
Genetics Responses to Hypoxia and Reoxygenation Stress in Larimichthys crocea Revealed via Transcriptome Analysis and Weighted Gene Co-Expression Network

Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Illumina Sequencing and Reads Mapping
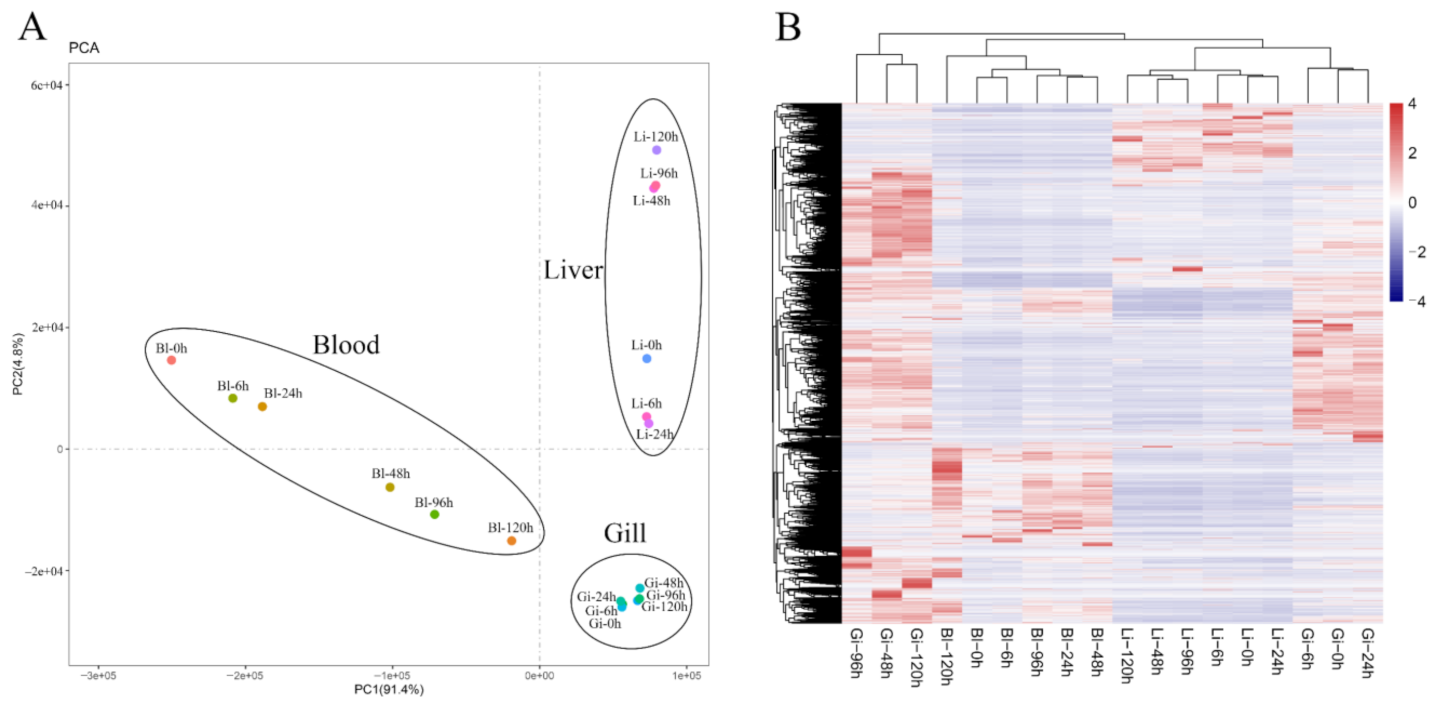
2.2. Correlation Analysis between Samples
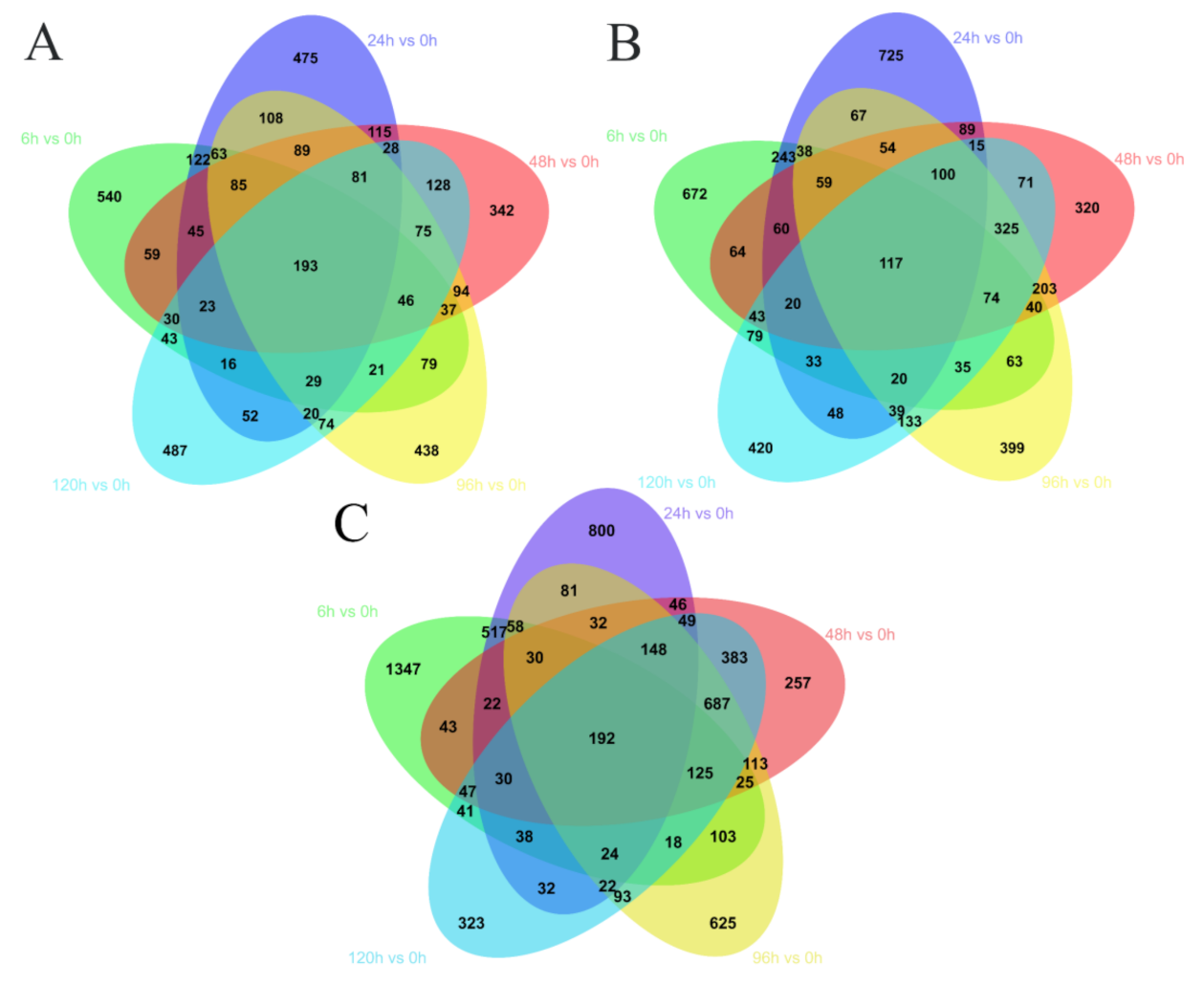
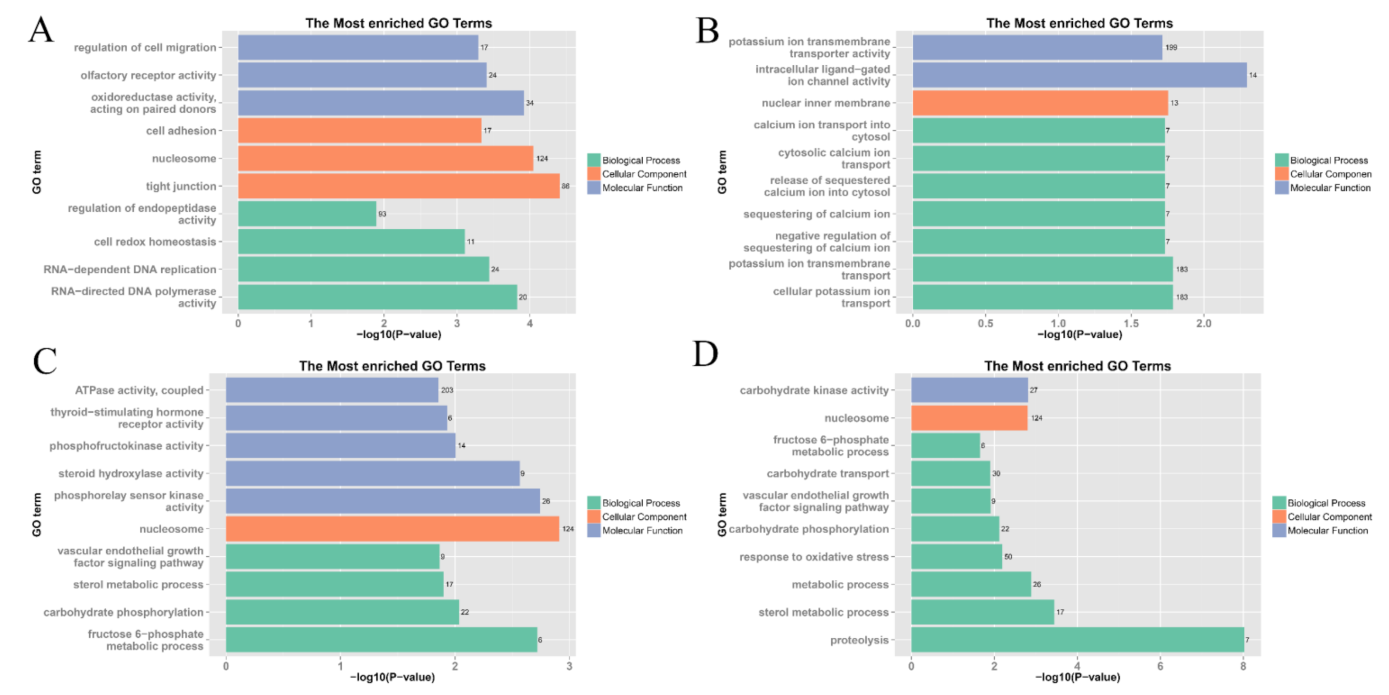
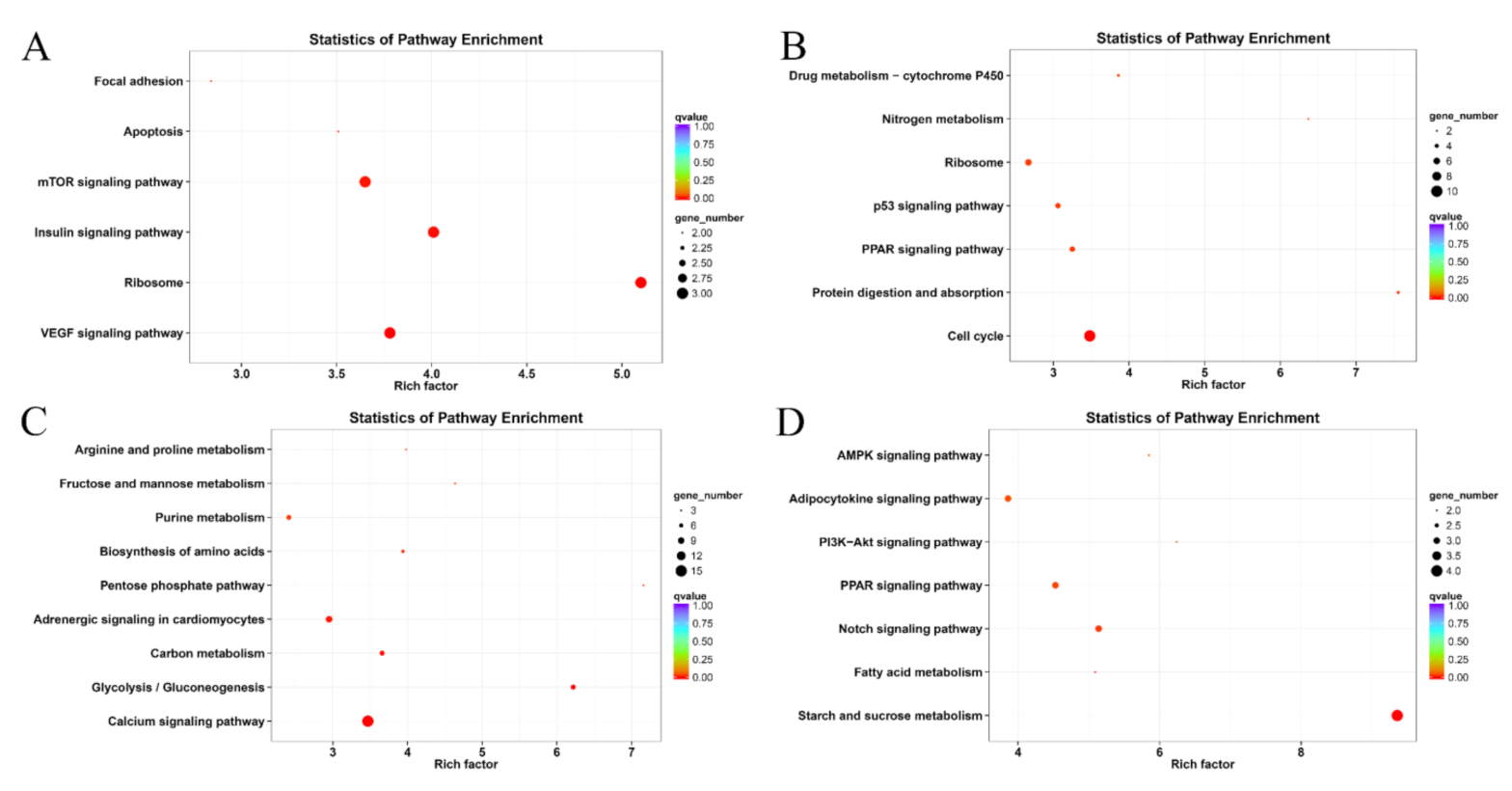
2.3. Differentially Expressed Genes (DEGs)
2.4. WGCNA
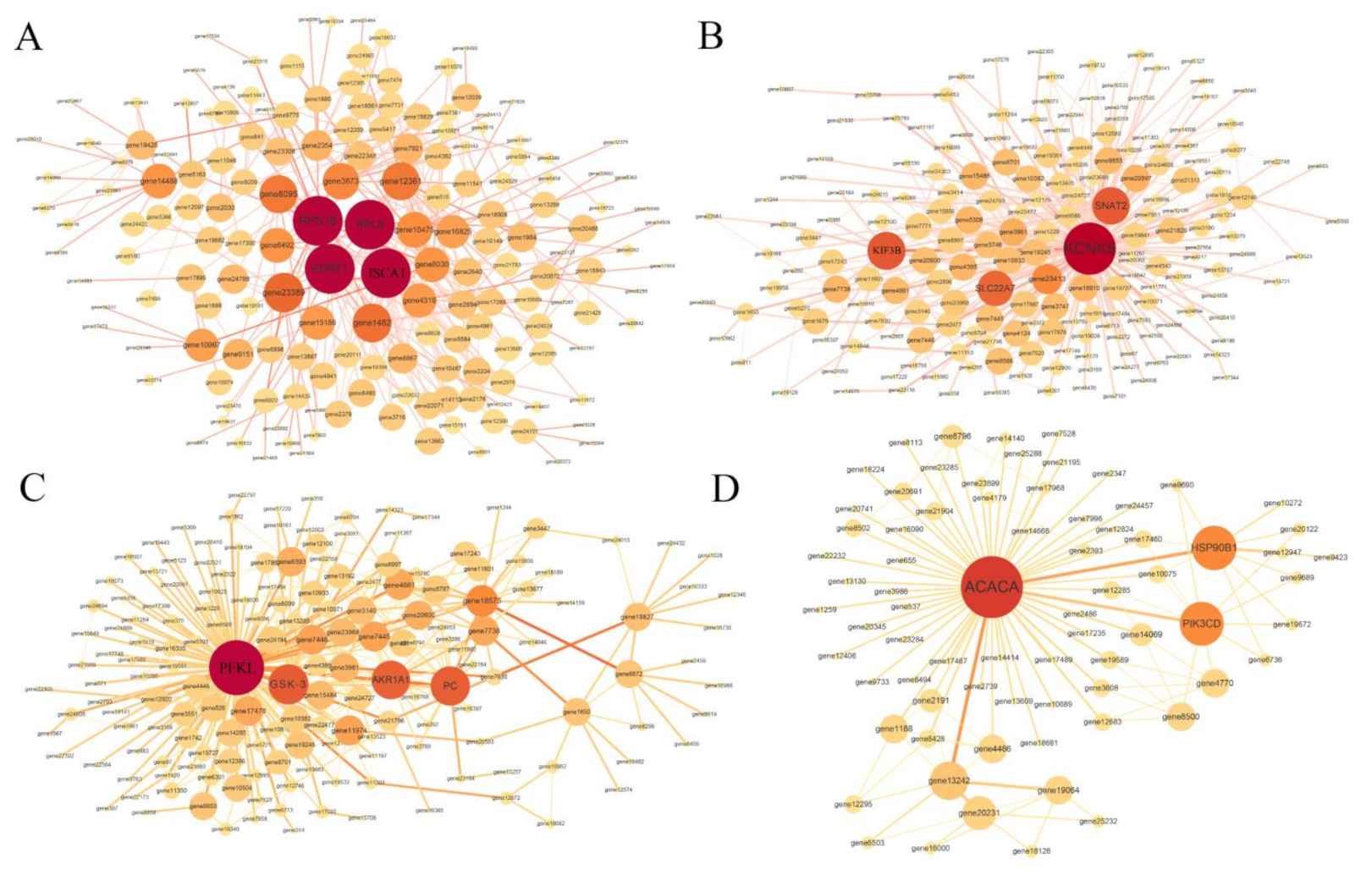
2.5. Hub Genes Selections
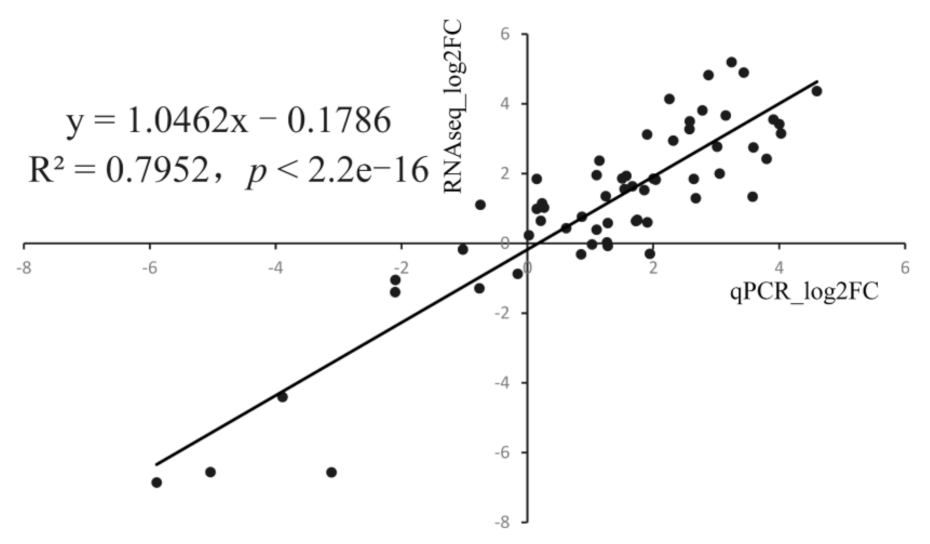
2.6. Quantitative PCR Validation
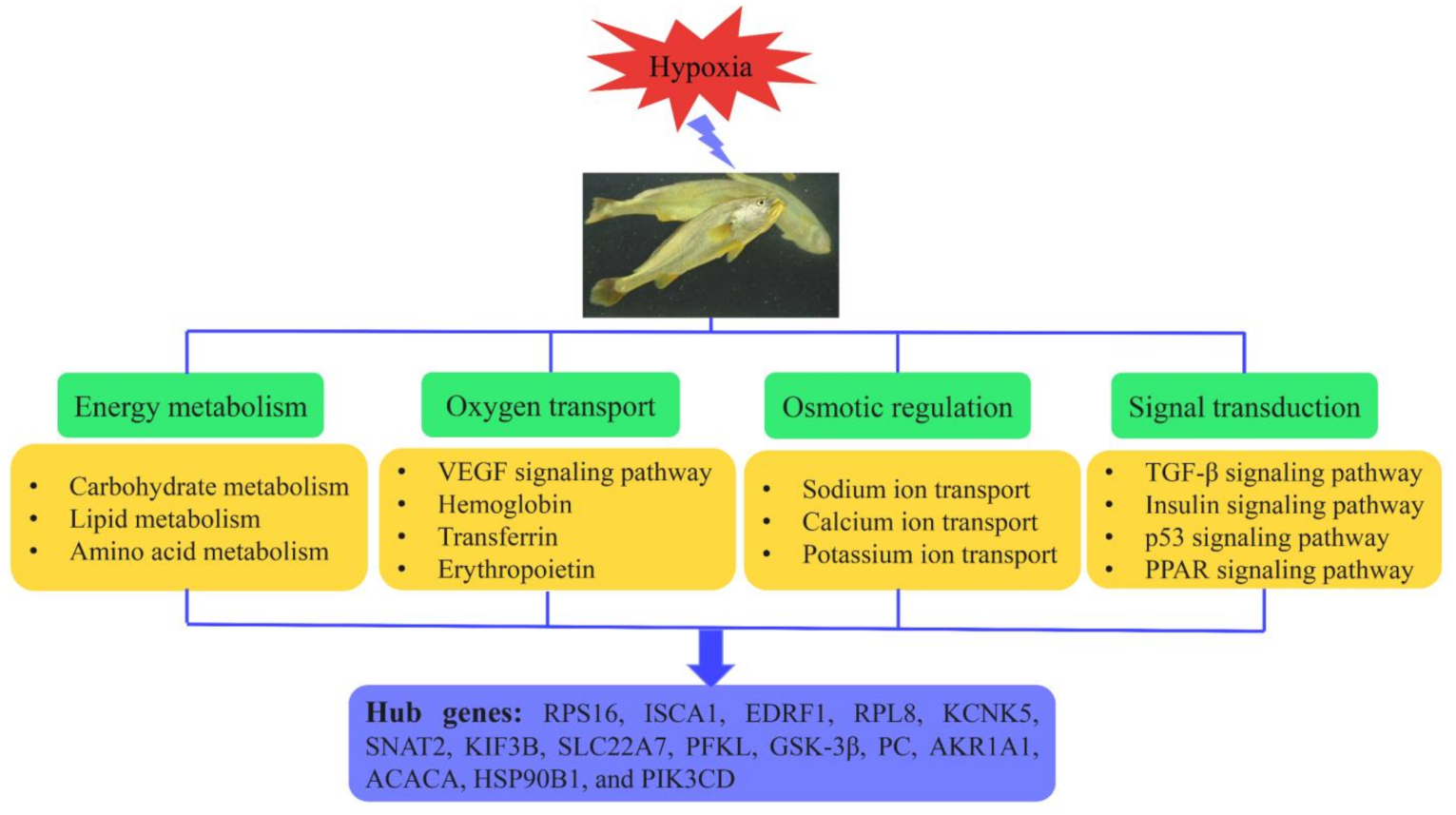
3. Discussion
3.1. Effects of Hypoxia and Reoxygenation on the L. crocea Transcriptome
3.2. Functional Analysis of Hub Genes
4. Materials and Methods
4.1. Experimental Materials and Hypoxia Experiments
4.2. RNA Sequencing
4.3. Data Processing and Analysis
4.4. Weighted Gene Co-Expression Network Analysis
4.5. Quantitative Real-Time PCR Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACACA | acetyl-CoA carboxylase alpha |
| AKR1A1 | alcohol dehydrogenase [NADP(+)] A isoform X1 |
| CS | citrate synthase |
| DEGs | differentially expressed genes |
| DO | dissolved oxygen |
| EDRF1 | erythroid differentiation-related factor 1 |
| EPO | erythropoietin |
| FDR | false discovery rate |
| GO | gene ontology |
| GSK3α/β | glycogen synthase kinase 3α/β |
| HIF-1α | hypoxia-inducible factor 1α |
| HK1 | hexokinase-1 |
| HSP90B1 | heat shock protein 90 kDa beta member 1 |
| ISCA1 | iron-sulfur cluster assembly 1 |
| KCNK5 | potassium channel subfamily K member 5 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KIF3B | kinesin superfamily protein 3B |
| LDHA | lactate dehydrogenase A |
| PC | pyruvate carboxylase |
| PCA | principal component analysis |
| PEPCK | phosphoenolpyruvate carboxykinase |
| PFKL | phosphofructokinase, liver type |
| PIK3CD | phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta |
| qRT-PCR | quantitative real-time PCR |
| RIN | relative intensity noise |
| RNA-seq | RNA sequencing |
| RPL8 | 60S ribosomal protein L8 |
| RPS16 | 40S ribosomal protein S16 |
| SDHB | succinate dehydrogenase complex subunits B |
| SLC22A7 | solute carrier family 22 member 7 |
| SNAT2 | sodium-coupled neutral amino acid transporter 2 |
| TF | transferrin |
| TGF-β | transforming growth factor-β |
| TPI | triosephosphate isomerase |
| VEGF | vascular endothelial growth factor |
| WGCNA | weighted gene co-expression network analysis |
References
- Conley, D.J.; Carstensen, J.; Vaquer-Sunyer, R.; Duarte, C.M. Ecosystem thresholds with hypoxia. Hydrobiologia 2009, 629, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.S.; Wen, H.S.; Li, J.F.; He, F.; Li, Y.; Qi, X. Environmental hypoxia causes growth retardation, osteoclast differentiation and calcium dyshomeostasis in juvenile rainbow trout (Oncorhynchus mykiss). Sci. Total Environ. 2020, 705, 135272. [Google Scholar] [CrossRef]
- Lee, J.; Park, K.-T.; Lim, J.-H.; Yoon, J.-E.; Kim, I.-N. Hypoxia in Korean Coastal Waters: A Case Study of the Natural Jinhae Bay and Artificial Shihwa Bay. Front. Mar. Sci. 2018, 5, 70, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Stow, C.A.; Qian, S.S.; Craig, J.K. Declining threshold for hypoxia in the Gulf of Mexico. Environ. Sci. Technol. 2005, 39, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Conley, D.J.; Carstensen, J.; Aigars, J.; Axe, P.; Bonsdorff, E.; Eremina, T.; Haahti, B.-M.; Humborg, C.; Jonsson, P.; Kotta, J.; et al. Hypoxia Is Increasing in the Coastal Zone of the Baltic Sea. Environ. Sci. Technol. 2011, 45, 6777–6783. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Song, X.; Yuan, Y.; Wang, W.; Cao, X.; Wu, Z.; Yu, Z. Main factors dominating the development, formation and dissipation of hypoxia off the Changjiang Estuary (CE) and its adjacent waters, China. Environ. Pollut. 2020, 265, 115066. [Google Scholar] [CrossRef]
- Wu, R.S.S. Hypoxia: From molecular responses to ecosystem responses. Mar. Pollut. Bull. 2002, 45, 35–45. [Google Scholar] [CrossRef]
- Ao, J.; Mu, Y.; Xiang, L.X.; Fan, D.; Feng, M.; Zhang, S.; Shi, Q.; Zhu, L.Y.; Li, T.; Ding, Y.; et al. Genome sequencing of the perciform fish Larimichthys crocea provides insights into molecular and genetic mechanisms of stress adaptation. PLoS Genet. 2015, 11, e1005118. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Liu, C.; Luo, S.; Zhang, Y.; Gao, X.; Wu, X.; Shen, W.; Zhu, J. Transcriptome and physiology analysis identify key metabolic changes in the liver of the large yellow croaker (Larimichthys crocea) in response to acute hypoxia. Ecotoxicol. Environ. Saf. 2020, 189, 109957. [Google Scholar] [CrossRef]
- Sun, P.; Bao, P.; Tang, B. Transcriptome analysis and discovery of genes involved in immune pathways in large yellow croaker (Larimichthys crocea) under high stocking density stress. Fish. Shellfish Immunol. 2017, 68, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.F.; Shen, W.L.; Hou, C.C.; Liu, C.; Wu, X.F.; Zhu, J.Q. Physiological responses and changes in gene expression in the large yellow croaker Larimichthys crocea following exposure to hypoxia. Chemosphere 2017, 169, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Hirono, I.; Kondo, H.; Hikima, J.-I.; Jung, T.S. Microarray technology is an effective tool for identifying genes related to the aquacultural improvement of Japanese flounder. Paralichthys Olivaceus. Comp. Biochem. Physiol. D Genom. Proteom. 2011, 6, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.E. Microarray studies of gene expression in fish. OMICS 2006, 10, 474–489. [Google Scholar] [CrossRef] [PubMed]
- Wenne, R.; Handschuh, L.; Pocwierz-Kotus, A.; Urbaniak, R.; Formanowicz, P.; Calkiewicz, J.; Brzozowska, K.; Figlerowicz, M.; Wegrzyn, G.; Wrobel, B. The application of microarray technology to the identification of Tc1-like element sequences in fish genomes. Mar. Biol. Res. 2011, 7, 466–477. [Google Scholar] [CrossRef]
- Xiaolin, L.; Lei, C.; Peng, X.; Guoqing, L.; Michael, W.; Xiaowen, S.; Songlin, C. Transcriptome analysis of crucian carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS ONE 2013, 8, e62308. [Google Scholar]
- Marques, I.J.; Leito, J.T.D.; Spaink, H.P.; Testerink, J.; Jaspers, R.T.; Witte, F.; Berg, S.V.D.; Bagowski, C.P. Transcriptome analysis of the response to chronic constant hypoxia in zebrafish hearts. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 2008, 178, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.; Yoon, D.; Lim, Y.; Roh, H.J.; Kim, S.; Park, C.-I.; Kim, H.-S.; Cha, H.-J.; Choi, Y.H.; Kim, D.-H. Co-Expression Network Analysis of Spleen Transcriptome in Rock Bream (Oplegnathus fasciatus) Naturally Infected with Rock Bream Iridovirus (RBIV). Int. J. Mol. Sci. 2020, 21, 1707. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yu, H.; Kong, L.; Li, Q. Gene Co-Expression Network Analysis Reveals the Correlation Patterns Among Genes in Euryhaline Adaptation of Crassostrea gigas. Mar. Biotechnol. 2016, 18, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Ma, A.; Yang, S.; Liu, X.; Zhao, T.; Zhang, J.; Wang, X.-A.; Sun, Z.; Liu, Z.; Xu, R. Transcriptome analysis and weighted gene co-expression network reveals potential genes responses to heat stress in turbot Scophthalmus maximus. Comp. Biochem. Physiol. D Genom. Proteom. 2020, 33, 100632. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Xin, G.; Zhao, L.M.; Huang, L.X.; Qin, Y.X.; Su, Y.Q.; Zheng, W.Q.; Wu, B.; Lin, N.; Yan, Q.P. Novel insights into host-pathogen interactions of large yellow croakers (Larimichthys crocea) and pathogenic bacterium Pseudomonas plecoglossicida using time-resolved dual RNA-seq of infected spleens. Zool. Res. 2020, 41, 314–327. [Google Scholar] [CrossRef]
- Jian Xu, Q.L.L.X. Shaolin Wang, Yanliang Jiang, Zixia Zhao, Yan Zhang, Jiongtang Li, Chuanju Dong, Peng Xu, Xiaowen Sun, Gene expression changes leading extreme alkaline tolerance in Amur ide (Leuciscus waleckii) inhabiting soda lake. BMC Genom. 2013, 14, 1–11. [Google Scholar]
- Guo, L.; Wang, Y.; Liang, S.; Lin, G.; Chen, S.; Yang, G. Tissue-overlapping response of half-smooth tongue sole (Cynoglossus semilaevis) to thermostressing based on transcriptome profiles. Gene 2016, 586, 97–104. [Google Scholar] [CrossRef]
- Yang, Y.; Fu, Q.; Wang, X.; Liu, Y.; Zeng, Q.; Li, Y.; Gao, S.; Bao, L.; Liu, S.; Gao, D.; et al. Comparative transcriptome analysis of the swimbladder reveals expression signatures in response to low oxygen stress in channel catfish, Ictalurus punctatus. Physiol. Genom. 2018, 50, 636–647. [Google Scholar] [CrossRef]
- Zhang, G.; Yin, S.; Mao, J.; Liang, F.; Zhao, C.; Li, P.; Zhou, G.; Chen, S.; Tang, Z. Integrated analysis of mRNA-seq and miRNA-seq in the liver of Pelteobagrus vachelli in response to hypoxia. Sci. Rep. 2016, 6, 22907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Teleman, A.A. Molecular mechanisms of metabolic regulation by insulin in Drosophila. Biochem. J. 2009, 425, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Eckel, J.; Reinauer, H. Insulin action on glucose transport in isolated cardiac myocytes: Signalling pathways and diabetes-induced alterations. Biochem. Soc. Trans. 1990, 18, 1125–1127. [Google Scholar] [CrossRef] [PubMed]
- Ahren, B. Glucagon--Early breakthroughs and recent discoveries. Peptides 2015, 67, 74–81. [Google Scholar] [CrossRef]
- Kular, J.K.; Basu, S.; Sharma, R.I. The extracellular matrix: Structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J. Tissue. Eng. 2014, 5, 1–17. [Google Scholar] [CrossRef]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J.; Schipani, E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res. 2010, 339, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-l.; Zhang, Z.-x.; Tan, Z. Mechanisms of focal adhesion kinase in the proliferation of human pulmonary artery smooth cells under hypoxia. Zhonghua Yi Xue Za Zhi 2011, 91, 2274–2277. [Google Scholar]
- Brown, R.C.; Mark, K.S.; Egleton, R.D.; Huber, J.D.; Burroughs, A.R.; Davis, T.P. Protection against hypoxia-induced increase in blood-brain barrier permeability: Role of tight junction proteins and NFκB. J. Cell Sci. 2003, 116, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matey, V.; Richards, J.G.; Wang, Y.; Wood, C.M.; Rogers, J.; Davies, R.; Murray, B.W.; Chen, X.Q.; Du, J.; Brauner, C.J. The effect of hypoxia on gill morphology and ionoregulatory status in the Lake Qinghai scaleless carp, Gymnocypris przewalskii. J. Exp. Biol. 2008, 211, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.M.; Kajimura, M.; Sloman, K.A.; Scott, G.R.; Walsh, P.J.; Almeida-Val, V.M.F.; Val, A.L. Rapid regulation of Na+ fluxes and ammonia excretion in response to acute environmental hypoxia in the Amazonian oscar, Astronotus ocellatus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R2048–R2058. [Google Scholar] [CrossRef] [Green Version]
- Boeuf, G.; Payan, P. How should salinity influence fish growth? Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2001, 130, 411–423. [Google Scholar] [CrossRef]
- Marshall, W.S.; Emberley, T.R.; Singer, T.D.; Bryson, S.E.; McCormick, S.D. Time course of salinity adaptation in a strongly euryhaline estuarine teleost, Fundulus heteroclitus: A multivariable approach. J. Exp. Biol. 1999, 202, 1535–1544. [Google Scholar] [CrossRef]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Amano, H.; Ito, Y.; Kashiwagi, S.; Yamazaki, Y.; Takeda, A.; Shibuya, M.; Kitasato, H.; Majima, M. Vascular endothelial growth factor receptor-1 (VEGFR-1) signaling enhances angiogenesis in a surgical sponge model. Biomed. Pharmacol. 2016, 78, 140–149. [Google Scholar] [CrossRef]
- Maybin, J.A.; Hirani, N.; Brown, P.; Jabbour, H.N.; Critchley, H.O.D. The Regulation of Vascular Endothelial Growth Factor by Hypoxia and Prostaglandin F-2 alpha during Human Endometrial Repair. J. Clin. Endocrinol. Metab. 2011, 96, 2475–2483. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, W.; Feng, S.; Wang, R. Research progress on mechanism in adaptation of hemoglobin to plateau hypoxia. Zhejiang Da Xue Xue Bao. Yi Xue Ban = J. Zhejiang Univ. Med. Sci. 2019, 48, 674–681. [Google Scholar]
- Frey, B.J.; Weber, R.E.; van Aardt, W.J.; Fago, A. The haemoglobin system of the mudfish, Labeo capensis: Adaptations to temperature and hypoxia. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 120, 735–742. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, Z.; Wang, R.; Sun, Y.; Xu, T. Miiuy Croaker Transferrin Gene and Evidence for Positive Selection Events Reveal Different Evolutionary Patterns. PLoS ONE 2012, 7, e43936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomme, P.T.; McCann, K.B. Transferrin: Structure, function and potential therapeutic actions. Drug Discov. Today 2005, 10, 267–273. [Google Scholar] [CrossRef]
- Li, Y.R.; Yu, P.; Chang, S.Y.; Wu, Q.; Yu, P.P.; Xie, C.C.; Wu, W.Y.; Zhao, B.L.; Gao, G.F.; Chang, Y.Z. Hypobaric Hypoxia Regulates Brain Iron Homeostasis in Rats. J. Cell. Biochem. 2017, 118, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Rolfs, A.; Marti, H.H.; Bauer, C.; Gassmann, M. Hypoxia, a novel inducer of acute-phase gene-expression in a human hepatoma-cell line. J. Biol. Chem. 1995, 270, 27865–27870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, Y.M.; Xie, L. Hypoxia-Inducible Factors Link Iron Homeostasis and Erythropoiesis. Gastroenterology 2014, 146, 630–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Zakharov, I.M. Sensitivity of cells to oxygen and erythropoietin production. Rossiiskii Fiziologicheskii Zhurnal Imeni IM Sechenova 2005, 91, 993–1004. [Google Scholar]
- Vetrovoy, O.V.; Rybnikova, E.A.; Glushchenko, T.S.; Baranova, K.A.; Samoilov, M.O. Mild hypobaric hypoxic postconditioning increases the expression of HIF-1 alpha and erythropoietin in the CA1 field of the hippocampus of rats that survive after severe hypoxia. Neurochem. J. 2014, 8, 103–108. [Google Scholar] [CrossRef]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted Gene Coexpression Network Analysis: State of the Art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef]
- Liu, W.; Li, L.; Ye, H.; Tu, W. Weighted gene co-expression network analysis in biomedicine research. Sheng Wu Gong Cheng Xue Bao/Chin. J. Biotechnol. 2017, 33, 1791–1801. [Google Scholar]
- Diao, J.; Yu, X.; Wang, X.; Fan, Y.; Wang, S.; Li, L.; Wang, Y.; Xu, L.; Gai, C.; Ye, H.; et al. Full-length transcriptome sequencing combined with RNA-seq analysis revealed the immune response of fat greenling (Hexagrammos otakii) to Vibrio harveyi in early infection. Microb. Pathog. 2020, 149, 104527. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.-L.; et al. MicroRNA-210 Regulates Mitochondrial Free Radical Response to Hypoxia and Krebs Cycle in Cancer Cells by Targeting Iron Sulfur Cluster Protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef] [Green Version]
- Korde, A.S.; Yadav, V.R.; Zheng, Y.-M.; Wang, Y.-X. Primary role of mitochondrial Rieske iron-sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic. Biol. Med. 2011, 50, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.M.; Yang, Z.; Joseph, L.; Song, T.; Wang, Y.X. Hypoxia-Induced Ryanodine Receptor-Medicated Ca2+ Release Causes Rieske Iron-Sulfur Protein-Dependent Mitochondrial Reactive Oxygen Species Production in Pulmonary Artery Myocytes. Am. J. Respir. Crit. Care Med. 2015, 191, A5278. [Google Scholar]
- Wang, D.C.; Li, Y.; Shen, B.F. Initial function analysis of a novel erythroid differentiation related gene EDRF1. Sci. China Ser. C Life Sci. 2001, 44, 489–496. [Google Scholar] [CrossRef]
- Wang, X.; Wang, D.C.; Chen, X.; Hu, M.R.; Wang, J.; Li, Y.; Guo, N.; Shen, B.F. cDNA cloning and function analysis of two novel erythroid differentiation related genes. Sci. China Ser. C Life Sci. 2001, 44, 99–105. [Google Scholar] [CrossRef]
- Wood, A.J.; Duff, R.J.; Oliver, M.J. The translational apparatus of Tortula ruralis: Polysomal retention of transcripts encoding the ribosomal proteins RPS14, RPS16 and RPL23 in desiccated and rehydrated gametophytes. J. Exp. Bot. 2000, 51, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.-r.; Hou, Y.-l.; Hao, Y.-z.; Zhang, T.; Peng, Z.-s.; Wu, Y.-S.; Wu, G.-f. cDNA, genomic sequence cloning and overexpression of ribosomal protein S16 gene (RPS16) from the Giant Panda. Afr. J. Biotechnol. 2010, 9, 2185–2191. [Google Scholar]
- Chowdhury, R.; Sekirnik, R.; Brissett, N.C.; Krojer, T.; Ho, C.-h.; Ng, S.S.; Clifton, I.J.; Ge, W.; Kershaw, N.J.; Fox, G.C.; et al. Ribosomal oxygenases are structurally conserved from prokaryotes to humans. Nature 2014, 510, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazier, S.P.; Mason, H.S.; Bateson, A.N.; Kemp, P.J. Cloning of the human TASK-2 (KCNK5) promoter and its regulation by chronic hypoxia. Biochem. Biophys. Res. Commun. 2005, 336, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Lin, H.; Zheng, H.; Kim, K.S.; Kim, J.Y.; Chun, Y.S.; Park, J.W.; Nam, J.H.; Kim, W.K.; Zhang, Y.H.; et al. HIF-1alpha-mediated upregulation of TASK-2 K(+) channels augments Ca(2)(+) signaling in mouse B cells under hypoxia. J. Immunol. 2014, 193, 4924–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelina, R.; Laura, C.B.; Francisco, Z.; Cecilio, G.; Maria Guadalupe, G.-A. Expression of the SNAT2 amino acid transporter during the development of rat cerebral cortex. Int. J. Dev. Neurosci. 2011, 29, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gameiro, A.; Grewer, C. Highly conserved asparagine 82 controls the interaction of Na+ with the sodium-coupled neutral amino acid transporter SNAT2. J. Biol. Chem. 2008, 283, 12284–12292. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Ohshiro, N.; Sakai, R.; Ohbayashi, M.; Kohyama, N.; Yamamoto, T. Transport mechanism and substrate specificity of human organic anion transporter 2 (hOat2 SLC22A7). J. Pharm. Pharmacol. 2005, 57, 573–578. [Google Scholar] [CrossRef]
- Gumy, L.F.; Chew, D.J.; Tortosa, E.; Katrukha, E.A.; Kapitein, L.C.; Tolkovsky, A.M.; Hoogenraad, C.C.; Fawcett, J.W. The Kinesin-2 Family Member KIF3C Regulates Microtubule Dynamics and Is Required for Axon Growth and Regeneration. J. Neurosci. 2013, 33, 11329–11345. [Google Scholar] [CrossRef]
- Sardella, M.; Navone, F.; Rocchi, M.; Rubartelli, A.; Viggiano, L.; Vignali, G.; Consalez, G.G.; Sitia, R.; Cabibbo, A. KIF3C, a novel member of the kinesin superfamily: Sequence, expression, and mapping to human chromosome 2 at 2p23. Genomics 1998, 47, 405–408. [Google Scholar] [CrossRef]
- Cota-Ruiz, K.; Leyva-Carrillo, L.; Peregrino-Uriarte, A.B.; Valenzuela-Soto, E.M.; Gollas-Galvan, T.; Gomez-Jimenez, S.; Hernandez, J.; Yepiz-Plascencia, G. Role of HIF-1 on phosphofructokinase and fructose 1, 6-bisphosphatase expression during hypoxia in the white shrimp Litopenaeus vannamei. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2016, 198, 1–7. [Google Scholar] [CrossRef]
- Cota-Ruiz, K.; Peregrino-Uriarte, A.B.; Felix-Portillo, M.; Martinez-Quintana, J.A.; Yepiz-Plascencia, G. Expression of fructose 1,6-bisphosphatase and phosphofructokinase is induced in hepatopancreas of the white shrimp Litopenaeus vannamei by hypoxia. Mar. Environ. Res. 2015, 106, 1–9. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional insights from cell biology and animal models. Front. Molec. Neurosci. 2011, 4, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Ahn, M.; Kim, J.; Park, C.; Cho, J.; Jee, Y.; Jung, K.; Moon, C.; Shin, T. Potential involvement of glycogen synthase kinase (GSK)-3beta in a rat model of multiple sclerosis: Evidenced by lithium treatment. Anat. Cell Biol. 2017, 50, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Fluegel, D.; Goerlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1 alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell. Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, J.C. My Favorite Pyruvate Carboxylase Feature. IUBMB Life 2010, 62, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; He, Y.; Wang, X.; Zhang, Y.; Hu, M.; Guo, W.; He, Y.; Zhang, T.; Lai, L.; Sun, Z.; et al. Targeting Pyruvate Carboxylase by a Small Molecule Suppresses Breast Cancer Progression. Adv. Sci. 2020, 7, 1903483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phannasil, P.; Thuwajit, C.; Warnnissorn, M.; Wallace, J.C.; MacDonald, M.J.; Jitrapakdee, S. Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells. PLoS ONE 2015, 10, e0129848. [Google Scholar] [CrossRef] [Green Version]
- Shang, H.; Zheng, J.; Tong, J. Integrated analysis of transcriptomic and metabolomic data demonstrates the significant role of pyruvate carboxylase in the progression of ovarian cancer. Aging 2020, 12, 21874–21889. [Google Scholar] [CrossRef]
- Holopainen, I.J.; Tonn, W.M.; Paszkowski, C.A. Tales of two fish: The dichotomous biology of crucian carp (Carassius carassius (L)) in northern Europe. Ann. Zool. Fenn. 1997, 34, 1–22. [Google Scholar]
- Shoubridge, E.A.; Hochachka, P.W. Ethanol: Novel end product of vertebrate anaerobic metabolism. Science 1980, 209, 308–309. [Google Scholar] [CrossRef]
- Johnston, I.A.; Bernard, L.M. Utilization of the ethanol pathway in carp following exposure to anoxia. J. Exp. Biol. 1983, 104, 73–78. [Google Scholar] [CrossRef]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. 2005, 62, 1784–1803. [Google Scholar] [CrossRef]
- Keenan, M.M.; Liu, B.; Tang, X.; Wu, J.; Cyr, D.; Stevens, R.D.; Ilkayeva, O.; Huang, Z.; Tollini, L.A.; Murphy, S.K.; et al. ACLY and ACC1 Regulate Hypoxia-Induced Apoptosis by Modulating ETV4 via alpha-ketoglutarate. PLoS Genet. 2015, 11, e1005599. [Google Scholar] [CrossRef] [Green Version]
- Zhai, R.; Gong, X.-Y.; Xiong, X.-C.; Jiang, Y.; Huang, Z.-J.; Fang, X. Progress in Molecular Chaperone Regulation of Heat Shock Protein 90 and Cancer. Chin. J. Anal. Chem. 2018, 46, 301–310. [Google Scholar]
- Prodromou, C.; Pearl, L.H. Structure and Functional Relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Daehwan, K.; Ben, L.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11(R14), 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Tao, C.J.G.O.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Gene Symbol | Gene ID | Description | Degree |
|---|---|---|---|---|
| darkorange | RPS16 | EH28_19390 | 40S ribosomal protein S16 | 42 |
| darkorange | ISCA1 | EH28_11232 | Iron-sulfur cluster assembly 1 | 38 |
| darkorange | EDRF1 | EH28_15699 | Erythroid differentiation-related factor 1 | 37 |
| darkorange | RPL8 | EH28_17632 | 60S ribosomal protein L8 | 37 |
| magenta | KCNK5 | EH28_04734 | Potassium channel subfamily K member 5 | 118 |
| magenta | SNAT2 | EH28_19256 | Sodium-coupled neutral amino acid transporter 2 | 58 |
| magenta | KIF3B | EH28_23591 | Kinesin-like protein KIF3B | 56 |
| magenta | SLC22A7 | EH28_16974 | Solute carrier family 22 member 7 | 48 |
| saddlebrown | PFKL | EH28_06053 | phosphofructokinase, liver type | 97 |
| saddlebrown | GSK-3β | EH28_10886 | Glycogen synthase kinase-3 beta | 42 |
| saddlebrown | PC | EH28_15368 | Pyruvate carboxylase, mitochondrial | 40 |
| saddlebrown | AKR1A1 | EH28_24096 | Alcohol dehydrogenase [NADP(+)] A isoform X1 | 38 |
| darkolivegreen | ACACA | EH28_08273 | Acetyl-CoA carboxylase alpha | 56 |
| darkolivegreen | HSP90B1 | EH28_10845 | Heat shock protein 90 kDa beta member 1 | 20 |
| darkolivegreen | PIK3CD | EH28_12563 | Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Ding, J.; Liu, C.; Luo, S.; Gao, X.; Wu, Y.; Wang, J.; Wang, X.; Wu, X.; Shen, W.; et al. Genetics Responses to Hypoxia and Reoxygenation Stress in Larimichthys crocea Revealed via Transcriptome Analysis and Weighted Gene Co-Expression Network. Animals 2021, 11, 3021. https://doi.org/10.3390/ani11113021
Zhang Y, Ding J, Liu C, Luo S, Gao X, Wu Y, Wang J, Wang X, Wu X, Shen W, et al. Genetics Responses to Hypoxia and Reoxygenation Stress in Larimichthys crocea Revealed via Transcriptome Analysis and Weighted Gene Co-Expression Network. Animals. 2021; 11(11):3021. https://doi.org/10.3390/ani11113021
Chicago/Turabian StyleZhang, Yibo, Jie Ding, Cheng Liu, Shengyu Luo, Xinming Gao, Yuanjie Wu, Jingqian Wang, Xuelei Wang, Xiongfei Wu, Weiliang Shen, and et al. 2021. "Genetics Responses to Hypoxia and Reoxygenation Stress in Larimichthys crocea Revealed via Transcriptome Analysis and Weighted Gene Co-Expression Network" Animals 11, no. 11: 3021. https://doi.org/10.3390/ani11113021
APA StyleZhang, Y., Ding, J., Liu, C., Luo, S., Gao, X., Wu, Y., Wang, J., Wang, X., Wu, X., Shen, W., & Zhu, J. (2021). Genetics Responses to Hypoxia and Reoxygenation Stress in Larimichthys crocea Revealed via Transcriptome Analysis and Weighted Gene Co-Expression Network. Animals, 11(11), 3021. https://doi.org/10.3390/ani11113021