
Selective Inner Hair Cell Loss in a Neonate Harbor Seal (Phoca vitulina)

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Inner Ear Analysis
2.2. Scanning Electron Microscopy (SEM)
2.3. Characterization of the Lesions
3. Results
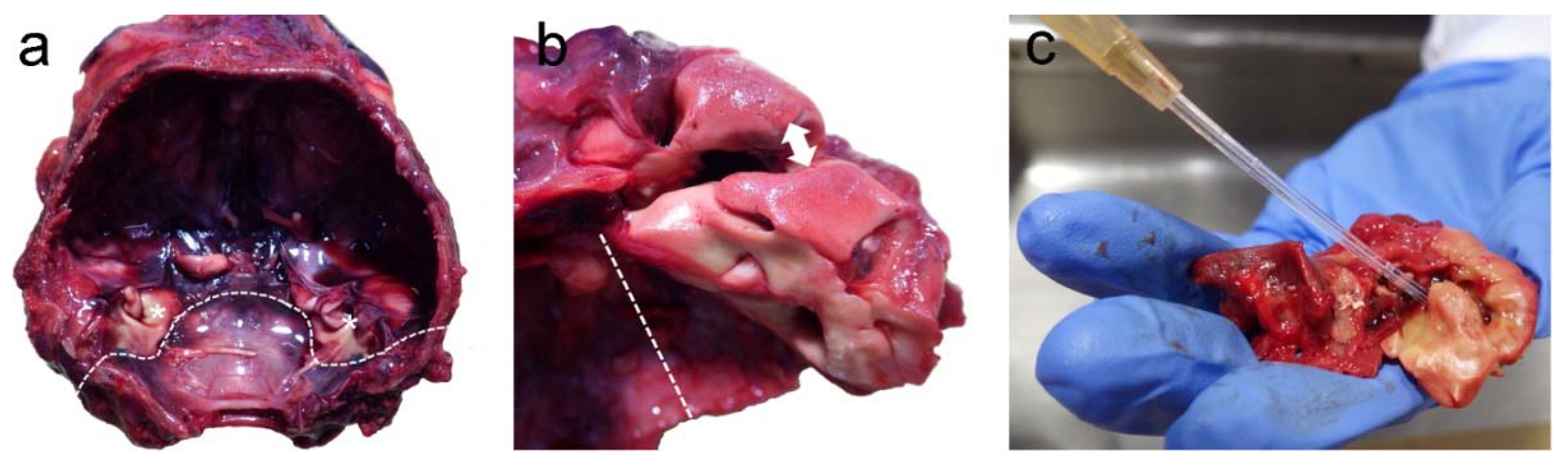
3.1. Post-Mortem Examination
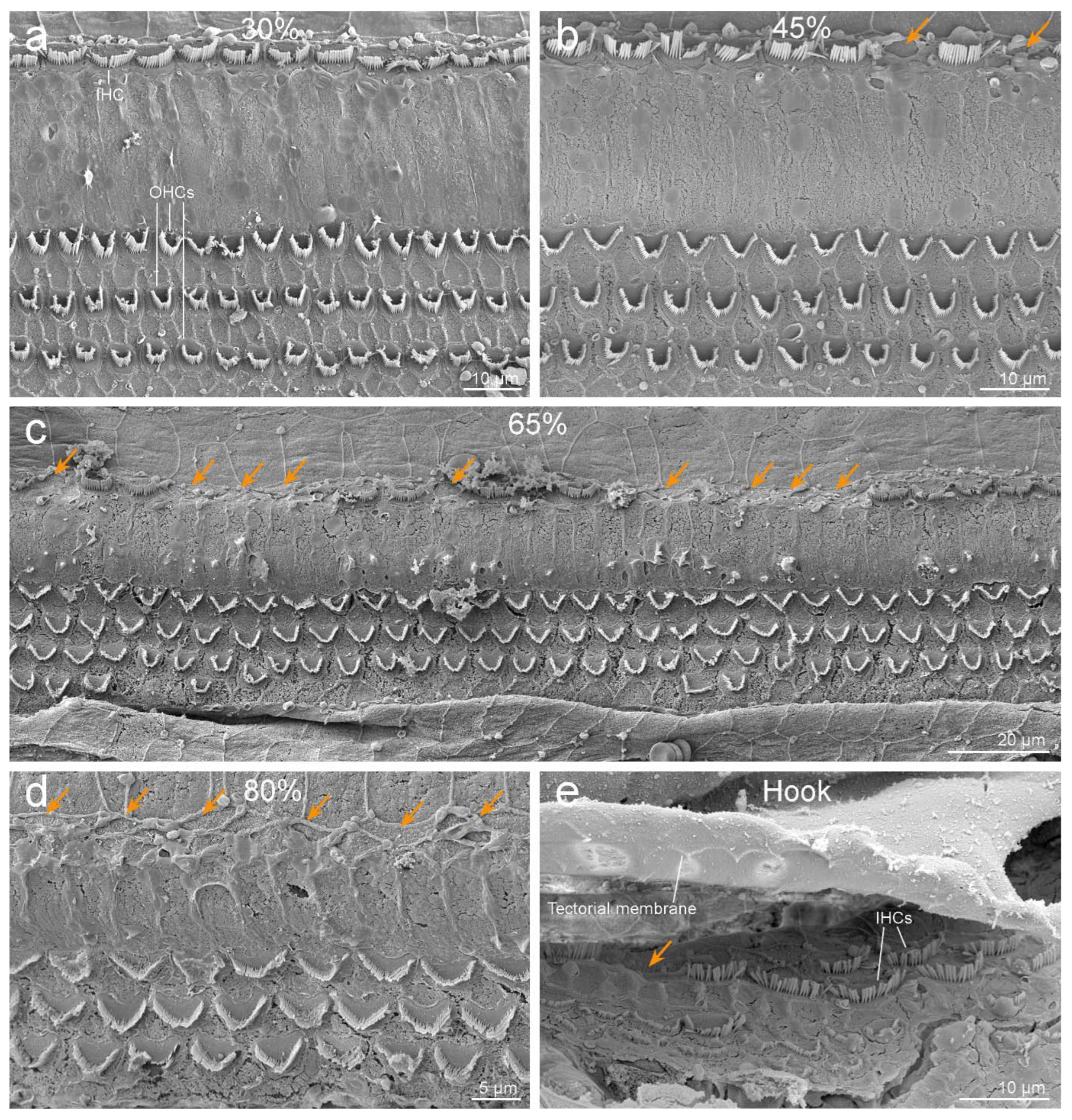
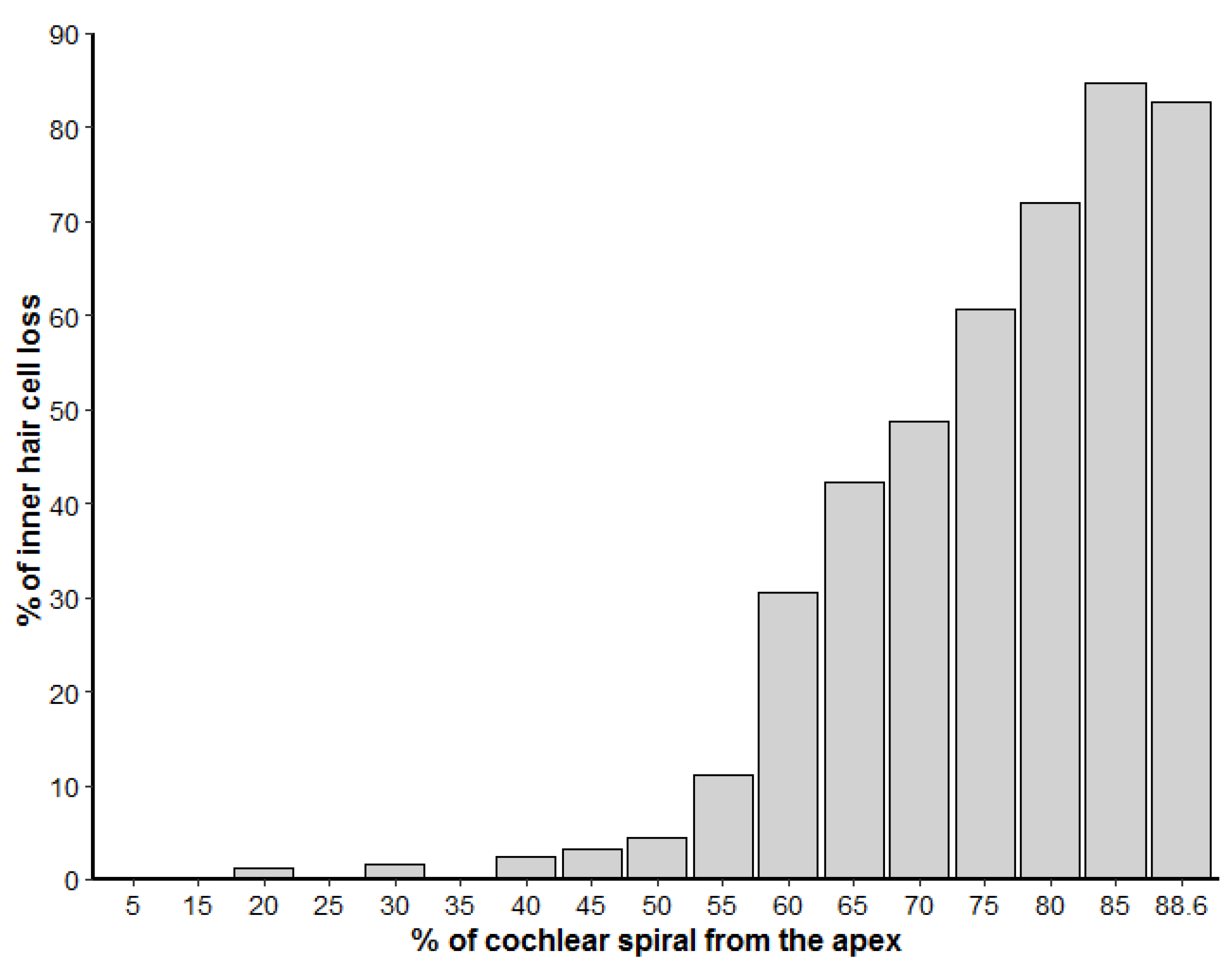
3.2. Inner Ear Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, R.; Bale, J.; White, K. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Belcher, R.; Virgin, F.; Duis, J.; Wootten, C. Genetic and non-genetic workup for pediatric congenital hearing loss. Front. Pediatr. 2021, 9, 536730. [Google Scholar] [CrossRef]
- Morton, N.E. Genetic epidemiology of hearing impairment. Ann. N. Y. Acad. Sci. USA 1991, 630, 16–31. [Google Scholar] [CrossRef]
- Raymond, M.; Walker, E.; Dave, I.; Dedhia, K. Genetic testing for congenital non-syndromic sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2019, 124, 68–75. [Google Scholar] [CrossRef]
- Kalatzis, V.; Petit, C. The fundamental and medical impacts of recent progress in research on hereditary hearing loss. Hum. Mol. Genet. 1998, 7, 1589–1597. [Google Scholar] [CrossRef]
- Farooq, R.; Hussain, K.; Tariq, M.; Farooq, A.; Mustafa, M. CRISPR/Cas9: Targeted genome editing for the treatment of hereditary hearing loss. J. Appl. Genet. 2020, 61, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Bredberg, G.; Ades, H.W.; Engström, H. Scanning electron microscopy of the normal and pathologically altered organ of Corti. Acta Oto-Laryngol. 1972, 73, 3–48. [Google Scholar] [CrossRef]
- Hu, B.H.; Guo, W.; Wang, P.Y.; Henderson, D.; Jiang, S.C. Intense noise-induced apoptosis in hair cells of guinea pig cochleae. Acta Oto-Laryngol. 2000, 120, 19–24. [Google Scholar] [CrossRef]
- Raphael, Y.; Altschuler, R.A. Reorganization of cytoskeletal and junctional proteins during cochlear hair cell degeneration. Cell Motil. Cytoskelet. 1991, 18, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Brownlow, A.; McGovern, B.; Raverty, S.A.; Shadwick, R.E.; André, M. Implementation of a method to visualize noise-induced hearing loss in mass stranded cetaceans. Sci. Rep. 2017, 7, 41848. [Google Scholar] [CrossRef]
- Suzuki, M.; Kishimoto, M.; Hayama, S.-I.; Ohtaishi, N.; Nakane, F. A case of cleft palate in a Kuril seal (Phoca vitulina stejnegeri), from Hokkaido, Japan. J. Wildl. Dis. 1992, 28, 490–493. [Google Scholar] [CrossRef] [PubMed]
- McKnight, C.A.; Reynolds, T.L.; Haulena, M.; Delahunta, A.; Gulland, F.M.D. Congenital hemicerebral anomaly in a stranded Pacific harbor seal (Phoca vitulina richardsi). J. Wildl. Dis. 2005, 41, 654–658. [Google Scholar] [CrossRef][Green Version]
- Dennison, S.E.; Forrest, L.J.; Fleetwood, M.L.; Gulland, F.M.D. Concurrent occipital bone malformation and atlantoaxial subluxation in a neonatal harbor seal (Phoca vitulina). J. Zoo Wildl. Med. 2009, 40, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Dennison, S.E.; Boor, M.; Fauquier, D.; Van Bonn, W.; Greig, D.J.; Gulland, F.M.D. Foramen ovale and ductus arteriosus patency in neonatal harbor seal (Phoca vitulina) pups in rehabilitation. Aquat. Mamm. 2011, 37, 161–166. [Google Scholar] [CrossRef]
- Harris, H.S.; Facemire, P.; Greig, D.J.; Colegrove, K.M.; Ylitalo, G.M.; Yanagida, G.K.; Nutter, F.B.; Fleetwood, M.; Gulland, F.M.D. Congenital neuroglial heterotopia in a neonatal harbor seal (Phoca vitulina richardsi ) with evidence of recent exposure to polycyclic aromatic hydrocarbons. J. Wildl. Dis. 2011, 47, 246–254. [Google Scholar] [CrossRef]
- Leger, J.A.S.; Nilson, E.M. Intestinal atresia in a harbor seal (Phoca vitulina) and a review of congenital conditions of the species. Aquat. Mamm. 2014, 40, 207–212. [Google Scholar] [CrossRef]
- D’Agnese, E.R.; Lambourn, D.M.; Olson, J.K.; Huggins, J.L.; Raverty, S.; Garner, M.M.; Calambokidis, J.; Scott, A.A.; Jeffries, S.J.; Gaydos, J.K. Congenital diseases in harbor seals (Phoca vitulina richardsii) from the Salish Sea. J. Wildl. Dis. 2021, 57, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Geraci, J.R.; Lounsbury, V.J. Marine Mammals Ashore: A Field Guide for Strandings, 2nd ed.; National Aquarium in Baltimore: Baltimore, MD, USA, 2005; p. 371. [Google Scholar]
- Raverty, S.A.; Duignan, P.J.; Jepson, P.D.; Morell, M. Gross Necropsy and Specimen Collection Protocols (Chapter 13). In CRC Handbook of Marine Mammal Medicine, 3rd ed.; Dierauf, L.A., Gulland, F.M., Eds.; CRC Press/Taylor & Francis Group: Boca Raton, FL, USA, 2018; pp. 249–266. [Google Scholar]
- Morell, M.; Lenoir, M.; Shadwick, R.E.; Jauniaux, T.; Dabin, W.; Begeman, L.; Ferreira, M.; Maestre, I.; Degollada, E.; Hernandez-Milian, G.; et al. Ultrastructure of the Odontocete organ of Corti: Scanning and transmission electron microscopy. J. Comp. Neurol. 2015, 523, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Raverty, S.A.; Mulsow, J.; Haulena, M.; Barret-Lennard, L.; Nordstrom, C.; Venail, F.; Shadwick, R.E. Combining cochlear analysis and auditory evoked potentials in a beluga whale with high-frequency hearing loss. Front. Vet. Sci. 2020, 7, 534917. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Ijsseldijk, L.L.; Piscitelli-Doshkov, M.; Ostertag, S.; Estrade, V.; Haulena, M.; Doshkov, P.; Bourien, J.; Raverty, S.A.; Siebert, U.; et al. Cochlear apical morphology in toothed whales: Using the pairing hair cell—Deiters’ cell as a marker to detect lesions. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2021, 1–12. [Google Scholar] [CrossRef]
- Girdlestone, C.D.; Ng, J.; Kössl, M.; Caplot, A.; Shadwick, R.E.; Morell, M. Correlating cochlear morphometrics from Parnell’s mustached bat (Pteronotus parnellii) with hearing. J. Assoc. Res. Otolaryngol. 2020, 21, 425–444. [Google Scholar] [CrossRef]
- Gibson, A.K.; Raverty, S.; Lambourn, D.M.; Huggins, J.; Magargal, S.L.; Grigg, M.E. Polyparasitism is associated with in-creased disease severity in Toxoplasma gondii-infected marine sentinel species. PLoS Negl. Trop. Dis. 2011, 5, e1142. [Google Scholar] [CrossRef]
- Bohne, A.B.; Harding, G.W. Degeneration in the cochlea after noise damage: Primary versus secondary events. Am. J. Otol. 2000, 21, 505–509. [Google Scholar]
- Huizing, E.H.; De Groot, J.C.M.J. Human cochlear pathology in aminoglycoside ototoxicity—A review. Acta Oto-Laryngologica 1987, 104, 117–125. [Google Scholar] [CrossRef]
- Takeno, S.; Harrison, R.V.; Mount, R.J.; Wake, M.; Harada, Y. Induction of selective inner hair cell damage by carboplatin. Scanning Microsc. 1994, 8, 97–106. [Google Scholar]
- Saito, T.; Saito, H.; Saito, K.; Wakui, S.; Manabe, Y.; Tsuda, G. Ototoxicity of carboplatin in guinea pigs. Auris Nasus Larynx 1989, 16, 13–21. [Google Scholar] [CrossRef]
- Karimi-Boroujeni, M.; Zahedi-Amiri, A.; Coombs, K.M. Embryonic origins of virus-induced hearing loss: Overview of molecular etiology. Viruses 2021, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Suzutani, T.; Baba, Y.; Koyano, S.; Nozawa, N.; Ishibashi, K.; Fujieda, K.; Inoue, N.; Omori, K. Etiology of severe sensorineural hearing loss in children: Independent impact of congenital cytomegalovirus infection and GJB2 mutations. J. Infect. Dis. 2007, 195, 782–788. [Google Scholar] [CrossRef]
- Grosse, S.D.; Ross, D.S.; Dollard, S.C. Congenital cytomegalovirus (CMV) infection as a cause of permanent bilateral hearing loss: A quantitative assessment. J. Clin. Virol. 2008, 41, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, M.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of congenital cytomegalovirus infection: Disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 2009, 22, 99–126. [Google Scholar] [CrossRef]
- Tsuprun, V.; Keskin, N.; Schleiss, M.R.; Schachern, P.; Cureoglu, S. Cytomegalovirus-induced pathology in human temporal bones with congenital and acquired infection. Am. J. Otolaryngol. 2019, 40, 102270. [Google Scholar] [CrossRef] [PubMed]
- Schachtele, S.J.; Mutnal, M.B.; Schleiss, M.R.; Lokensgard, J.R. Cytomegalovirus-induced sensorineural hearing loss with persistent cochlear inflammation in neonatal mice. J. NeuroVirology 2011, 17, 201–211. [Google Scholar] [CrossRef]
- Nomura, Y.; Kurata, T.; Saito, K. Cochlear changes after Herpes simplex virus infection. Acta Oto-Laryngol. 1985, 99, 419–427. [Google Scholar] [CrossRef]
- Cohen, B.E.; Durstenfeld, A.; Roehm, P.C. Viral causes of hearing loss: A review for hearing health professionals. Trends Hear. 2014, 18. [Google Scholar] [CrossRef]
- Hemenway, W.G.; Sando, I.; McChesney, D. Temporal bone pathology following maternal rubella. Arch. Klin. Exp. Ohr. Nas. Kehlk. Heilk. 1969, 193, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Pujol, R. Age-related structural investigation of the Bronx waltzer mutant mouse cochlea: Scanning and transmission electron microscopy. Hear. Res. 1984, 13, 123–134. [Google Scholar] [CrossRef]
- Liberman, M.C.; Tartaglini, E.; Fleming, J.C.; Neufeld, E.J. Deletion of SLC19A2, the high affinity Thiamine transporter, causes selective inner hair cell loss and an auditory neuropathy phenotype. J. Assoc. Res. Otolaryngol. 2006, 7, 211–217. [Google Scholar] [CrossRef]
- Schrott, A.; Stephan, K.; Spoendlin, H. Hearing with selective inner hair cell loss. Hear. Res. 1989, 40, 213–219. [Google Scholar] [CrossRef]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Zelante, L.; Gasparini, P.; Estivill, X.; Melchionda, S.; D’Agruma, L.; Govea, N.; Mila, M.; Monica, M.D.; Lutfi, J.; Shohat, M.; et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997, 6, 1605–1609. [Google Scholar] [CrossRef]
- Chen, S.; Sun, Y.; Lin, X.; Kong, W. Down regulated connexin26 at different postnatal stage displayed different types of cellular degeneration and formation of organ of Corti. Biochem. Biophys. Res. Commun. 2014, 445, 71–77. [Google Scholar] [CrossRef] [PubMed]
- De Siati, R.D.; Rosenzweig, F.; Gersdor, G.; Gregoire, A.; Rombaux, P.; Deggouj, N. Auditory neuropathy spectrum disorders: From diagnosis to treatment: Literature review and case reports. J. Clin. Med. 2020, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.; Starr, A. Auditory neuropathy–neural and synaptic mechanisms. Nat. Rev. Neurol. 2016, 12, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Hansen, M.R. Auditory synaptopathy, auditory neuropathy, and cochlear implantation. Laryngoscope Investig. Otolaryngol. 2019, 4, 429–440. [Google Scholar] [CrossRef]
- Foerst, A.; Beutner, D.; Lang-Roth, R.; Huttenbrink, K.B.; von Wedel, H.; Walger, M. Prevalence of auditory neuropathy/synaptopathy in a population of children with profound hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2006, 70, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Amatuzzi, M.; Liberman, M.C.; Northrop, C. Selective inner hair cell loss in prematurity: A temporal bone study of infants from a neonatal intensive care unit. J. Assoc. Res. Otolaryngol. 2011, 12, 595–604. [Google Scholar] [CrossRef]
- Soons, J.A.M.; Ricci, A.J.; Steele, C.R.; Puria, S. Cytoarchitecture of the mouse organ of Corti from base to apex, determined using in situ two-photon imaging. J. Assoc. Res. Otolaryngol. 2014, 16, 47–66. [Google Scholar] [CrossRef]
- Lobarinas, E.; Salvi, R.; Ding, D. Selective inner hair cell dysfunction in chinchillas impairs hearing-in-noise in the absence of outer hair cell loss. J. Assoc. Res. Otolaryngol. 2015, 17, 89–101. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morell, M.; Rojas, L.; Haulena, M.; Busse, B.; Siebert, U.; Shadwick, R.E.; Raverty, S.A. Selective Inner Hair Cell Loss in a Neonate Harbor Seal (Phoca vitulina). Animals 2022, 12, 180. https://doi.org/10.3390/ani12020180
Morell M, Rojas L, Haulena M, Busse B, Siebert U, Shadwick RE, Raverty SA. Selective Inner Hair Cell Loss in a Neonate Harbor Seal (Phoca vitulina). Animals. 2022; 12(2):180. https://doi.org/10.3390/ani12020180
Chicago/Turabian StyleMorell, Maria, Laura Rojas, Martin Haulena, Björn Busse, Ursula Siebert, Robert E. Shadwick, and Stephen A. Raverty. 2022. "Selective Inner Hair Cell Loss in a Neonate Harbor Seal (Phoca vitulina)" Animals 12, no. 2: 180. https://doi.org/10.3390/ani12020180
APA StyleMorell, M., Rojas, L., Haulena, M., Busse, B., Siebert, U., Shadwick, R. E., & Raverty, S. A. (2022). Selective Inner Hair Cell Loss in a Neonate Harbor Seal (Phoca vitulina). Animals, 12(2), 180. https://doi.org/10.3390/ani12020180