
Mitochondrial DNA of Sardinian and North-West Italian Populations Revealed a New Piece in the Mosaic of Phylogeography and Phylogeny of Salariopsis fluviatilis (Blenniidae)


 ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
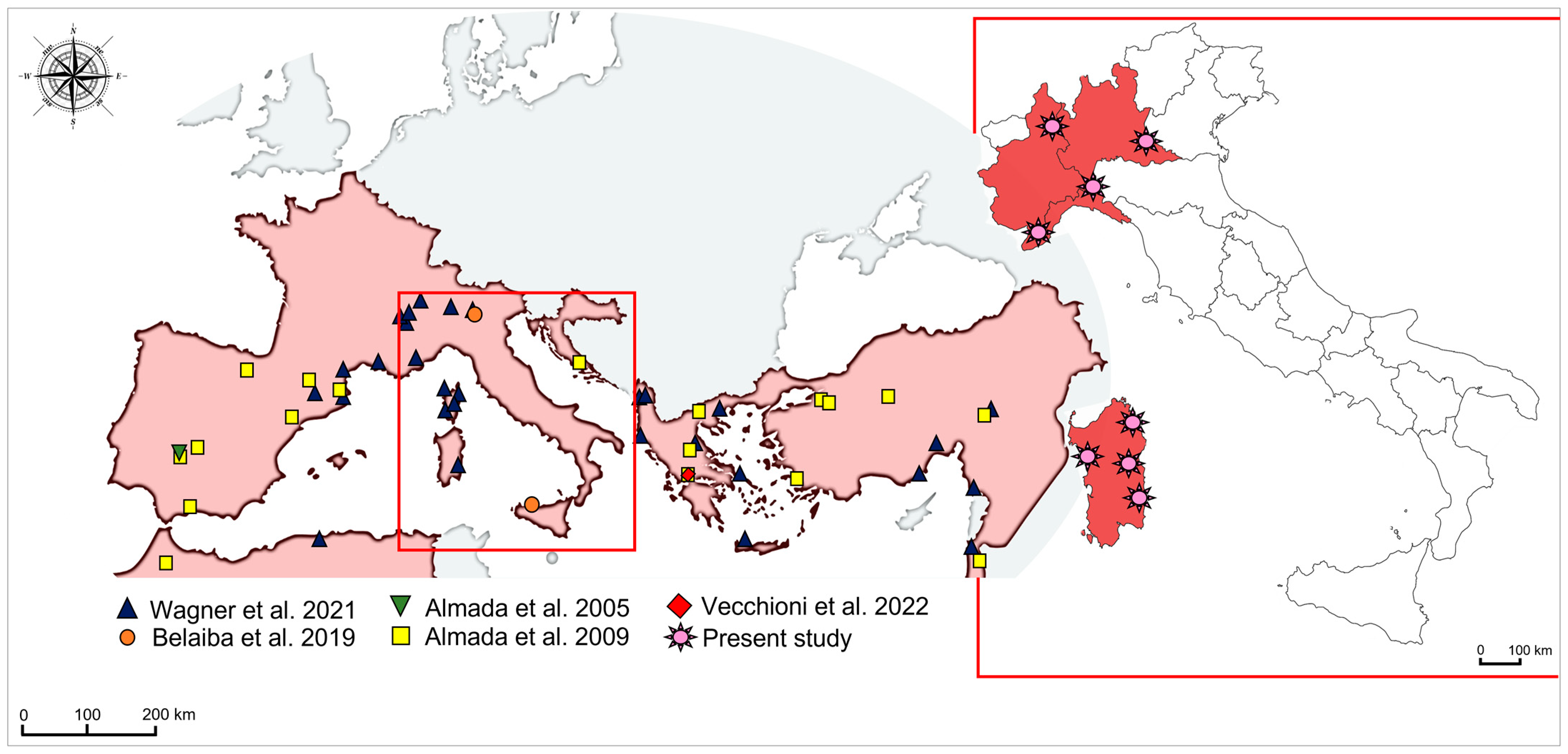
2.1. Sample Collection
2.2. Implemented Molecular Analysis
2.3. Phylogenetic and Phylogeographic Analyses
2.4. Estimation of Divergence Time Analyses
3. Results
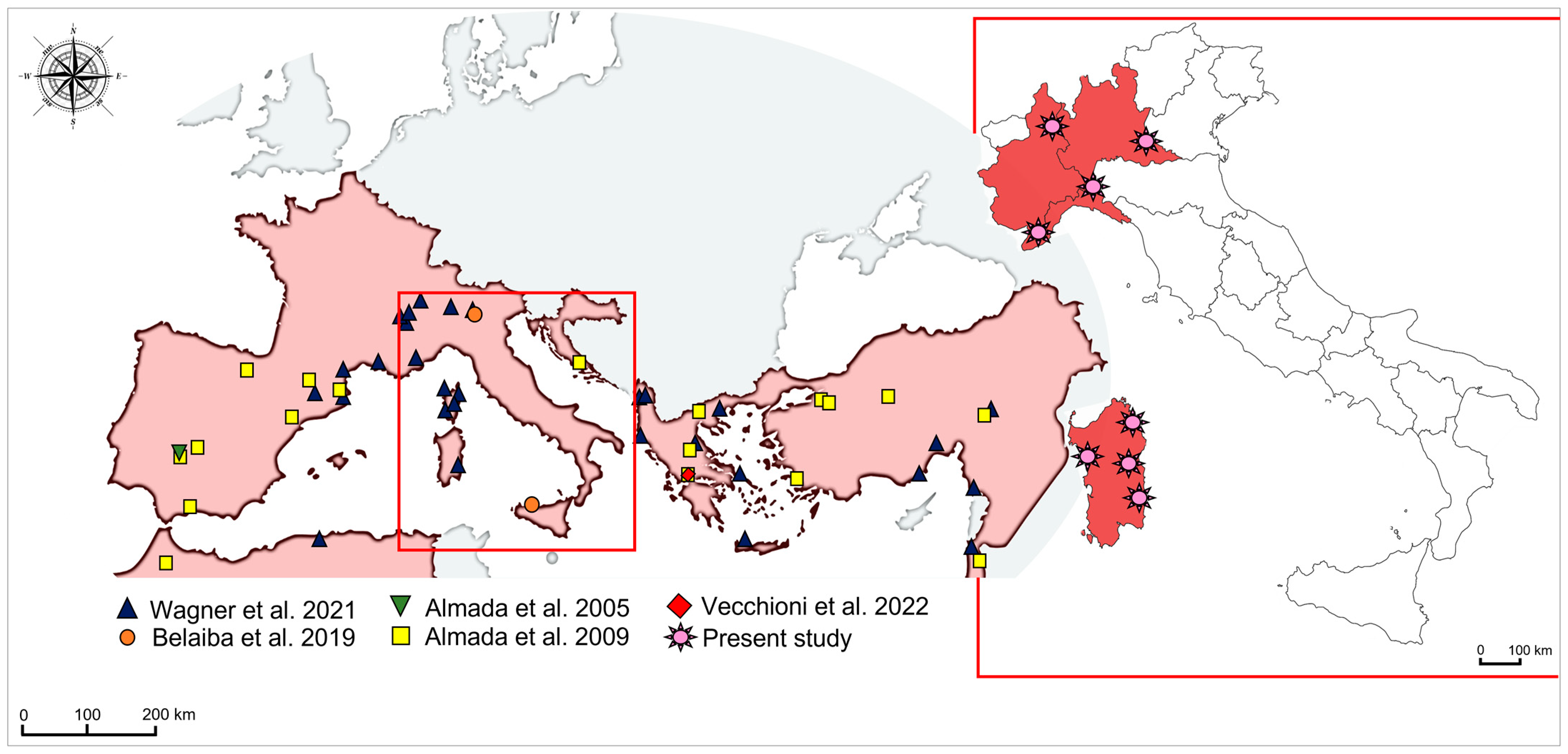
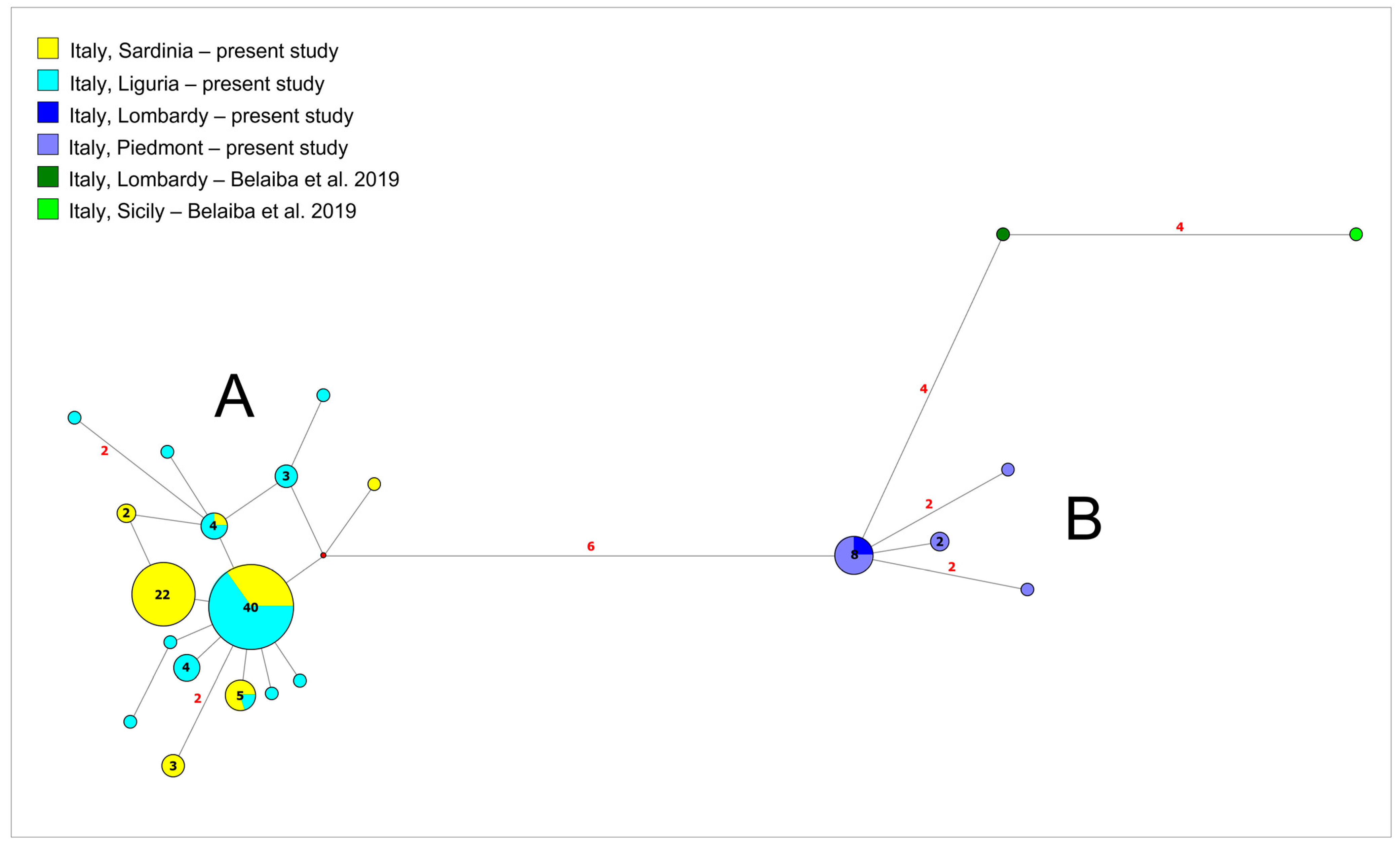
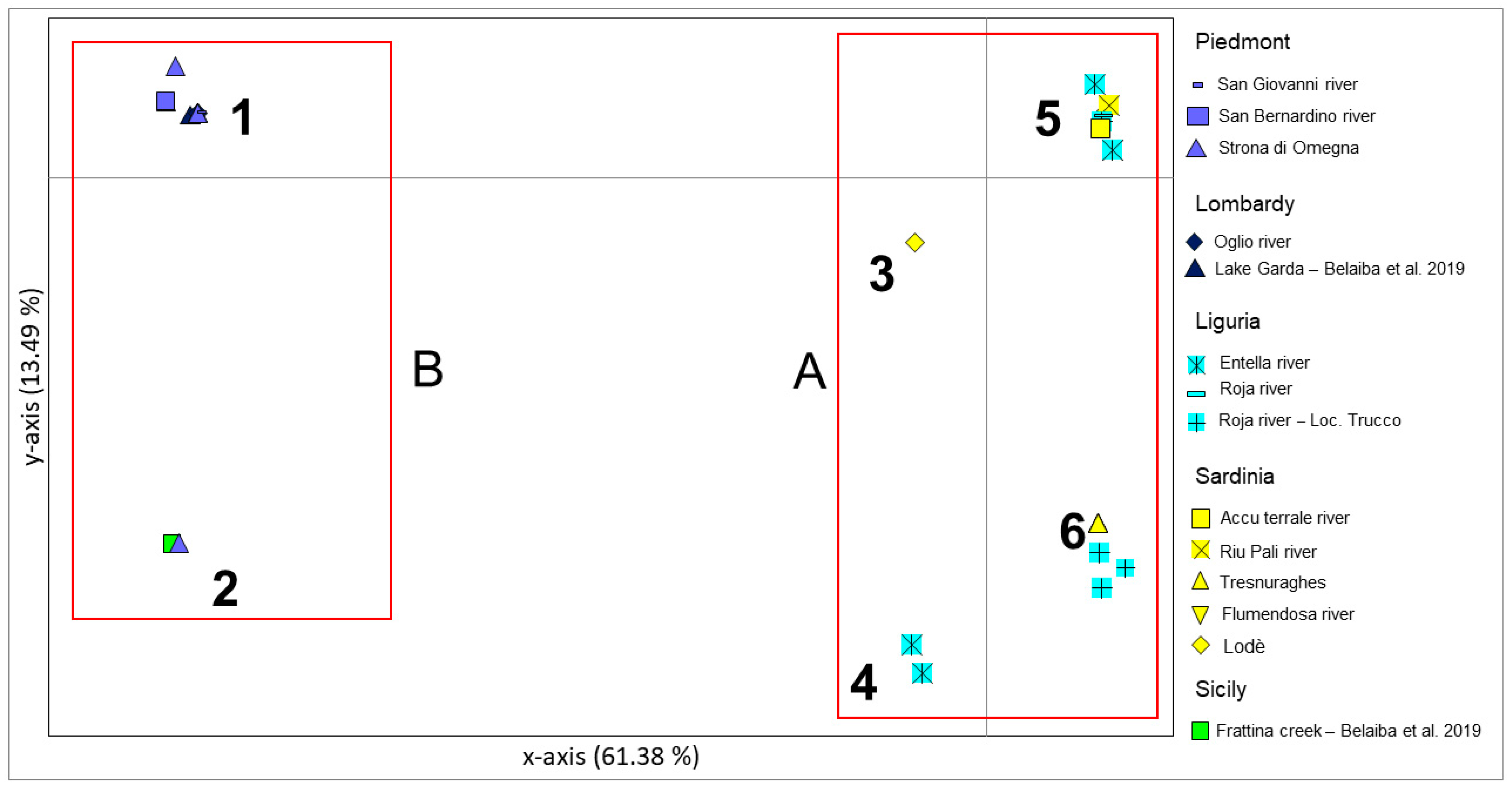
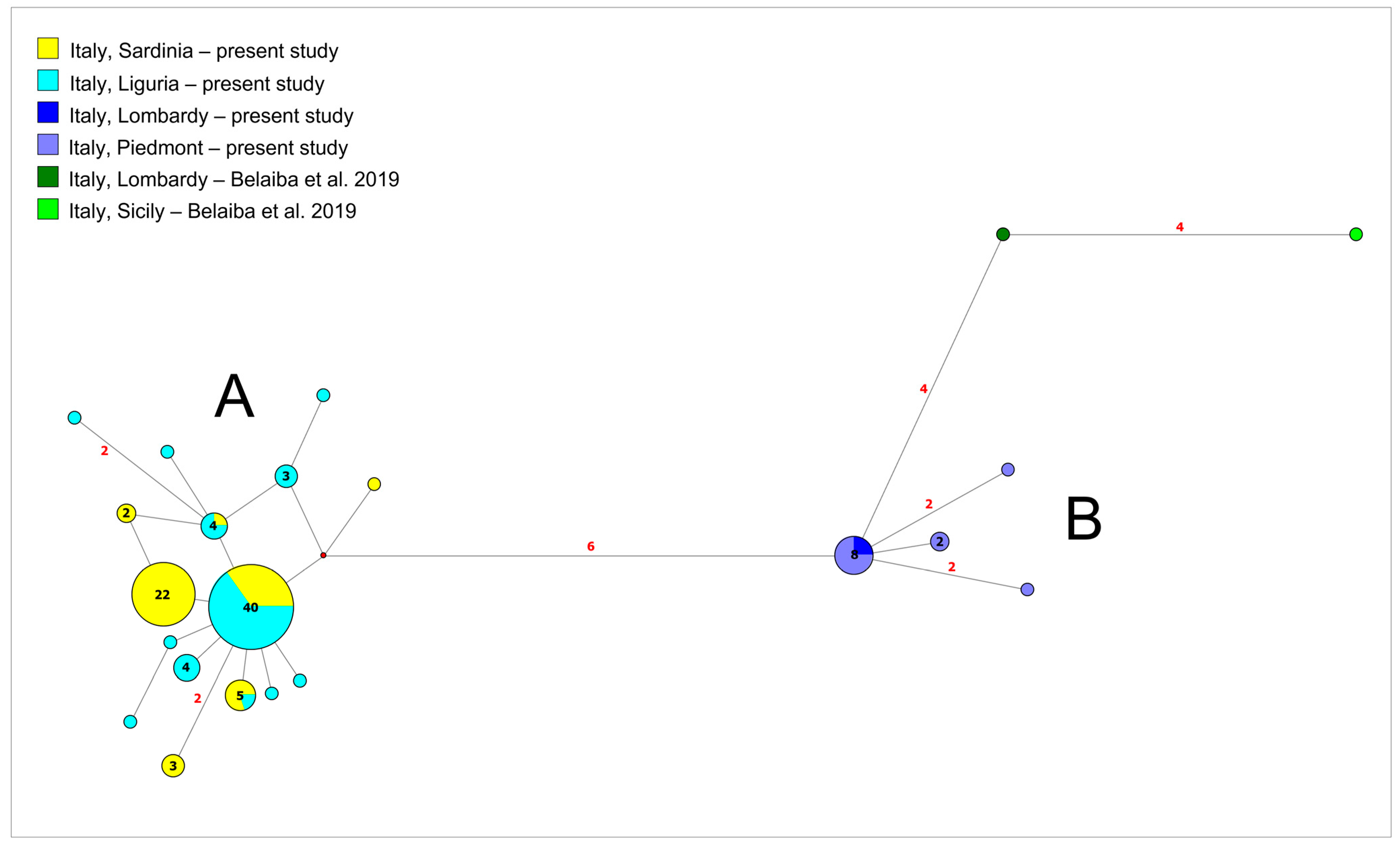
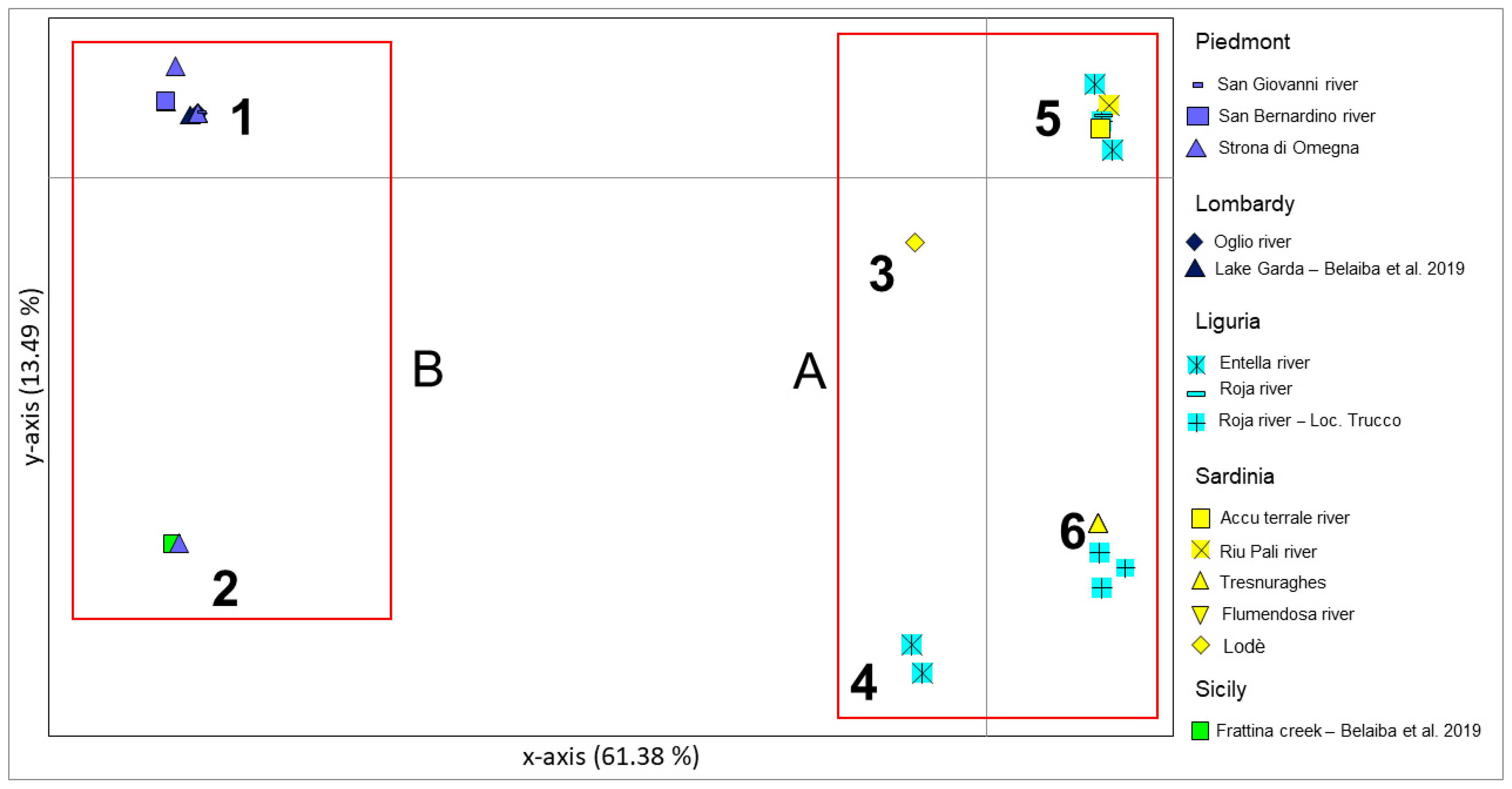
3.1. Phylogeographic Relationships among New Samples from North-West Italy and Sardinia Island
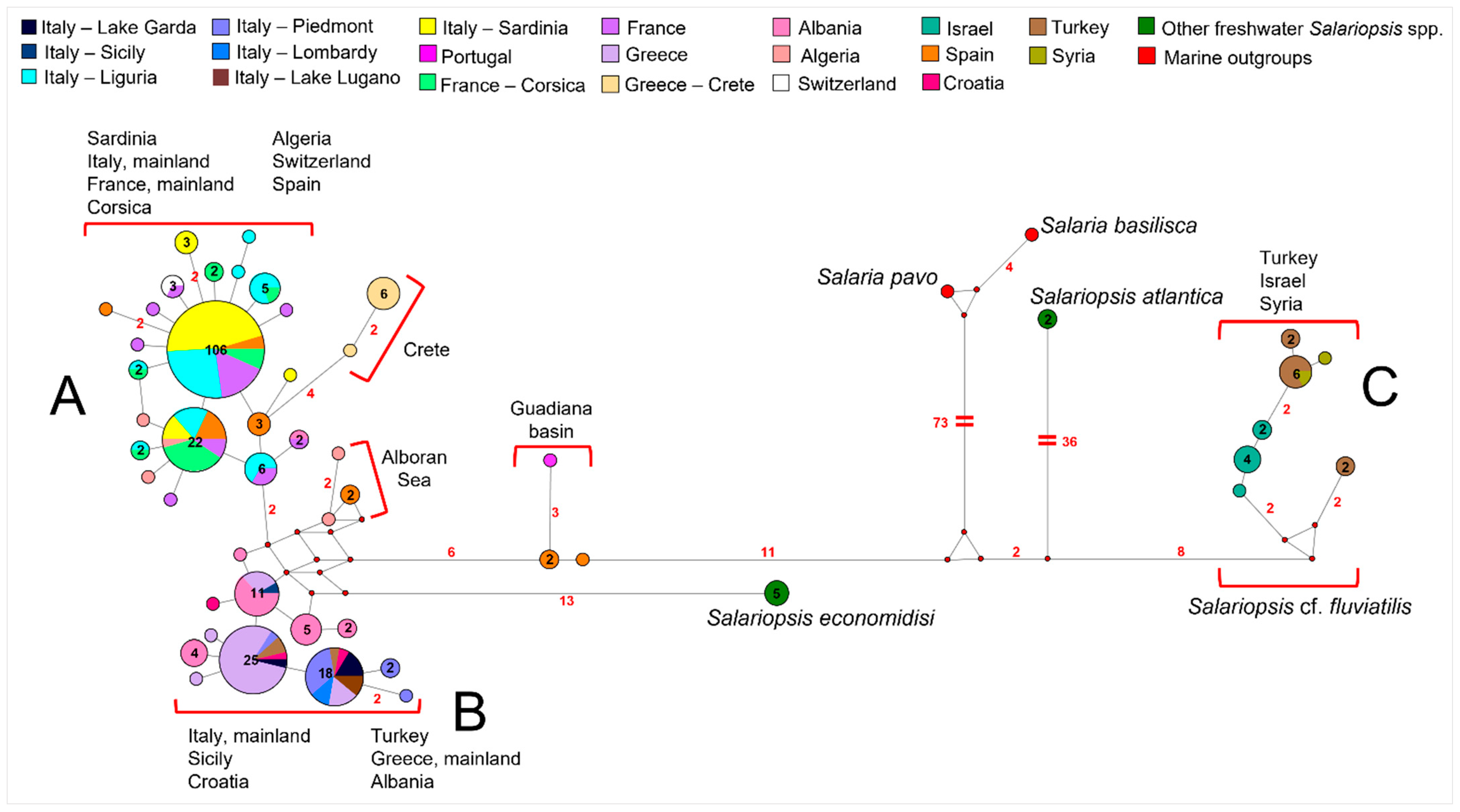
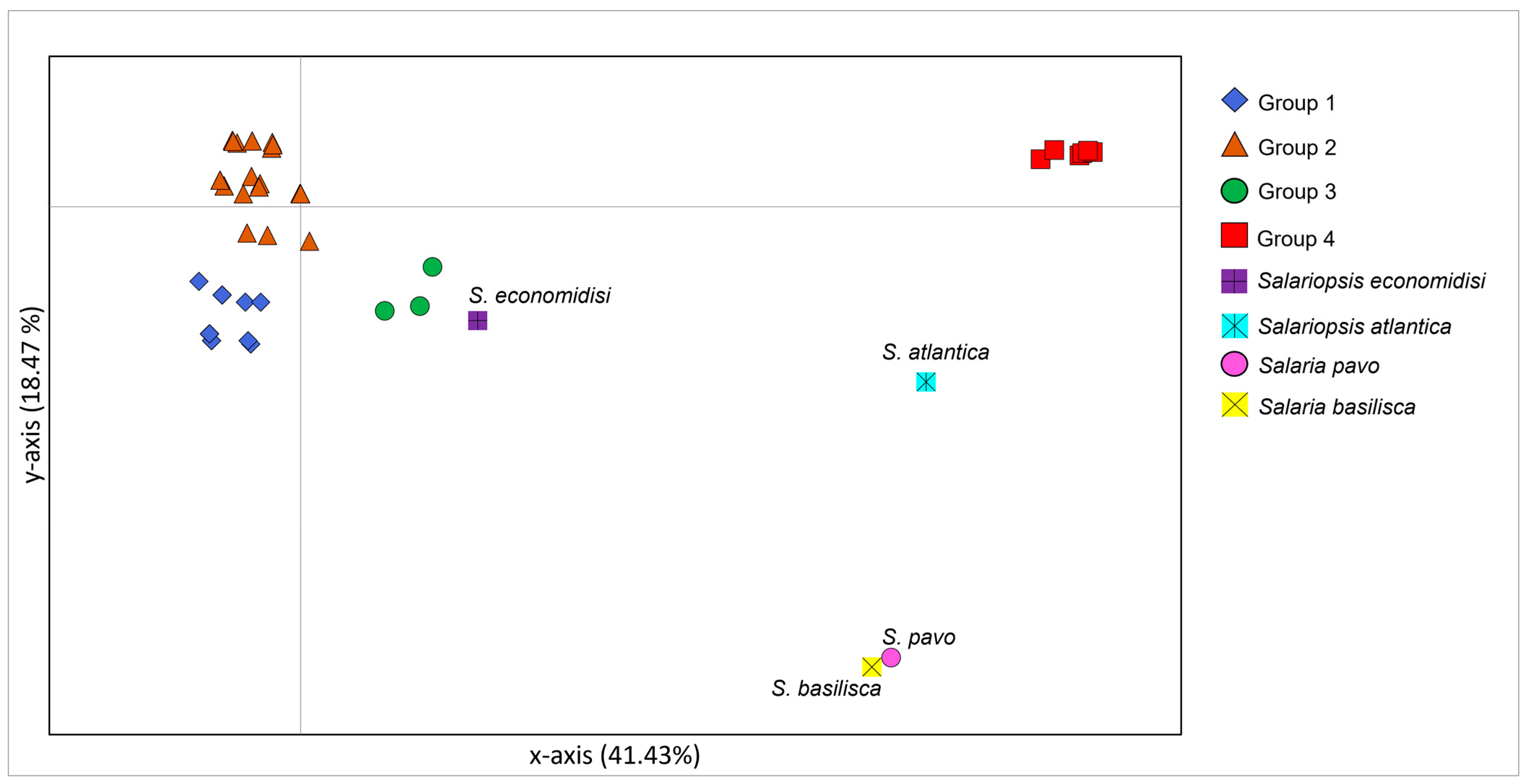
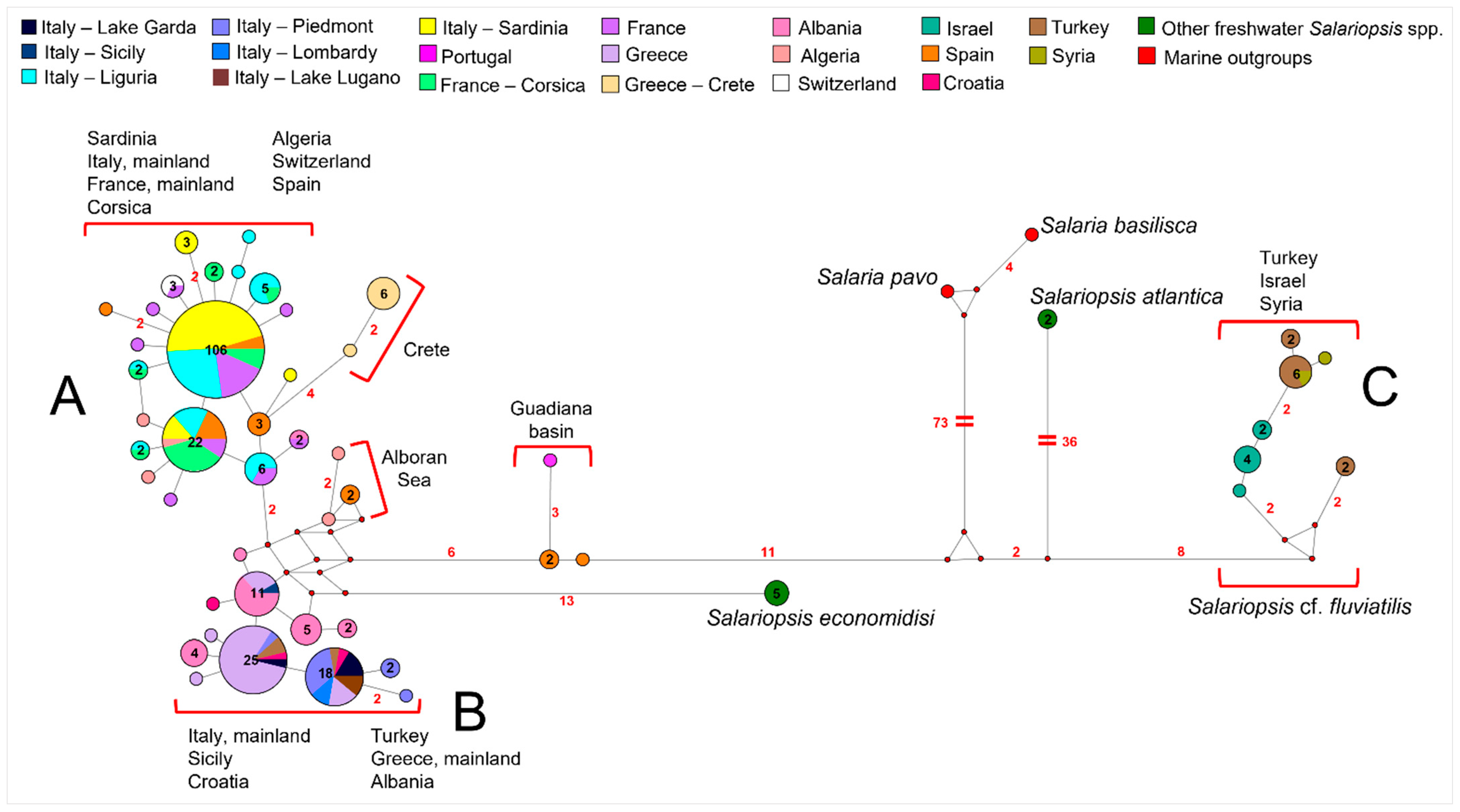
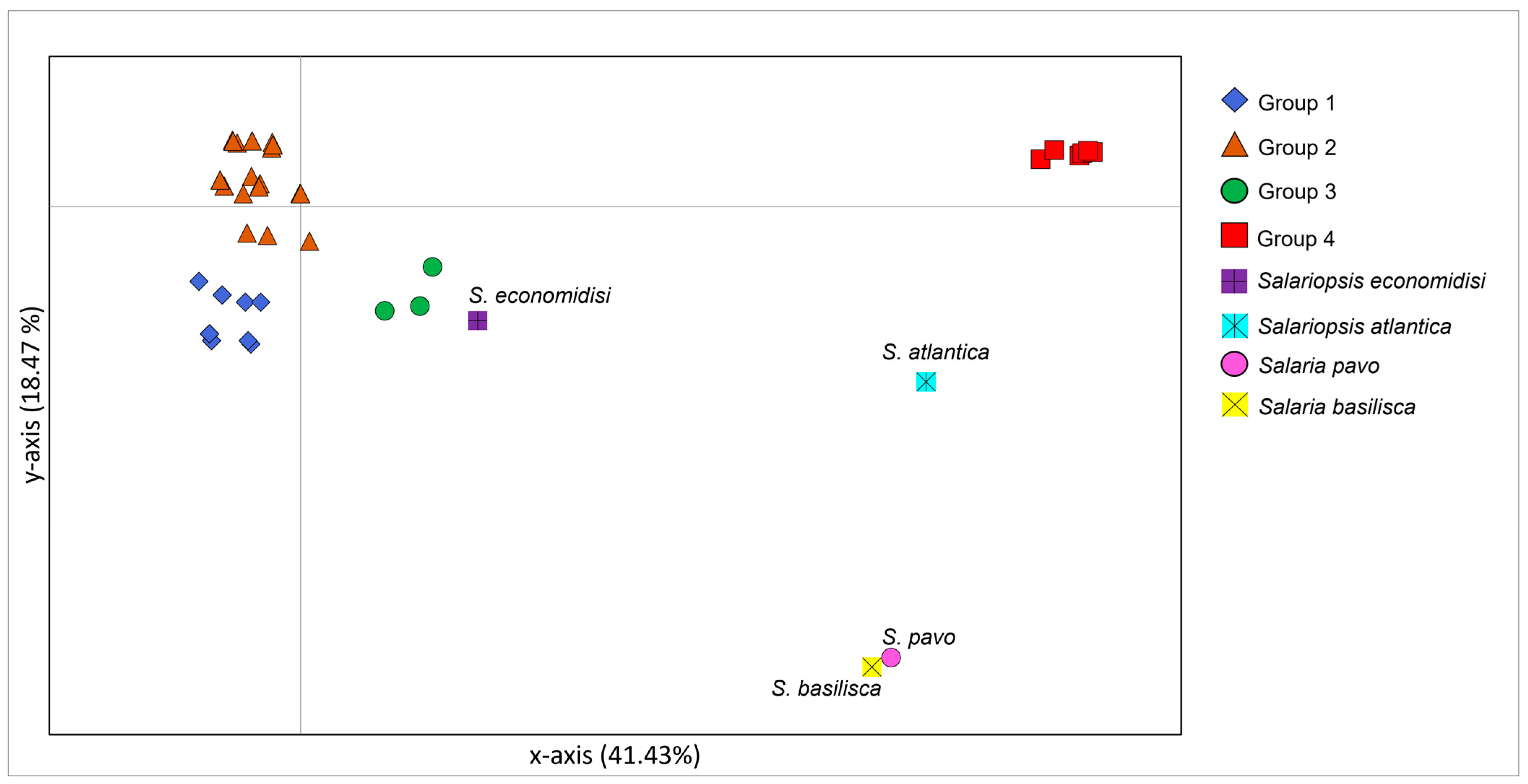
3.2. Phylogenetic Reconstruction and Species Delimitation Based on Control Region
3.3. Phylogenetic Reconstruction Based on 16s Gene
4. Discussion
4.1. Genetic Structuring and Phylogeographic Patterns of Salariopsis fluviatilis in Italy
4.2. Reconstruction of Phylogeography of Salariopsis fluviatilis in the Sardinia Island
4.3. Uprising of Distinct Taxonomic Entities within Salariopsis fluviatilis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vecchioni, L.; Ching, A.C.; Marrone, F.; Arculeo, M.; Hundt, P.J.; Simons, A.M. Multi-locus phylogenetic analyses of the Almadablennius clade reveals inconsistencies with the present taxonomy of blenniid fishes. Diversity 2022, 14, 53. [Google Scholar] [CrossRef]
- Kottelat, M. Salaria economidisi, a new species of freshwater fish from Lake Trichonis, Greece, with comments on variation in S. fluviatilis (Teleostei: Blenniidae). Rev. Suisse Zool. 2004, 111, 121–137. [Google Scholar] [CrossRef]
- Zander, C.D. Blenniidae. In Fishes of the North-Eastern Atlantic and the Mediterranean; Whitehead, P.J.P., Bauchot, M.-L., Hureau, J.-C., Nielsen, J., Tortonese, E., Eds.; UNESCO: Paris, France, 1986; pp. 1096–1112. [Google Scholar]
- Duquenne-Delobel, E.; Doadrio, I.; Denys, G.P. Revalidation of the genus Ichthyocoris Bonaparte, 1840 (Actinopterygii: Blenniiformes: Blenniidae). Acta Ichthyol. Piscat. 2022, 52, 35–41. [Google Scholar] [CrossRef]
- Vecchioni, L.; Arculeo, M.; Hundt, P.J.; Marrone, F. The valid genus name of the European freshwater blennies, Ichthyocoris or Salariopsis (Teleostei: Blenniidae)? Zootaxa 2022, 5162, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Neat, F.C.; Lengkeek, W.; Westerbeek, E.P.; Laarhoven, B.; Videler, J.J. Behavioural and morphological differences between lake and river populations of Salaria fluviatilis. J. Fish Biol. 2003, 63, 374–387. [Google Scholar] [CrossRef]
- Perdices, A.; Doadrio, I.; Côté, I.M.; Machordom, A.; Economidis, P.; Reynolds, J.D. Genetic divergence and origin of Mediterranean populations of the river blenny Salaria fluviatilis (Teleostei: Blenniidae). Copeia 2000, 2000, 723–731. [Google Scholar] [CrossRef]
- Almada, V.C.; Robalo, J.I.; Levy, A.; Freyhof, J.; Bernardi, G.; Doadrio, I. Phylogenetic analysis of peri-Mediterranean blennies of the genus Salaria: Molecular insights on the colonization of freshwaters. Mol. Phylogenet. Evol. 2009, 52, 424–431. [Google Scholar] [CrossRef]
- Laporte, M.; Perrier, C.; Magnan, P.; Berrebi, P. Genetic evidence of recent migration among isolated-by-sea populations of the freshwater blenny (Salaria fluviatilis). Conserv. Genet. 2016, 17, 389–399. [Google Scholar] [CrossRef]
- Plaut, I. Comparison of salinity tolerance and osmoregulation in two closely related species of blennies from different habitats. Fish Physiol. Biochem. 1998, 19, 181–188. [Google Scholar] [CrossRef]
- Vinyoles, D.; Côté, I.M.; De Sostoa, A. Nest orientation patterns in Salaria fluviatilis. J. Fish Biol. 2002, 61, 405–416. [Google Scholar] [CrossRef]
- Magnan, P.; Proulx, R.; Berrebi, P.; Blondel, J.; Perret, P.; Roché, B. Morphological variation in the freshwater blenny Salaria fluviatilis from Corsican rivers: Adaptive divergence, phenotypic plasticity or both? J. Fish Biol. 2014, 84, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Claude, J.; Berrebi, P.; Perret, P.; Magnan, P. Shape plasticity in response to water velocity in the freshwater blenny Salaria fluviatilis. J. Fish Biol. 2016, 88, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Elvira, B.; Nicola, G.G.; Ayllón, D.; Almodóvar, A. Determinants of large-scale spatial distribution and seasonal microhabitat selection patterns of the endangered freshwater blenny Salaria fluviatilis in the Ebro River basin, Spain. Aquat. Conserv. Mar. Freshw. Ecosyst. 2021, 31, 3261–3275. [Google Scholar] [CrossRef]
- Belaiba, E.; Marrone, F.; Vecchioni, L.; Bahri-Sfar, L.; Arculeo, M. An exhaustive phylogeny of the combtooth blenny genus Salaria (Pisces, Blenniidae) shows introgressive hybridization and lack of reciprocal mtDNA monophyly between the marine species Salaria basilisca and Salaria pavo. Mol. Phylogenet. Evol. 2019, 135, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Zogaris, S.; Berrebi, P.; Freyhof, J.; Koblmüller, S.; Magnan, P.; Laporte, M. Diversity and biogeography of Mediterranean freshwater blennies (Blenniidae, Salaria). Divers. Distrib. 2021, 27, 1832–1847. [Google Scholar] [CrossRef]
- Doadrio, I.; Perea, S.; Yahyaoui, A. Morphological and molecular analyses of freshwater blennids: A new species of the genus Salaria Forsskål, 1775 (Actinopterygii, Blennidae) in Morocco. Graellsia 2011, 67, 151–173. [Google Scholar] [CrossRef] [Green Version]
- Cote, I.M.; Vinyoles, D.; Reynolds, J.D.; Doadrio, I.; Perdices, A. Potential impacts of gravel extraction on Spanish populations of river blennies Salaria fluviatilis (Pisces, Blenniidae). Biol. Conserv. 1999, 87, 359–367. [Google Scholar] [CrossRef]
- Hernandez, R.; Lacomba, R.T.; Uviñtas, Y.N.; Oltra, R. Distribution pattern of river blennies in the Júcar River basin (eastern Spain). J. Fish Biol. 2000, 57, 250–254. [Google Scholar] [CrossRef]
- Méndez, L.; Perdices, A.; Machordom, A. Genetic structure and diversity of the Iberian populations of the freshwater blenny Salaria fluviatilis (Asso, 1801) and its conservation implications. Conserv. Genet. 2019, 20, 1223–1236. [Google Scholar] [CrossRef]
- Freeman, M.C.; Viñolas, D.; Grossman, G.D.; De Sostoa, A. Microhabitat use by Blennius fluviatilis in the Río Matarraña, Spain. Freshw. Biol. 1990, 24, 335–345. [Google Scholar] [CrossRef]
- Alp, A.; Kara, C. Distribution pattern and morphological differences between the sexes of river Blenny, Salaria fluviatilis (Asso, 1801), in the Ceyhan river basin, Turkey. Turk. J. Zool. 2007, 31, 113–120. [Google Scholar]
- Tutman, P.; Glamuzina, B. Detection and Confirmation of Freshwater Blenny Salaria fluviatilis (Actinopterygii: Blenniidae) in Bosnia and Herzegovina. Croat. J. Fish. 2021, 79, 75–82. [Google Scholar] [CrossRef]
- Almada, F.; Almada, V.C.; Guillemaud, T.; Wirtz, P. Phylogenetic relationships of the north-eastern Atlantic and Mediterranean blenniids. Biol. J. Linn. Soc. 2005, 86, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Laporte, M.; Leblois, R.; Coulon, A.; Bonhomme, F.; Magnan, P.; Berrebi, P. Genetic structure of a vulnerable species, the freshwater blenny (Salaria fluviatilis). Conserv. Genet. 2015, 16, 571–581. [Google Scholar] [CrossRef]
- Zava, B.; Violani, C. Contributi alla conoscenza dell’ittiofauna delle acque interne siciliane. I. Sulla presenza in Sicilia di Salaria fluviatilis (Asso, 1801) (Pisces, Blenniidae). Boll. Mus. Reg. Sci. Nat. Torino 1991, 9, 313–324. [Google Scholar]
- Gallo, L.; Lucadamo, L.; Mezzotero, A.; Morisi, A.; Battegazzore, M.; Fenoglio, S. First data on the freshwater fish fauna of Calabria (southern Italy). Ital. J. Zool. 2012, 79, 246–251. [Google Scholar] [CrossRef]
- Lorenzoni, M.; Borghesan, F.; Carosi, A.; Ciuffardi, L.; De Curtis, O.; Delmastro, G.; Di Tizio, L.; Franzoi, P.; Maio, G.; Mojetta, A.; et al. Check-list dell’ittiofauna delle acque dolci italiane. Italy J. Freshw. Ichthyol. 2019, 5, 239–254. [Google Scholar]
- Fricke, R.; Eschmeyer, W.N.; Van der Laan, R. Catalog of Fishes: Genera, Species, References; California Academy of Sciences: San Francisco, CA, USA, 2018. [Google Scholar]
- Orrù, F.; Deiana, A.M.; Cau, A. Introduction and distribution of alien freshwater fishes on the island of Sardinia (Italy): An assessment on the basis of existing data sources. J. Appl. Ichthyol. 2010, 26, 46–52. [Google Scholar] [CrossRef]
- Orrù, F.; Deiana, A.M.; Cau, A. Conservazione della biodiversità e specie alloctone invasive: Il gambero Procambarus clarkii nelle acque interne della Sardegna. Studi Trentini Sci. Nat. 2009, 86, 162. [Google Scholar]
- Orrù, F.; Niffoi, A.; Sanna, D.; Varcasia, A.; Buscarinu, P.; Casu, M. Prima segnalazione di Leucos aula (Teleostei: Cyprinidae) in Sardegna basata su analisi morfologiche e DNA barcoding. Italy J. Freshw. Ichthyol. 2019, 5, 166–172. [Google Scholar]
- Casu, M.; Scarpa, F.; Cossu, P.; Lai, T.; Curini-Galletti, M.; Varcasia, A.; Sanna, D. First record of Esox cisalpinus (Teleostea: Esocidae) in Sardinia with insight on its mitochondrial DNA genetic variability. Ital. J. Zool. 2016, 83, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Sanna, D.; Azzena, I.; Scarpa, F.; Cossu, P.; Pira, A.; Gagliardi, F.; Casu, M. First Record of the Alien Species Procambarus virginalis Lyko, 2017 in FreshWaters of Sardinia and Insight into Its Genetic Variability. Life 2021, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Nabholz, B.; Glémin, S.; Galtier, N. Strong variations of mitochondrial mutation rate across mammals—The longevity hypothesis. Mol. Biol. Evol. 2008, 25, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Wan, Q.H.; Wu, H.; Fujihara, T.; Fang, S.G. Which genetic marker for which conservation genetics issue? Electrophoresis 2004, 25, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Primmer, C.R. From conservation genetics to conservation genomics. Ann. N. Y. Acad. Sci. 2009, 1162, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Nabholz, B.; Glémin, S.; Hurst, G.D. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Biologici, I.G.D.L.M. Metodi Biologici per Le Acque Superficiali Interne. In Manuali e Linee Guida; ISPRA-Settore Editoria: Roma, Italy, 2014; ISBN 978-88-448-0651. [Google Scholar]
- Sanna, D.; Scarpa, F.; Lai, T.; Cossu, P.; Falautano, M.; Castriota, L.; Andaloro, F.; Follesa, M.C.; Francalacci, P.; Curini-Galletti, M.; et al. Fistularia commersonii (Teleostea: Fistulariidae): Walking through the Lessepsian paradox of mitochondrial DNA. Ital. J. Zool. 2015, 82, 499–512. [Google Scholar] [CrossRef]
- Sanna, D.; Biagi, F.; Alaya, H.B.; Maltagliati, F.; Addis, A.; Romero, A.; De Juan, J.; Quignard, J.P.; Castelli, A.; Franzoi, P.; et al. Mitochondrial DNA variability of the pipefish Syngnathus abaster. J. Fish Biol. 2013, 82, 856–876. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. In Multiple Sequence Alignment Methods; Russell, D., Ed.; Humana Press: Totowa, NJ, USA, 2014; Volume 1079, pp. 105–116. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; Von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, F.; Cossu, P.; Sanna, D.; Lai, T.; Casu, M.; Curini-Galletti, M. New insights on the genus Otoplana Du Plessis, 1889 (Platyhelminthes: Proseriata), with description of two new species from the Canary Islands. Mar. Biodivers. 2019, 49, 2075–2087. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Cossu, P.; Lai, T.; Casu, M.; Curini-Galletti, M. How to achieve internal fertilization without a vagina: The study case of the genus Archilina Ax, 1959 (Platyhelminthes, Proseriata) from Canary Islands. Mar. Biodivers. 2019, 49, 2057–2073. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Scarpa, F.; Cossu, P.; Sanna, D.; Lai, T.; Norenburg, J.L.; Curini-Galletti, M.; Casu, M. An 18S and 28S-based clock calibration for marine Proseriata (Platyhelminthes). J. Exp. Mar. Biol. Ecol. 2015, 463, 22–31. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Gelman, A.; Rubin, D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992, 7, 457–472. [Google Scholar] [CrossRef]
- Ezard, T.H.; Pearson, P.N.; Purvis, A. Algorithmic approaches to aid species’ delimitation in multidimensional morphospace. BMC Evol. Biol. 2010, 10, 175. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, F.; Cossu, P.; Lai, T.; Sanna, D.; Curini-Galletti, M.; Casu, M. Meiofaunal cryptic species challenge species delimitation: The case of the Monocelis lineata (Platyhelminthes: Proseriata) species complex. Contrib. Zool. 2016, 85, 123–145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [Green Version]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2020, 21, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Cossu, P.; Delogu, V.; Lai, T.; Sanna, D.; Leasi, F.; Norenburg, J.L.; Curini-Galletti, M.; Casu, M. Molecular support for morphology-based family-rank taxa: The contrasting cases of two families of Proseriata (Platyhelminthes). Zool. Scr. 2017, 46, 753–766. [Google Scholar] [CrossRef]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; Dewaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Tran Thi, G.; Azzena, I.; Scarpa, F.; Cossu, P.; Danh Le, C.; Ton Nu, P.A.; Chau Ngo, T.M.; Sanna, D.; Casu, M. Molecular Identification and Appraisal of the Genetic Variation of Taenia saginata in Central Regions of Vietnam. Life 2022, 12, 70. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, e214. [Google Scholar] [CrossRef] [Green Version]
- Koblmüller, S.; Steinwender, B.; Weiß, S.; Sefc, K.M. Gene flow, population growth and a novel substitution rate estimate in a subtidal rock specialist, the black-faced blenny Tripterygion delaisi (Perciformes, Blennioidei, Tripterygiidae) from the Adriatic Sea. J. Zool. Syst. Evol. 2015, 53, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6, 2014. Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 19 October 2022).
- Strimmer, K.; von Haeseler, A. Quartet Puzzling: A quartet Maximum-Likelihood method for reconstructing tree topologies. Mol. Biol. Evol. 1996, 13, 964–969. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; von Haeseler, A. Phylogenetic inference using maximum likelihood methods. In The Phylogenetic Handbook, 5th ed.; Lemey, P., Salemi, M., Vandamme, A.M., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 181–209. [Google Scholar]
- Hwang, U.W.; Kim, W. General properties and phylogenetic utilities of nuclear ribosomal DNA and mitochondrial DNA commonly used in molecular systematics. KJP 1999, 37, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, F.; Galli, P.; Zaccara, S.; Crosa, G. Genetic variability and phylogeography of the cyprinid Telestes muticellus within the Italian peninsula as revealed by mitochondrial DNA. J. Zool. Syst. Evol. Res. 2004, 42, 323–331. [Google Scholar] [CrossRef]
- Ciuffardi, L.; Maldini, M.; Balduzzi, A.; Arillo, A. Caratterizzazione meristica e genetica di Ciprinidi atipici presenti nel bacino spezzino del Magra-Vara-Atti XII Conv. naz. A.I.I.A.D., San Michele all’Adige (TN). Studi Trentini Sci. Nat.-Acta Biol. 2010, 87, 171–174. [Google Scholar]
- Ciuffardi, L.; Balduzzi, A.; Nonnis Marzano, F.; Arillo, A. Analisi Morfometrica di Leuciscini Atipici Presenti Nel Bacino del Vara-Magra (La Spezia, Italia Nord-Occidentale) (Pisces, Osteichthyes, Cyprinidae); Doriana: Genova, Italy, 2011; Volume 8, pp. 1–10. [Google Scholar]
- Ciuffardi, L.; Monaci, E.; Civitella, D.; Balduzzi, A.; Palanti, E.; Nonnis Marzano, F.; Arillo, A. Indagini Sulla Genetica e Sulla Biologia Riproduttiva di Ciprinidi Atipici, Appartenenti Alla Sottofamiglia Leuciscinae, Presenti in Provincia Della Spezia-Università Degli Studi di Genova e Polizia Provinciale Della Spezia Sez. Faunistica 2011.
- Nonnis Marzano, F. Caratterizzazione Genetica di Ciprinidi Atipici Della Provincia di La Spezia-Spin off “Gen-Tech” Università degli Studi di Parma 2010.
- Vamberger, M.; Stuckas, H.; Sacco, F.; D’Angelo, S.; Arculeo, M.; Cheylan, M.; Corti, C.; Lo Valvo, M.; Marrone, F.; Wink, M.; et al. Differences in gene flow in a twofold secondary contact zone of pond turtles in southern Italy (Testudines: Emydidae: Emys orbicularis galloitalica, E. o. hellenica, E. trinacris). Zool. Scr. 2015, 44, 233–249. [Google Scholar] [CrossRef]
- Marrone, F.; Nardi, G.; Cianfanelli, S.; GovediČ, M.; Barra, S.A.; Arculeo, M.; Bodon, M. Diversity and taxonomy of the genus Unio Philipsson in Italy, with the designation of a neotype for Unio elongatulus C. Pfeiffer (Mollusca, Bivalvia, Unionidae). Zootaxa 2019, 4545, 339–374. [Google Scholar] [CrossRef]
- Cossu, P.; Dedola, G.L.; Scarpa, F.; Sanna, D.; Lai, T.; Maltagliati, F.; Curini-Galletti, M. Casu, M. Patterns of spatial genetic variation in Patella ulyssiponensis: Insights from the western Mediterranean marine ecoregion. Hydrobiologia 2015, 755, 39–55. [Google Scholar] [CrossRef]
- Casu, M.; Rivera-Ingraham, G.A.; Cossu, P.; Lai, T.; Sanna, D.; Dedola, G.L.; Sussarellu, R.; Sella, G.; Cristo, B.; Curini-Galletti, M.; et al. Patterns of spatial genetic structuring in the endangered limpet Patella ferruginea: Implications for the conservation of a Mediterranean endemic. Genetica 2011, 139, 1293–1308. [Google Scholar] [CrossRef]
- Maltagliati, F.; Lupi, L.; Castelli, A.; Pannacciulli, F.G. The genetic structure of the exotic ascidian Styela plicata (Tunicata) from Italian ports, with a re-appraisal of its worldwide genetic pattern. Mar. Ecol. 2016, 37, 492–502. [Google Scholar] [CrossRef]
- Drábková, M.; Jachníková, N.; Tyml, T.; Sehadová, H.; Ditrich, O.; Myšková, E.; Hypša, V.; Štefka, J. Population codivergence in common cuttlefish (Sepia officinalis) and its dicyemid parasite in the Mediterranean Sea. Sci. Rep. 2019, 9, 14300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marić, S.; Sušnik, S.; Simonović, P.; Snoj, A. Phylogeographic study of brown trout from Serbia, based on mitochondrial DNA control region analysis. Genet. Sel. Evol. 2006, 38, 411. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, L. The evolutionary history of brown trout (Salmo trutta L.) inferred from phylogeographic, nested clade, and mismatch analyses of mitochondrial DNA variation. Evolution 2001, 55, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Splendiani, A.; Fioravanti, T.; Giovannotti, M.; Negri, A.; Ruggeri, P.; Olivieri, L.; Cerioni, P.N.; Lorenzoni, M.; Caputo Barucchi, V. The effects of paleoclimatic events on Mediterranean trout: Preliminary evidences from ancient DNA. PLoS ONE 2016, 11, e0157975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Splendiani, A.; Palmas, F.; Sabatini, A.; Caputo Barucchi, V. The name of the trout: Considerations on the taxonomic status of the Salmo trutta L., 1758 complex (Osteichthyes: Salmonidae) in Italy. Eur. Zool. J. 2019, 86, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Splendiani, A.; Berrebi, P.; Tougard, C.; Righi, T.; Reynaud, N.; Fioravanti, T.; Lo Conte, P.; Delmastro, G.B.; Baltieri, M.; Ciuffardi, L.; et al. The role of the south-western Alps as a unidirectional corridor for Mediterranean brown trout (Salmo trutta complex) lineages. Biol. J. Linn. Soc. 2020, 131, 909–926. [Google Scholar] [CrossRef]
- Sanz, N.; García-Marín, J.L. Pla, Divergence of brown trout (Salmo trutta) within glacial refuge. Can. J. Fish. Aquat. 2000, 57, 2201–2210. [Google Scholar] [CrossRef]
- Grill, A.; Casula, P.; Lecis, R.; Menken, S. Endemism in Sardinia. In Phylogeography of Southern European Refugia; Weiss, S., Ferrand, N., Eds.; Springer: Dordrecht, The Netherland, 2007; pp. 273–296. [Google Scholar] [CrossRef]
- Gauthier, A.; Berrebi, P. La Colonisation de l’Ile par les différentes Souches de Truite. In Contribution à la Gestion des Populations de Truites en Corse; Muracciole, S., Ed.; Fédération Corse pour la Pêche et la Protection du Milieu Aquatique: Corte, France, 2007; pp. 4–10. [Google Scholar]
- Doadrio, I.; Perea, S.; Garzón-Heydt, P.; González, Y.J.L. Ictiofauna continental española. In Bases Para su Seguimiento. DG Medio Natural y Política Forestal; MARM: Madrid, Spain, 2011; 616p. [Google Scholar]
- Doadrio, I. Atlas y Libro Rojo de Los Peces Continentales de España; MNCN-CSIC: Madrid, Spain, 2001; p. 364. [Google Scholar]
- Frey, J.K. Modes of peripheral isolate formation and speciation. Syst. Biol. 1993, 42, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Marrone, F.; Vecchioni, L.; Deidun, A.; Mabrouki, Y.; Arab, A.; Arculeo, M. DNA taxonomy of the potamid freshwater crabs from Northern Africa (Decapoda, Potamidae). Zool. Scr. 2020, 49, 473–487. [Google Scholar] [CrossRef]
- Segherloo, I.H.; Freyhof, J.; Berrebi, P.; Ferchaud, A.L.; Geiger, M.; Laroche, J.; Levin, B.A.; Normandeau, E.; Bernatchez, L. A genomic perspective on an old question: Salmo trouts or Salmo trutta (Teleostei: Salmonidae)? Mol. Phylogenet. Evol. 2021, 162, 107204. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Region Dataset | ||||||
|---|---|---|---|---|---|---|
| N | bp | H | S | h | π | |
| Present study | 103 | 304 | 14 | 16 | 0.554 | 0.00461 |
| Sardinia | 47 | 304 | 4 | 4 | 0.272 | 0.00046 |
| Liguria | 44 | 304 | 8 | 7 | 0.581 | 0.00298 |
| Piedmont | 10 | 304 | 4 | 4 | 0.644 | 0.00317 |
| Lombardy | 2 | 304 | 1 | 0 | 0.000 | 0.00000 |
| 16s Dataset | ||||||
| N | bp | H | S | h | π | |
| Present study | 103 | 599 | 8 | 7 | 0.620 | 0.00073 |
| Sardinia | 47 | 599 | 3 | 2 | 0.581 | 0.00029 |
| Liguria | 44 | 599 | 5 | 4 | 0.216 | 0.00041 |
| Piedmont | 10 | 599 | 2 | 1 | 0.200 | 0.00037 |
| Lombardy | 2 | 599 | 1 | 0 | 0.000 | 0.00000 |
| Concatenated Dataset | ||||||
| N | bp | H | S | h | π | |
| Present study | 103 | 849 | 20 | 23 | 0.797 | 0.00210 |
| Sardinia | 47 | 849 | 7 | 6 | 0.692 | 0.00038 |
| Liguria | 44 | 849 | 12 | 11 | 0.644 | 0.00132 |
| Piedmont | 10 | 849 | 4 | 5 | 0.644 | 0.00136 |
| Lombardy | 2 | 849 | 1 | 0 | 0.000 | 0.00000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azzena, I.; Scarpa, F.; Locci, C.; Cossu, P.; Niffoi, A.; Orrù, F.; Bovero, S.; Sotgiu, G.; Sanna, D.; Casu, M. Mitochondrial DNA of Sardinian and North-West Italian Populations Revealed a New Piece in the Mosaic of Phylogeography and Phylogeny of Salariopsis fluviatilis (Blenniidae). Animals 2022, 12, 3403. https://doi.org/10.3390/ani12233403
Azzena I, Scarpa F, Locci C, Cossu P, Niffoi A, Orrù F, Bovero S, Sotgiu G, Sanna D, Casu M. Mitochondrial DNA of Sardinian and North-West Italian Populations Revealed a New Piece in the Mosaic of Phylogeography and Phylogeny of Salariopsis fluviatilis (Blenniidae). Animals. 2022; 12(23):3403. https://doi.org/10.3390/ani12233403
Chicago/Turabian StyleAzzena, Ilenia, Fabio Scarpa, Chiara Locci, Piero Cossu, Alessio Niffoi, Flavio Orrù, Stefano Bovero, Giuseppe Sotgiu, Daria Sanna, and Marco Casu. 2022. "Mitochondrial DNA of Sardinian and North-West Italian Populations Revealed a New Piece in the Mosaic of Phylogeography and Phylogeny of Salariopsis fluviatilis (Blenniidae)" Animals 12, no. 23: 3403. https://doi.org/10.3390/ani12233403
APA StyleAzzena, I., Scarpa, F., Locci, C., Cossu, P., Niffoi, A., Orrù, F., Bovero, S., Sotgiu, G., Sanna, D., & Casu, M. (2022). Mitochondrial DNA of Sardinian and North-West Italian Populations Revealed a New Piece in the Mosaic of Phylogeography and Phylogeny of Salariopsis fluviatilis (Blenniidae). Animals, 12(23), 3403. https://doi.org/10.3390/ani12233403