
Hybrid Sequencing in Different Types of Goat Skeletal Muscles Reveals Genes Regulating Muscle Development and Meat Quality

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Management and Sample Collection
2.2. Meat Quality Analysis
2.3. Short-Read RNA Sequencing
2.4. Full-Length RNA Sequencing
2.5. Analysis of Short-Read Sequencing Data
2.6. Analysis of Full-Length Sequencing Raw Data
2.7. Identification and Functional Annotation of Novel Genes and Isoforms
2.8. Identification of lncRNAs and Novel Isoforms’ Open Reading Frames
2.9. Quantitative Real-Time PCR (qPCR) and Data Analysis
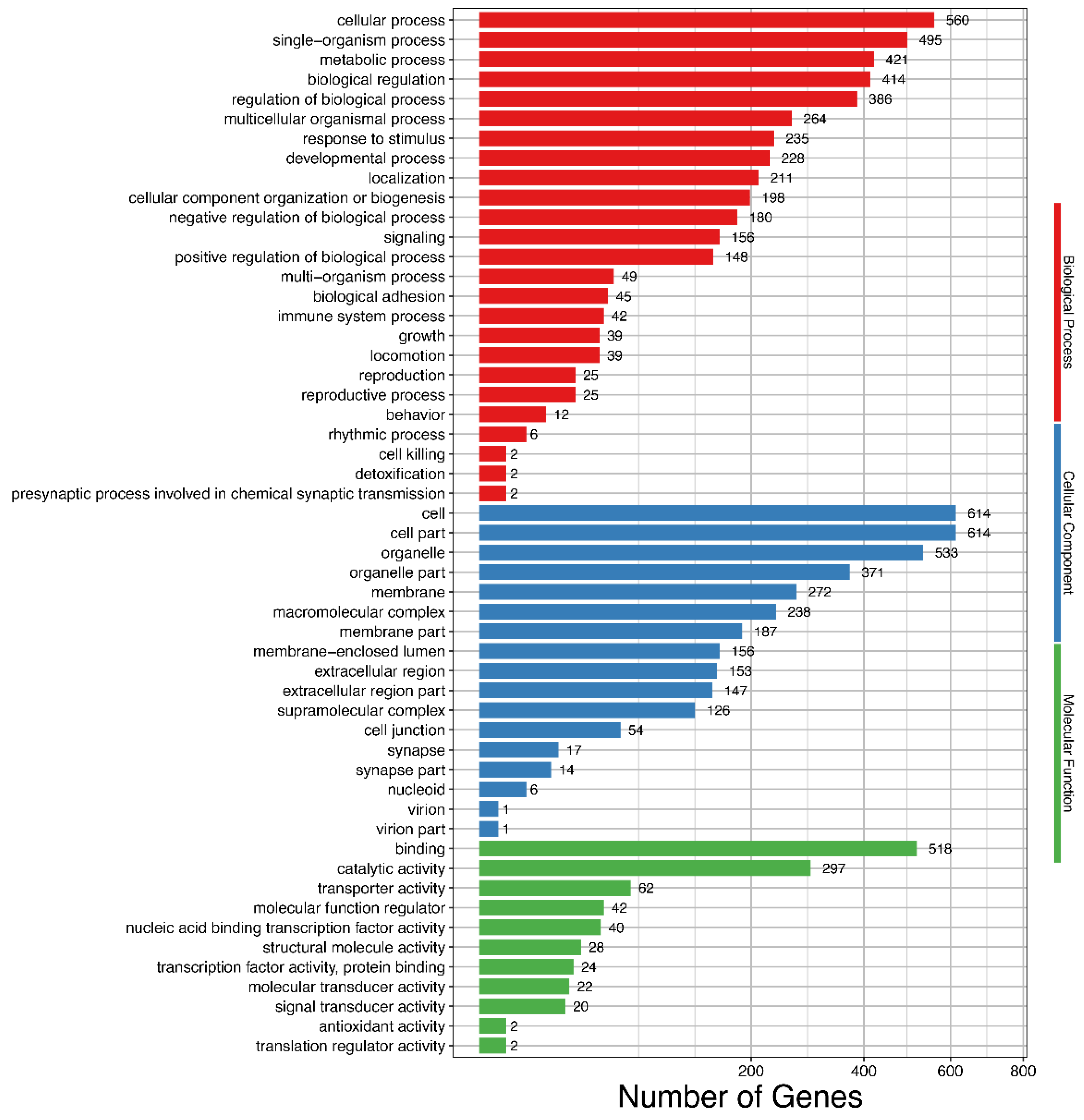
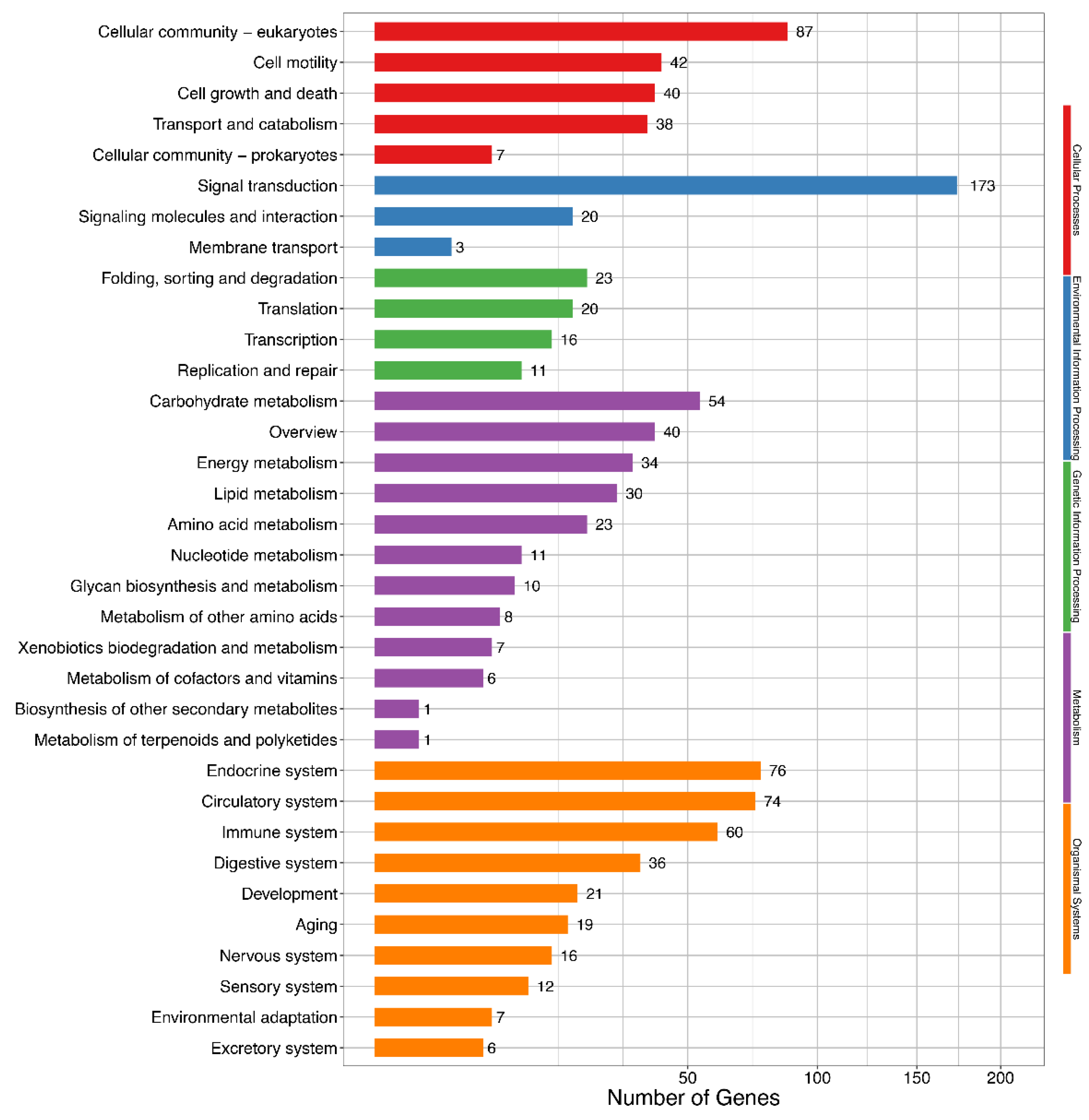
3. Results
3.1. Slaughter Performance and Meat Quality
3.2. Transcriptome Profiling of Goat Longissimus Dorsi and Biceps Femoris Muscles
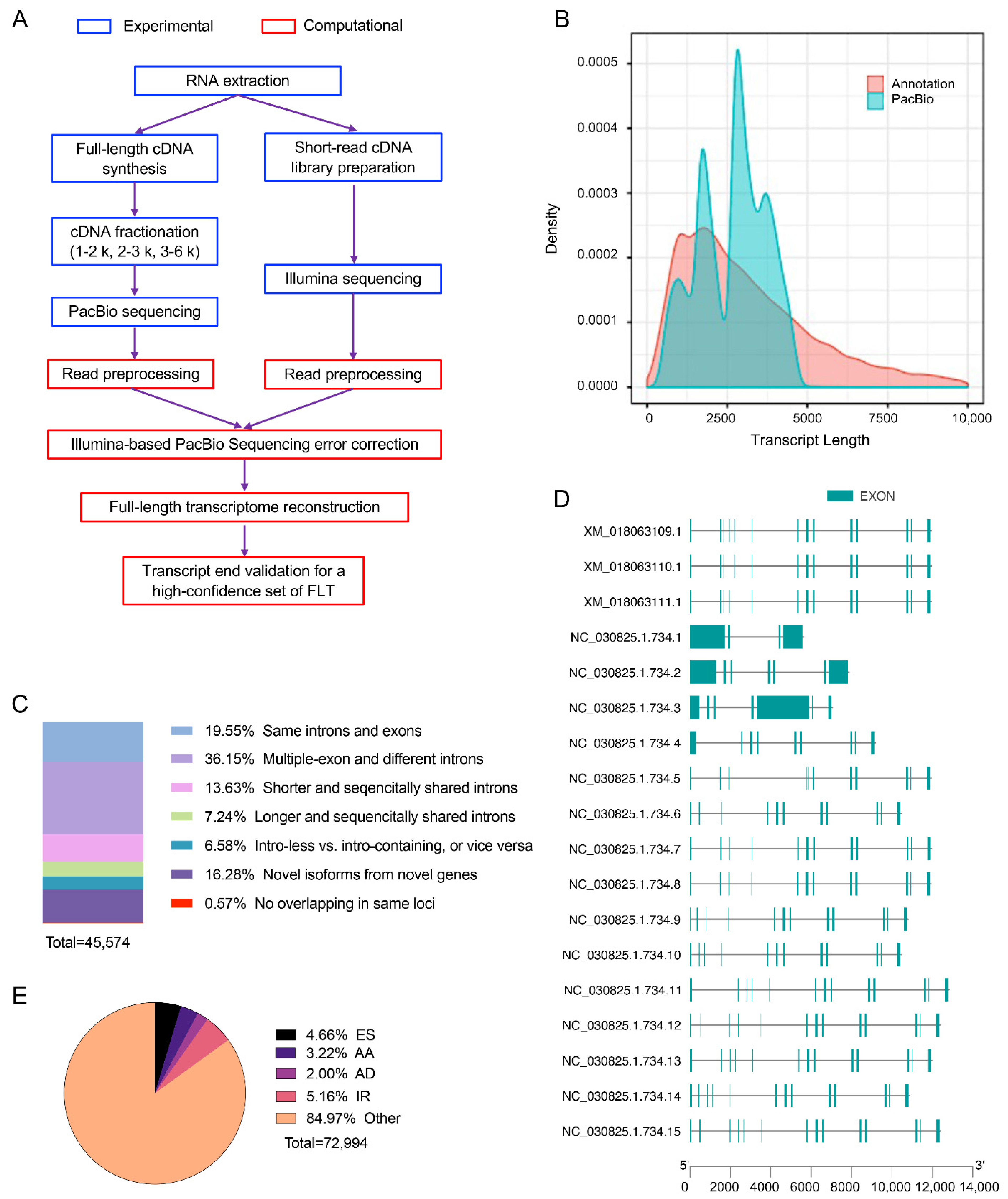
3.3. Characterization of Novel Isoforms
3.4. Alternative Splicing Events Analysis
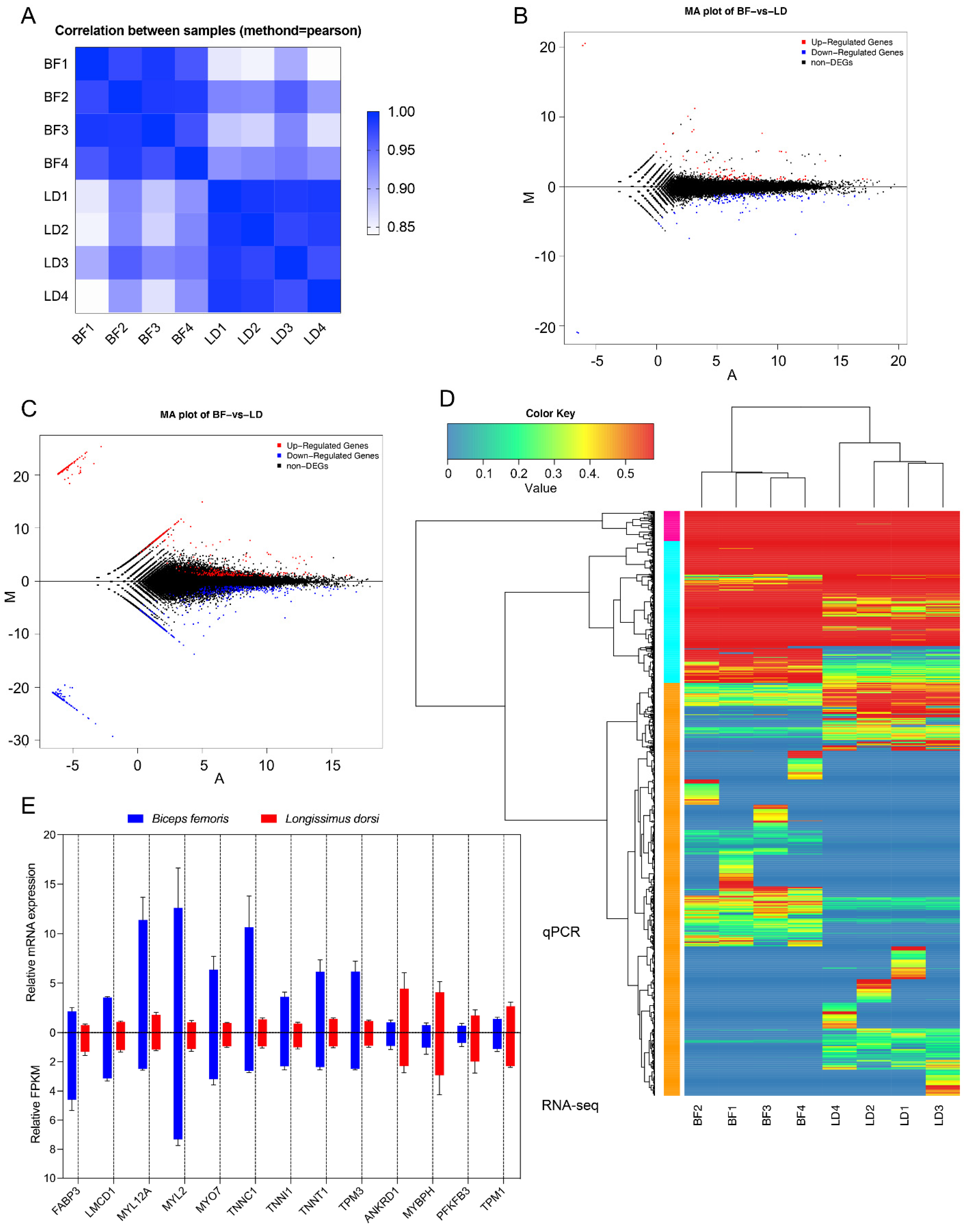
3.5. Overall Gene Expression Level
3.6. Differentially Expressed Genes and Isoforms in Different Muscles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chebli, Y.; El Otmani, S.; Chentouf, M.; Hornick, J.L.; Bindelle, J.; Cabaraux, J.F. Foraging Behavior of Goats Browsing in Southern Mediterranean Forest Rangeland. Animals 2020, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Pophiwa, P.; Webb, E.C.; Frylinck, L. A review of factors affecting goat meat quality and mitigating strategies. Small Rumin. Res. 2020, 183, 106035. [Google Scholar] [CrossRef]
- Shen, J.; Hao, Z.; Wang, J.; Hu, J.; Liu, X.; Li, S.; Ke, N.; Song, Y.; Lu, Y.; Hu, L.; et al. Comparative Transcriptome Profile Analysis of Longissimus dorsi Muscle Tissues from Two Goat Breeds with Different Meat Production Performance Using RNA-Seq. Front. Genet. 2020, 11, 619399. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Orlic, D.; Kajstura, J.; Chimenti, S.; Jakoniuk, I.; Anderson, S.M.; Li, B.; Pickel, J.; McKay, R.; Nadal-Ginard, B.; Bodine, D.M.; et al. Bone marrow cells regenerate infarcted myocardium. Nature 2001, 410, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef]
- Webb, E.C.; Casey, N.H.; Simela, L. Goat meat quality. Small Rumin. Res. 2005, 60, 153–166. [Google Scholar] [CrossRef]
- Banskalieva, V.V.; Sahlu, T.; Goetsch, A.L. Fatty acid composition of goat muscles and fat depots: A review. Small Rumin. Res. 2000, 37, 255–268. [Google Scholar] [CrossRef]
- Miller, R. Drivers of Consumer Liking for Beef, Pork, and Lamb: A Review. Foods 2020, 9, 428. [Google Scholar] [CrossRef] [Green Version]
- Klont, R.E.; Brocks, L.; Eikelenboom, G. Muscle fibre type and meat quality. Meat Sci. 1998, 49 (Suppl. S1), S219–S229. [Google Scholar] [CrossRef]
- Ramalingam, V.; Hwang, I. Deciphering the significant role of various gene expression in modulating the toughness of bovine muscle. Biocatal. Agric. Biotechnol. 2021, 31, 101642. [Google Scholar] [CrossRef]
- Lin, Y.; Zhu, J.; Wang, Y.; Li, Q.; Lin, S. Identification of differentially expressed genes through RNA sequencing in goats (Capra hircus) at different postnatal stages. PLoS ONE 2017, 12, e0182602. [Google Scholar] [CrossRef] [Green Version]
- Sacca, E.; Corazzin, M.; Bovolenta, S.; Piasentier, E. Meat quality traits and the expression of tenderness-related genes in the loins of young goats at different ages. Animal 2019, 13, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, K.; Su, R.; Liu, W.; Ren, Y.; Zhang, C.; Du, M.; Zhang, J. Effect of dietary Tartary buckwheat extract supplementation on growth performance, meat quality and antioxidant activity in ewe lambs. Meat Sci. 2017, 134, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhang, T.; Cao, Y.; Deng, B.; Zhang, J.; Zhao, J. Effects of dietary sea buckthorn pomace supplementation on skeletal muscle mass and meat quality in lambs. Meat Sci. 2020, 166, 108141. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiao, L.; An, L.; Wang, W.; Liu, J.; Ren, Y.; Pan, Y.; Jing, J.; Liu, W. Transcriptome analysis of adipose tissues from two fat-tailed sheep breeds reveals key genes involved in fat deposition. BMC Genom. 2018, 19, 338. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Liu, H.; Ren, J.; Ye, X.; Feng, H.; Liu, Z. Single-molecule real-time sequencing facilitates the analysis of transcripts and splice isoforms of anthers in Chinese cabbage (Brassica rapa L. ssp. pekinensis). BMC Plant Biol. 2019, 19, 517. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Jung, E.; Bairoch, A. SWISS-PROT: Connecting biomolecular knowledge via a protein database. Curr. Issues Mol. Biol. 2001, 3, 47–55. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inf. 2009, 23, 205–211. [Google Scholar]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Wang, X.; You, X.T.; Langer, J.D.; Hou, J.Y.; Rupprecht, F.; Vlatkovic, I.; Quedenau, C.; Tushev, G.; Epstein, I.; Schaefke, B.; et al. Full-length transcriptome reconstruction reveals a large diversity of RNA and protein isoforms in rat hippocampus. Nat. Commun. 2019, 10, 5009–5015. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kumar, V.; Olson, A.; Ware, D. Reviving the Transcriptome Studies: An Insight into the Emergence of Single-Molecule Transcriptome Sequencing. Front. Genet. 2019, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Joo, S.T.; Kim, G.D.; Hwang, Y.H.; Ryu, Y.C. Control of fresh meat quality through manipulation of muscle fiber characteristics. Meat Sci. 2013, 95, 828–836. [Google Scholar] [CrossRef]
- Ramalingam, V.; Hwang, I. Identification of Meat Quality Determining Marker Genes in Fibroblasts of Bovine Muscle Using Transcriptomic Profiling. J. Agric. Food Chem. 2021, 69, 3776–3786. [Google Scholar] [CrossRef] [PubMed]
- Gunning, P.W.; Ghoshdastider, U.; Whitaker, S.; Popp, D.; Robinson, R.C. The evolution of compositionally and functionally distinct actin filaments. J. Cell Sci. 2015, 128, 2009–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murgia, M.; Toniolo, L.; Nagaraj, N.; Ciciliot, S.; Vindigni, V.; Schiaffino, S.; Reggiani, C.; Mann, M. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19, 2396–2409. [Google Scholar] [CrossRef] [Green Version]
- Veltri, T.; Landim-Vieira, M.; Parvatiyar, M.S.; Gonzalez-Martinez, D.; Dieseldorff Jones, K.M.; Michell, C.A.; Dweck, D.; Landstrom, A.P.; Chase, P.B.; Pinto, J.R. Hypertrophic Cardiomyopathy Cardiac Troponin C Mutations Differentially Affect Slow Skeletal and Cardiac Muscle Regulation. Front. Physiol. 2017, 8, 221. [Google Scholar] [CrossRef] [Green Version]
- Kazmierczak, K.; Liang, J.; Yuan, C.C.; Yadav, S.; Sitbon, Y.H.; Walz, K.; Ma, W.; Irving, T.C.; Cheah, J.X.; Gomes, A.V.; et al. Slow-twitch skeletal muscle defects accompany cardiac dysfunction in transgenic mice with a mutation in the myosin regulatory light chain. FASEB J. 2019, 33, 3152–3166. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Jin, J.P. TNNT1, TNNT2, and TNNT3: Isoform genes, regulation, and structure-function relationships. Gene 2016, 582, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.J.; Jin, J.P. TNNI1, TNNI2 and TNNI3: Evolution, regulation, and protein structure-function relationships. Gene 2016, 576, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Suthon, S.; Perkins, R.S.; Bryja, V.; Miranda-Carboni, G.A.; Krum, S.A. WNT5B in Physiology and Disease. Front. Cell Dev. Biol. 2021, 9, 667581. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, A.; Tsukada, S.; Kamiyama, M.; Yanagimoto, T.; Nakajima, M.; Maeda, S. Wnt5b partially inhibits canonical Wnt/beta-catenin signaling pathway and promotes adipogenesis in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2005, 330, 505–510. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, C.; Zhang, Y.; Dai, M.; Jiang, Y.; Wang, H.; Yu, M.; Jing, W.; Tian, W. Wnt5a regulates the cell proliferation and adipogenesis via MAPK-independent pathway in early stage of obesity. Cell Biol. Int. 2018, 42, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, B.; Fu, C.; Hao, R. Cloning and characterization of adipogenin and its overexpression enhances fat accumulation of bovine myosatellite cells. Gene 2017, 601, 27–35. [Google Scholar] [CrossRef]
- Ren, G.; Eskandari, P.; Wang, S.; Smas, C.M. Expression, regulation and functional assessment of the 80 amino acid Small Adipocyte Factor 1 (Smaf1) protein in adipocytes. Arch. Biochem. Biophys. 2016, 590, 27–36. [Google Scholar] [CrossRef]
- Eckel, R.H. Lipoprotein lipase: A multifunctional enzyme relevant to common metabolic diseases. N. Engl. J. Med. 1989, 320, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Zyla, P.A.; Barnett, R.M.; Beringer, J.; Dahl, O.; Dwyer, D.A.; Groom, D.E.; Lin, C.J.; Lugovsky, K.S.; Pianori, E.; Robinson, D.J.; et al. Review of Particle Physics. Prog. Theor. Exp. Phys. 2020, 2020, 83C01. [Google Scholar] [CrossRef]
- Faerman, A.; Shani, M. The expression of the regulatory myosin light chain 2 gene during mouse embryogenesis. Development 1993, 118, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.; Xu, Y.; Xie, H.Q.; Li, X.Q.; Wei, Y.Q.; Yang, Z.M. The possible role of myosin light chain in myoblast proliferation. Biol. Res. 2009, 42, 121–132. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Muscle Type | p-Value | |
|---|---|---|---|
| LD | BF | ||
| pH45 min | 6.71 ± 0.13 | 6.94 ± 0.13 | 0.26 |
| pH24 h | 5.72 ± 0.06 | 5.83 ± 0.08 | 0.28 |
| Lightness, L | 35.35 ± 1.59 | 35.23 ± 1.46 | 0.96 |
| Redness, a | 11.63 ± 0.75 b | 14.08 ± 0.41 a | 0.03 |
| Yellowness, b | 1.28 ± 0.43 | 0.53 ± 0.37 | 0.23 |
| Water loss (%) | 33.02 ± 2.12 | 28.14 ± 5.22 | 0.42 |
| Water hold (%) | 48.44 ± 1.98 | 46.33 ± 0.75 | 0.32 |
| Shear force (N) | 29.06 ± 2.54 b | 48.65 ± 7.2 a | 0.04 |
| Marbling score | 6.00 ± 0.00 a | 5.25 ± 0.25 b | 0.02 |
| Type of Reads | Library | ||
|---|---|---|---|
| 1–2 kb | 2–3 kb | 3–6 kb | |
| CCS 1 | 144,625 | 133,687 | 135,310 |
| 5′ reads | 100,807 | 90,964 | 79,482 |
| 3′ reads | 105,218 | 95,303 | 84,285 |
| Poly-A reads | 102,657 | 93,083 | 80,576 |
| Filtered short reads | 10,454 | 3043 | 6894 |
| Non-full-length reads | 46,175 | 50,806 | 60,436 |
| Full-length reads | 87,996 | 79,838 | 67,980 |
| FLNC 2 reads | 87,230 | 77,147 | 61,032 |
| Average FLNC reads | 1592 | 2604 | 2523 |
| Category | Pre-Correction | Post-Correction | Merge |
|---|---|---|---|
| Unmapped | 2645 (1.17%) | 1159 (0.51%) | 1144 (0.51%) |
| Multiple-best | 1467 (0.65%) | 1506 (0.67%) | 1333 (0.59%) |
| Low pid | 14,116 (6.26%) | 11,041 (4.90%) | 9935 (4.41%) |
| High quality map | 207,181 (91.91%) | 211,703 (93.92%) | 212,997 (94.49%) |
| Category | Annotation in RefSeq | Annotation in PacBio Sequences |
|---|---|---|
| Total Loci | 22,570 | 18,491 |
| Loci < 1 K | 4284 (18.98%) | 1214 (6.57%) |
| Loci 1–2 K | 5770 (25.56%) | 3254 (17.60%) |
| Loci 2–3 K | 4401 (19.50%) | 4637 (25.08%) |
| Loci ≥ 3 K | 8115 (35.95%) | 9386 (50.76%) |
| Total isoforms | 46,472 | 45,574 |
| Gene | BF (Mean FPKM) | LD (Mean FPKM) | log2 (Fold Change) | Enriched Terms |
|---|---|---|---|---|
| ANKRD1 | 158.10 | 447.93 | 1.49 | 1, 4, 6 |
| ANKRD2 | 89.54 | 23.71 | −1.88 | 1, 4, 8, 9 |
| PITX1 | 13.10 | 0.48 | −4.76 | 1, 7, 9 |
| MYL2 | 11,534.42 | 1795.08 | −2.66 | 2, 6, 10, 11 |
| LOC106502520 | 3.71 | 1.10 | −1.88 | 2, 6, 12, 13, 14 |
| HOXD9 | 4.94 | 14.39 | 1.56 | 1 |
| TPM1 | 8470.62 | 17,679.59 | 1.07 | 3, 6 |
| FGF1 | 2.45 | 1.12 | −1.12 | 3 |
| FGF9 | 0.93 | 0.18 | −2.38 | 3 |
| PROX1 | 4.61 | 1.44 | −1.66 | 5, 6 |
| TNNT1 | 7761.57 | 3153.20 | −1.29 | 12, 13 |
| LOC102181869 | 4269.46 | 1419.53 | −1.78 | 12, 13, 14 |
| WNT5B | 0.92 | 0.33 | −1.45 | 15, 16, 19 |
| ADIG | 1.70 | 4.83 | 1.11 | 15, 16, 18, 19 |
| LPL | 90.98 | 28.05 | −1.67 | 17, 21 |
| LOC106502520 | 3.71 | 1.10 | −1.88 | 22, 23, 24 |
| LOC102181869 | 4269.46 | 1419.53 | −1.78 | 22, 23, 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Chen, S.; Niu, S.; Bi, X.; Qiao, L.; Yang, K.; Liu, J.; Liu, W. Hybrid Sequencing in Different Types of Goat Skeletal Muscles Reveals Genes Regulating Muscle Development and Meat Quality. Animals 2021, 11, 2906. https://doi.org/10.3390/ani11102906
Pan Y, Chen S, Niu S, Bi X, Qiao L, Yang K, Liu J, Liu W. Hybrid Sequencing in Different Types of Goat Skeletal Muscles Reveals Genes Regulating Muscle Development and Meat Quality. Animals. 2021; 11(10):2906. https://doi.org/10.3390/ani11102906
Chicago/Turabian StylePan, Yangyang, Sijia Chen, Shu Niu, Xilin Bi, Liying Qiao, Kaijie Yang, Jianhua Liu, and Wenzhong Liu. 2021. "Hybrid Sequencing in Different Types of Goat Skeletal Muscles Reveals Genes Regulating Muscle Development and Meat Quality" Animals 11, no. 10: 2906. https://doi.org/10.3390/ani11102906
APA StylePan, Y., Chen, S., Niu, S., Bi, X., Qiao, L., Yang, K., Liu, J., & Liu, W. (2021). Hybrid Sequencing in Different Types of Goat Skeletal Muscles Reveals Genes Regulating Muscle Development and Meat Quality. Animals, 11(10), 2906. https://doi.org/10.3390/ani11102906