 | 0.468 | 2.7 | ns | −0.044 | 11 | 1 | 0 | 0 |
| 4 | Denmark-3 | Netherlands | 14 | 2.8 | 0.410 | 0.355 | 2.6 | ns | −0.162 | 10 | 1 | 0 | 0 |
| 5 | Denmark-4 | Hungary | 73 | 8.2 | 0.717 | 0.725 | 5.5 | ns | 0.011 | 10 | 2 | 0.200 | 0.105 |
| 6 | Denmark-5 | Denmark | 19 | 3.1 | 0.398 | 0.428 | 2.7 | ns | 0.072 | 10 | 1 | 0 | 0 |
| 7 | Germany | Germany | 46 | 5.7 | 0.550 | 0.563 | 3.9 | ns | 0.023 | 9 | 3 | 0.667 | 0.214 |
| 8 | Finland-1 | Finland | 31 | 3.7 | 0.582 | 0.534 | 3.1 | ns | −0.091 | 11 | 1 | 0 | 0 |
| 9 | Finland-2 | Finland | 20 | 2.8 | 0.603 | 0.487 | 2.6 | * | −0.248 | 10 | 1 | 0 | 0 |
| 10 | France-1 | France | 63 | 5.4 | 0.591 | 0.599 | 3.9 | ns | 0.013 | 10 | 1 | 0 | 0 |
| 11 | Belgium-1 | Germany | 100 | 7.2 | 0.810 | 0.726 | 5.1 | * | −0.116 | 11 | 2 | 0.182 | 0.099 |
| 12 | Belgium-2 | Netherlands | 100 | 4.7 | 0.646 | 0.619 | 3.6 | ns | −0.045 | 10 | 2 | 0.200 | 0.035 |
| 13 | Sweden | Sweden | 30 | 4.4 | 0.582 | 0.534 | 3.6 | ns | −0.090 | 9 | 1 | 0 | 0 |
| 14 | Hungary-1 | Hungary | 53 | 6.2 | 0.747 | 0.690 | 4.9 | ns | −0.084 | 12 | 2 | 0.409 | 0.143 |
| 15 | Tunisiaint | Unknown | 59 | 3.7 | 0.359 | 0.405 | 2.7 | ns | 0.115 | 14 | 1 | 0 | 0 |
| 16 | France-2int | Unknown | 51 | 4.6 | 0.671 | 0.598 | 4.0 | * | −0.122 | 13 | 2 | 0.154 | 0.027 |
| 17 | Czech Rep. | Czech Rep. | 70 | 3.8 | 0.438 | 0.473 | 2.9 | ns | 0.074 | 10 | 1 | 0 | 0 |
| 18 | Poland-1 | Poland | 14 | 4.6 | 0.564 | 0.598 | 4.2 | ns | 0.058 | 6 | 3 | 0.600 | 0.234 |
| 19 | Poland-2 | Poland | 11 | 4.2 | 0.676 | 0.644 | 4.1 | ns | −0.052 | 4 | 2 | 0.500 | 0.350 |
| 20 | Finland-3 | Finland | 32 | 4.8 | 0.600 | 0.604 | 3.6 | ns | 0.008 | 11 | 1 | 0 | 0 |
| 21 | Finland-4 | Finland | 31 | 4.7 | 0.534 | 0.613 | 3.9 | ns | 0.131 | 11 | 1 | 0 | 0 |
| Total/overall | 958 | 15.4 | 0.611 | 0.760 | * | 0.198 | 212 | 5 | 0.544 | 0.249 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsaparis, D.; Lecocq, T.; Kyriakis, D.; Oikonomaki, K.; Fontaine, P.; Tsigenopoulos, C.S. Assessing Genetic Variation in Wild and Domesticated Pikeperch Populations: Implications for Conservation and Fish Farming. Animals 2022, 12, 1178. https://doi.org/10.3390/ani12091178
Tsaparis D, Lecocq T, Kyriakis D, Oikonomaki K, Fontaine P, Tsigenopoulos CS. Assessing Genetic Variation in Wild and Domesticated Pikeperch Populations: Implications for Conservation and Fish Farming. Animals. 2022; 12(9):1178. https://doi.org/10.3390/ani12091178
Chicago/Turabian StyleTsaparis, Dimitrios, Thomas Lecocq, Dimitrios Kyriakis, Katerina Oikonomaki, Pascal Fontaine, and Costas S. Tsigenopoulos. 2022. "Assessing Genetic Variation in Wild and Domesticated Pikeperch Populations: Implications for Conservation and Fish Farming" Animals 12, no. 9: 1178. https://doi.org/10.3390/ani12091178
APA StyleTsaparis, D., Lecocq, T., Kyriakis, D., Oikonomaki, K., Fontaine, P., & Tsigenopoulos, C. S. (2022). Assessing Genetic Variation in Wild and Domesticated Pikeperch Populations: Implications for Conservation and Fish Farming. Animals, 12(9), 1178. https://doi.org/10.3390/ani12091178