
Genetic Diversity and Selection Signatures in Jianchang Black Goats Revealed by Whole-Genome Sequencing Data
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Whole-Genome Sequencing Data
2.2. Alignment of Short-Reads and Variant Calling
2.3. Phylogenetic and Population Structure Analysis
2.4. Genetic Diversity, Linkage Disequilibrium, and Genome-Wide Detection of ROHs
2.5. Estimation of Effective Population Size and Genomic Inbreeding Coefficients
2.6. Genome-Wide Identification of ROH Islands and Selection Signals
3. Results
3.1. Abundant Genomic Variants in Three Black Goat Breeds and Bezoars
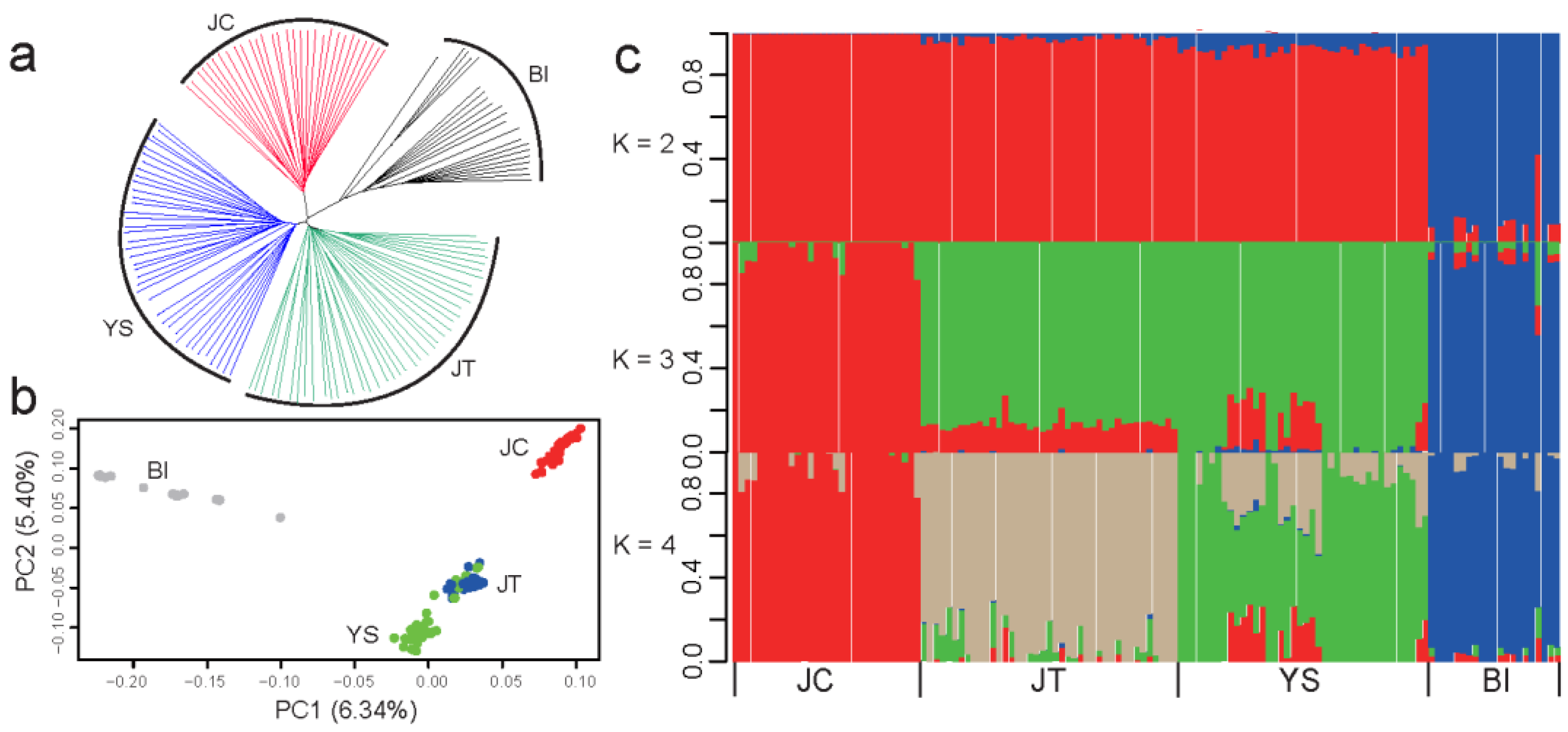
3.2. JC Was Genetically Distinct from Two Other Black Breeds in Southwest China
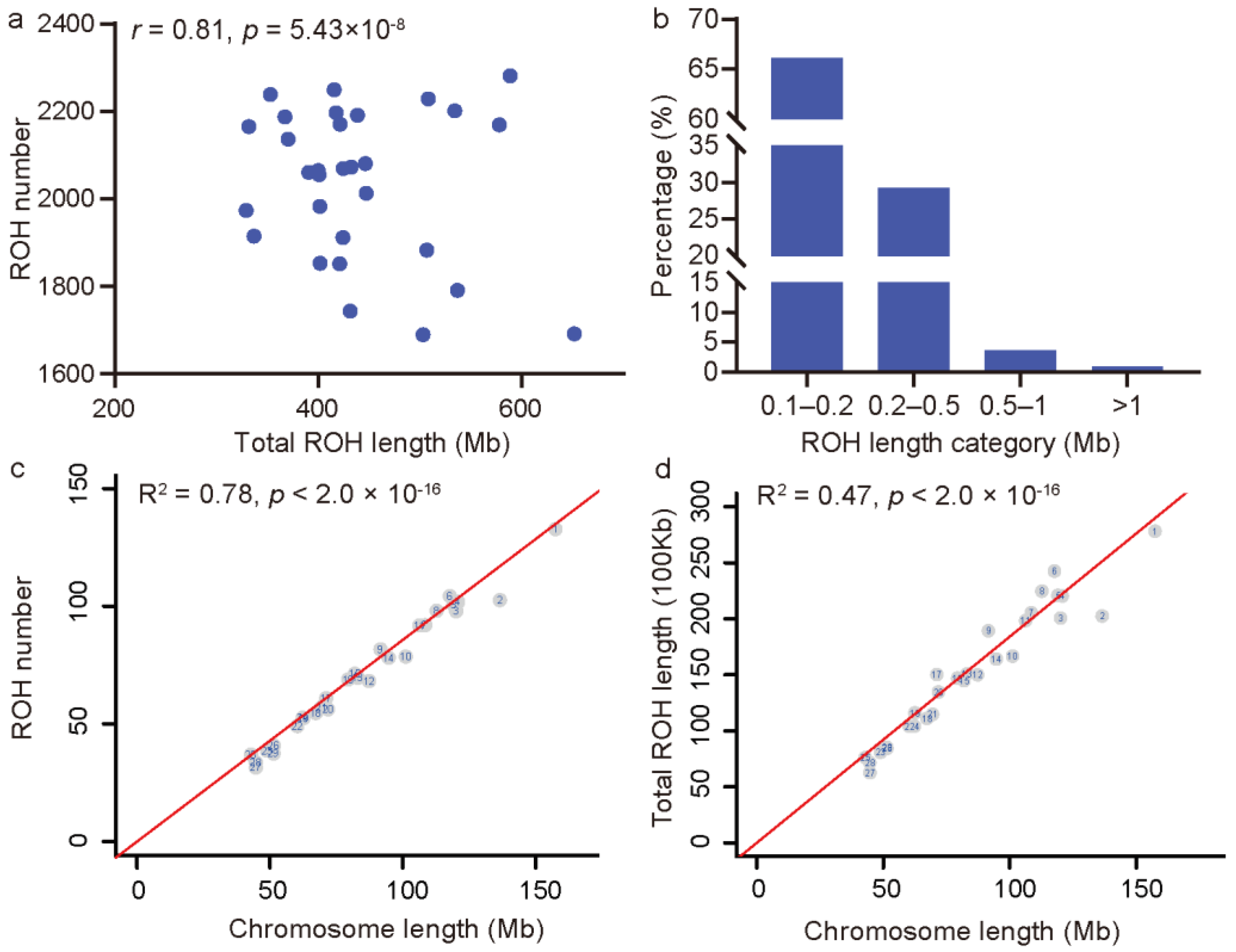
3.3. Genetic Diversity Metrics and the Genome-Wide Pattern of ROH in JC Goats
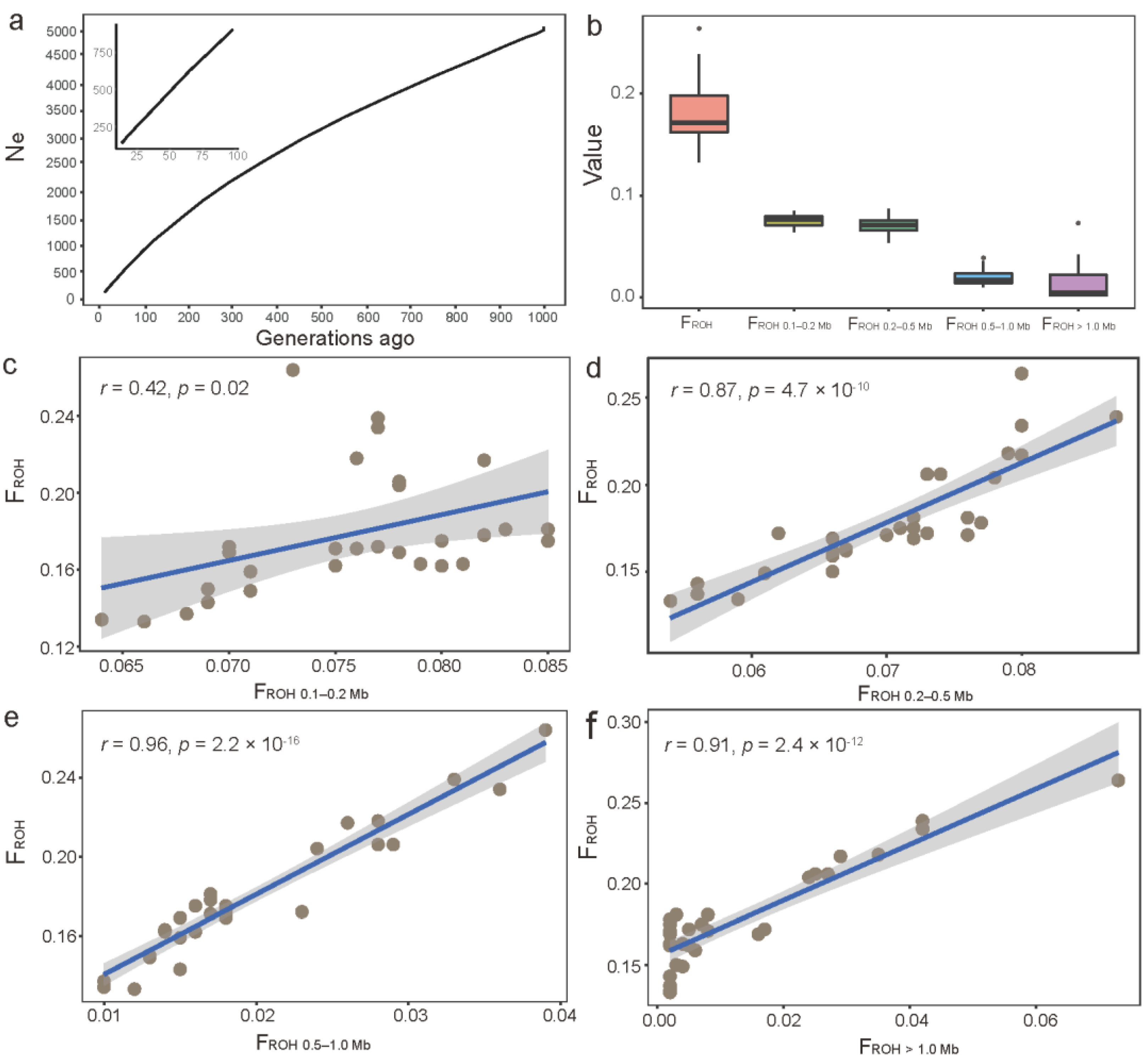
3.4. Effective Population Size and Genomic-Based Inbreeding Coefficients in JC Goats
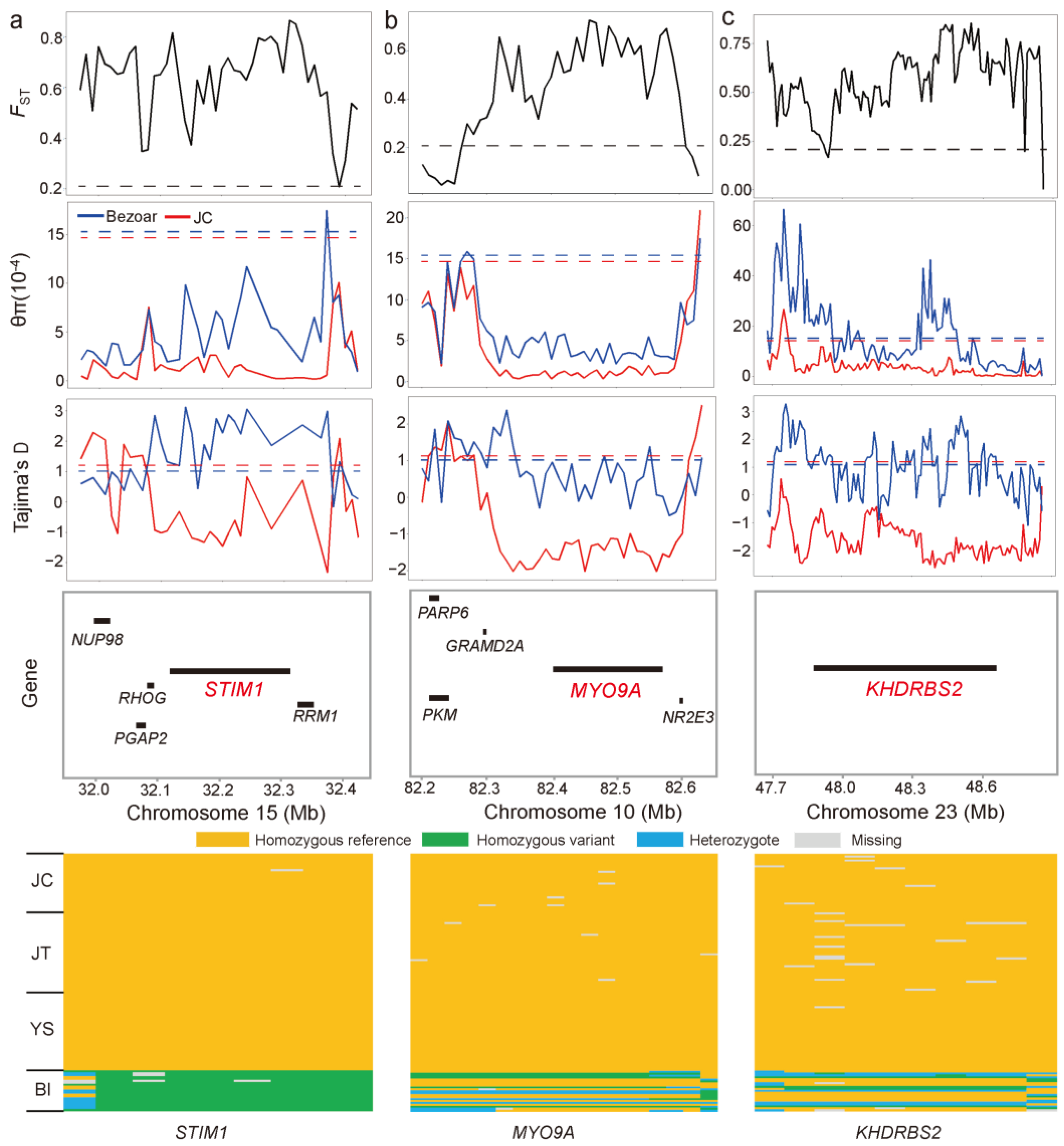
3.5. Detection of ROH Islands and Selection Signals in JC Goats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amills, M.; Capote, J.; Tosser-Klopp, G. Goat domestication and breeding: A jigsaw of historical, biological and molecular data with missing pieces. Anim. Genet. 2017, 48, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Colli, L.; Milanesi, M.; Talenti, A.; Bertolini, F.; Chen, M.; Crisà, A.; Daly, K.G.; Del Corvo, M.; Guldbrandtsen, B.; Lenstra, J.A.; et al. Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet. Sel. Evol. 2018, 50, 58. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Ghadikolaei, A.; Mehrabani-Yeganeh, H.; Miarei-Aashtiani, S.R.; Staiger, E.A.; Rashidi, A.; Huson, H.J. Genome-Wide Association Studies Identify Candidate Genes for Coat Color and Mohair Traits in the Iranian Markhoz Goat. Front. Genet. 2018, 9, 00105. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Z.; He, X.; Jiang, Y.; Ouyang, Y.; Hong, Q.; Chu, M. Differentially Expressed Circular RNA Profile Signatures Identified in Prolificacy Trait of Yunshang Black Goat Ovary at Estrus Cycle. Front. Physiol. 2022, 13, 820459. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; He, X.Y.; Jiang, Y.T.; Lan, R.; Li, M.; Li, Z.M.; Yang, W.F.; Hong, Q.H.; Chu, M.X. Combined approaches to reveal genes associated with litter size in Yunshang black goats. Anim. Genet. 2020, 51, 924–934. [Google Scholar] [CrossRef]
- Broman, K.W.; Weber, J.L. Long Homozygous Chromosomal Segments in Reference Families from the Centre d’Étude du Polymorphisme Humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Villanueva, B.; Fernández, A.; Saura, M.; Caballero, A.; Fernández, J.; Morales-González, E.; Toro, M.A.; Pong-Wong, R. The value of genomic relationship matrices to estimate levels of inbreeding. Genet. Sel. Evol. 2021, 53, 42. [Google Scholar] [CrossRef]
- Wu, F.; Sun, H.; Lu, S.; Gou, X.; Yan, D.; Xu, Z.; Zhang, Z.; Qadri, Q.R.; Zhang, Z.; Wang, Z.; et al. Genetic Diversity and Selection Signatures Within Diannan Small-Ear Pigs Revealed by Next-Generation Sequencing. Front. Genet. 2020, 11, 00733. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.D.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shi, L.; Li, Y.; Chen, L.; Garrick, D.; Wang, L.; Zhao, F. Estimates of genomic inbreeding and identification of candidate regions that differ between Chinese indigenous sheep breeds. J. Anim. Sci. Biotechnol. 2021, 12, 95. [Google Scholar] [CrossRef]
- Cortellari, M.; Bionda, A.; Negro, A.; Frattini, S.; Mastrangelo, S.; Somenzi, E.; Lasagna, E.; Sarti, F.M.; Ciani, E.; Ciampolini, R.; et al. Runs of homozygosity in the Italian goat breeds: Impact of management practices in low-input systems. Genet. Sel. Evol. 2021, 53, 92. [Google Scholar] [CrossRef] [PubMed]
- Signer-Hasler, H.; Henkel, J.; Bangerter, E.; Bulut, Z.; Drögemüller, C.; Leeb, T.; Flury, C. Runs of homo-zygosity in Swiss goats reveal genetic changes associated with domestication and modern selection. Genet. Sel. Evol. 2022, 54, 6. [Google Scholar] [CrossRef]
- Yuan, J.; Li, S.; Sheng, Z.; Zhang, M.; Liu, X.; Yuan, Z.; Yang, N.; Chen, J. Genome-wide run of homozygosity analysis reveals candidate genomic regions associated with environmental adaptations of Tibetan native chickens. BMC Genom. 2022, 23, 91. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhong, J.; Li, L.; Zhong, T.; Wang, L.; Song, T.; Zhang, H. Comparative genome analyses reveal the unique genetic composition and selection signals underlying the phenotypic characteristics of three Chinese domestic goat breeds. Genet. Sel. Evol. 2019, 51, 70. [Google Scholar] [CrossRef]
- Guo, J.; Jiang, R.; Mao, A.; Liu, G.E.; Zhan, S.; Li, L.; Zhong, T.; Wang, L.; Cao, J.; Chen, Y.; et al. Genome-wide association study reveals 14 new SNPs and confirms two structural variants highly associated with the horned/polled phenotype in goats. BMC Genom. 2021, 22, 769. [Google Scholar] [CrossRef]
- Alberto, F.J.; Boyer, F.; Orozco-Terwengel, P.; Streeter, I.; Servin, B.; de Villemereuil, P.; Benjelloun, B.; Librado, P.; Biscarini, F.; Colli, L.; et al. Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 2018, 9, 813. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2018, 35, 1786–1788. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, W.-R.; Lv, F.-H.; He, S.-G.; Tian, S.-L.; Peng, W.-F.; Sun, Y.-W.; Zhao, Y.-X.; Tu, X.-L.; Zhang, M.; et al. Whole-Genome Sequencing of Native Sheep Provides Insights into Rapid Adaptations to Extreme Environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Orozco-Terwengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. DetectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. 2018. Available online: https://orca.cardiff.ac.uk/id/eprint/108906 (accessed on 4 May 2022).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.; Szpiech, Z.A.; Hernandez, R.D. Human demographic history has amplified the effects of background selection across the genome. PLoS Genet. 2018, 14, e1007387. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Hernandez, R.D. selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef]
- Bertolini, F.; Cardoso, T.F.; Marras, G.; Nicolazzi, E.L.; Rothschild, M.F.; Amills, M.; the AdaptMap consortium. Genome-wide patterns of homozygosity provide clues about the population history and adaptation of goats. Genet. Sel. Evol. 2018, 50, 59. [Google Scholar] [CrossRef]
- Islam, R.; Li, Y.; Liu, X.; Berihulay, H.; Abied, A.; Gebreselassie, G.; Ma, Q.; Ma, Y. Genome-Wide Runs of Homozygosity, Effective Population Size, and Detection of Positive Selection Signatures in Six Chinese Goat Breeds. Genes 2019, 10, 938. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Di Gerlando, R.; Sardina, M.T.; Sutera, A.M.; Moscarelli, A.; Tolone, M.; Cortellari, M.; Marletta, D.; Crepaldi, P.; Portolano, B. Genome-Wide Patterns of Homozygosity Reveal the Conservation Status in Five Italian Goat Populations. Animals 2021, 11, 1510. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Druml, T.; Neuditschko, M.; Dobretsberger, M.; Horna, M.; Brem, G. High-resolution population structure and runs of homozygosity reveal the genetic architecture of complex traits in the Lipizzan horse. BMC Genom. 2019, 20, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulim, H.A.; Brito, L.F.; Pinto, L.F.B.; Ferraz, J.B.S.; Grigoletto, L.; Silva, M.R.; Pedrosa, V.B. Characterization of runs of homozygosity, heterozygosity-enriched regions, and population structure in cattle populations selected for different breeding goals. BMC Genom. 2022, 23, 209. [Google Scholar] [CrossRef] [PubMed]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Sun, H.; Zhang, Z.; Zhao, Q.; Olasege, B.S.; Li, Q.; Yue, Y.; Ma, P.; Zhang, X.; Wang, Q.; et al. Assessment of Autozygosity Derived from Runs of Homozygosity in Jinhua Pigs Disclosed by Sequencing Data. Front. Genet. 2019, 10, 00274. [Google Scholar] [CrossRef]
- Macciotta, N.P.P.; Colli, L.; Cesarani, A.; Ajmone-Marsan, P.; Low, W.Y.; Tearle, R.; Williams, J.L. The distribution of runs of homozygosity in the genome of river and swamp buffaloes reveals a history of adaptation, migration and crossbred events. Genet. Sel. Evol. 2021, 53, 20. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef]
- Gurgul, A.; Szmatoła, T.; Topolski, P.; Jasielczuk, I.; Żukowski, K.; Bugno-Poniewierska, M. The use of runs of homozygosity for estimation of recent inbreeding in Holstein cattle. J. Appl. Genet. 2016, 57, 527–530. [Google Scholar] [CrossRef]
- Kim, E.-S.; Cole, J.B.; Huson, H.; Wiggans, G.R.; Van Tassell, C.P.; Crooker, B.A.; Liu, G.; Da, Y.; Sonstegard, T.S. Effect of Artificial Selection on Runs of Homozygosity in U.S. Holstein Cattle. PLoS ONE 2013, 8, e80813. [Google Scholar] [CrossRef]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of Inbreeding Due to Distant Ancestors and Its Detection Using Dense Single Nucleotide Polymorphism Data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemu, S.W.; Kadri, N.K.; Harland, C.; Faux, P.; Charlier, C.; Caballero, A.; Druet, T. An evaluation of inbreeding measures using a whole-genome sequenced cattle pedigree. Heredity 2021, 126, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Z.; Sun, J.; Huang, Y.; Hanif, Q.; Liao, Y.; Lei, C. Identification of Genomic Characteristics and Selective Signals in a Du’an Goat Flock. Animals 2020, 10, 994. [Google Scholar] [CrossRef] [PubMed]
- Onzima, R.B.; Upadhyay, M.R.; Doekes, H.P.; Brito, L.F.; Bosse, M.; Kanis, E.; Groenen, M.A.; Crooijmans, R.P. Genome-Wide Characterization of Selection Signatures and Runs of Homozygosity in Ugandan Goat Breeds. Front. Genet. 2018, 9, 00318. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wang, X.; Li, M.; Li, Y.; Yang, Z.; Wang, X.; Pan, X.; Gong, M.; Zhang, Y.; Guo, Y.; et al. The origin of domestication genes in goats. Sci. Adv. 2020, 6, eaaz5216. [Google Scholar] [CrossRef]
- Singh, V.; Gowda, C.P.; Singh, V.; Ganapathy, A.S.; Karamchandani, D.M.; Eshelman, M.A.; Yochum, G.S.; Nighot, P.; Spiegelman, V.S. The mRNA-binding protein IGF2BP1 maintains intestinal barrier function by up-regulating occludin expression. J. Biol. Chem. 2020, 295, 8602–8612. [Google Scholar] [CrossRef]
- Wang, K.; Hu, H.; Tian, Y.; Li, J.; Scheben, A.; Zhang, C.; Li, Y.; Wu, J.; Yang, L.; Fan, X.; et al. The Chicken Pan-Genome Reveals Gene Content Variation and a Promoter Region Deletion in IGF2BP1 Affecting Body Size. Mol. Biol. Evol. 2021, 38, 5066–5081. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, M.; Cheng, H.; Fan, W.; Yuan, Z.; Gao, Q.; Xu, Y.; Guo, Z.; Zhang, Y.; Hu, J.; et al. An intercross population study reveals genes associated with body size and plumage color in ducks. Nat. Commun. 2018, 9, 2648. [Google Scholar] [CrossRef]
- Daly, K.G.; Delser, P.M.; Mullin, V.E.; Scheu, A.; Mattiangeli, V.; Teasdale, M.D.; Hare, A.J.; Burger, J.; Verdugo, M.P.; Collins, M.J.; et al. Ancient goat genomes reveal mosaic domestication in the Fertile Crescent. Science 2018, 361, 85–88. [Google Scholar] [CrossRef]
- Bertolini, F.; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhiere, I.; Crisa, A.; Catillo, G.; Steri, R.; et al. Signatures of selection and environmental adaptation across the goat genome post-domestication. Genet. Sel. Evol. 2018, 50, 57. [Google Scholar] [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Neto, L.R.P.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Tao, H.; Li, P.; Li, L.; Zhong, T.; Wang, L.; Ma, J.; Chen, X.; Song, T.; Zhang, H. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 2018, 8, 10405. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Liu, X.; Gebreselassie, G.; Abied, A.; Ma, Q.; Ma, Y. Genome-wide association analysis reveals the genetic locus for high reproduction trait in Chinese Arbas Cashmere goat. Genes Genom. 2020, 42, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Reverter, A.; Porto-Neto, L.R.; Fortes, M.R.S.; McCulloch, R.; Lyons, R.E.; Moore, S.; Nicol, D.; Henshall, J.; Lehnert, S.A. Genomic analyses of tropical beef cattle fertility based on genotyping pools of Brahman cows with unknown pedigree. J. Anim. Sci. 2016, 94, 4096–4108. [Google Scholar] [CrossRef]
- Verardo, L.L.; Silva, F.F.; Lopes, M.S.; Madsen, O.; Bastiaansen, J.W.M.; Knol, E.F.; Kelly, M.; Varona, L.; Lopes, P.S.; Guimarães, S.E.F. Revealing new candidate genes for reproductive traits in pigs: Combining Bayesian GWAS and functional pathways. Genet. Sel. Evol. 2016, 48, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Guo, J.; Li, L.; Zhong, T.; Wang, L.; Zhan, S.; Lu, J.; Wang, D.; Dai, D.; Liu, G.E.; et al. Genetic Diversity and Selection Signatures in Jianchang Black Goats Revealed by Whole-Genome Sequencing Data. Animals 2022, 12, 2365. https://doi.org/10.3390/ani12182365
Sun X, Guo J, Li L, Zhong T, Wang L, Zhan S, Lu J, Wang D, Dai D, Liu GE, et al. Genetic Diversity and Selection Signatures in Jianchang Black Goats Revealed by Whole-Genome Sequencing Data. Animals. 2022; 12(18):2365. https://doi.org/10.3390/ani12182365
Chicago/Turabian StyleSun, Xueliang, Jiazhong Guo, Li Li, Tao Zhong, Linjie Wang, Siyuan Zhan, Juan Lu, Decheng Wang, Dinghui Dai, George E. Liu, and et al. 2022. "Genetic Diversity and Selection Signatures in Jianchang Black Goats Revealed by Whole-Genome Sequencing Data" Animals 12, no. 18: 2365. https://doi.org/10.3390/ani12182365
APA StyleSun, X., Guo, J., Li, L., Zhong, T., Wang, L., Zhan, S., Lu, J., Wang, D., Dai, D., Liu, G. E., & Zhang, H. (2022). Genetic Diversity and Selection Signatures in Jianchang Black Goats Revealed by Whole-Genome Sequencing Data. Animals, 12(18), 2365. https://doi.org/10.3390/ani12182365