Nanoparticles from Equine Fetal Bone Marrow-Derived Cells Enhance the Survival of Injured Chondrocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Bone Marrow Cells (BMCs)
2.2. Culture of Equine Chondrocytes
2.3. Co-Culture of Equine Chondrocytes with BMCs
2.4. Flow Cytometry
2.5. Cytokine Treatment in BMCs and qRT-PCR
2.6. In Vitro Differentiation of BMCs
2.7. Establishment of In Vitro Model of Chondrocyte Injury
2.8. CCK-8 Assay
2.9. Collection of NPs
2.10. Cryo-TEM
2.11. NTA Assessment
2.12. Apoptosis Assay
2.13. Immunobolotting
2.14. BMC-NPs Uptake Study
2.15. Statistical Analysis
3. Results
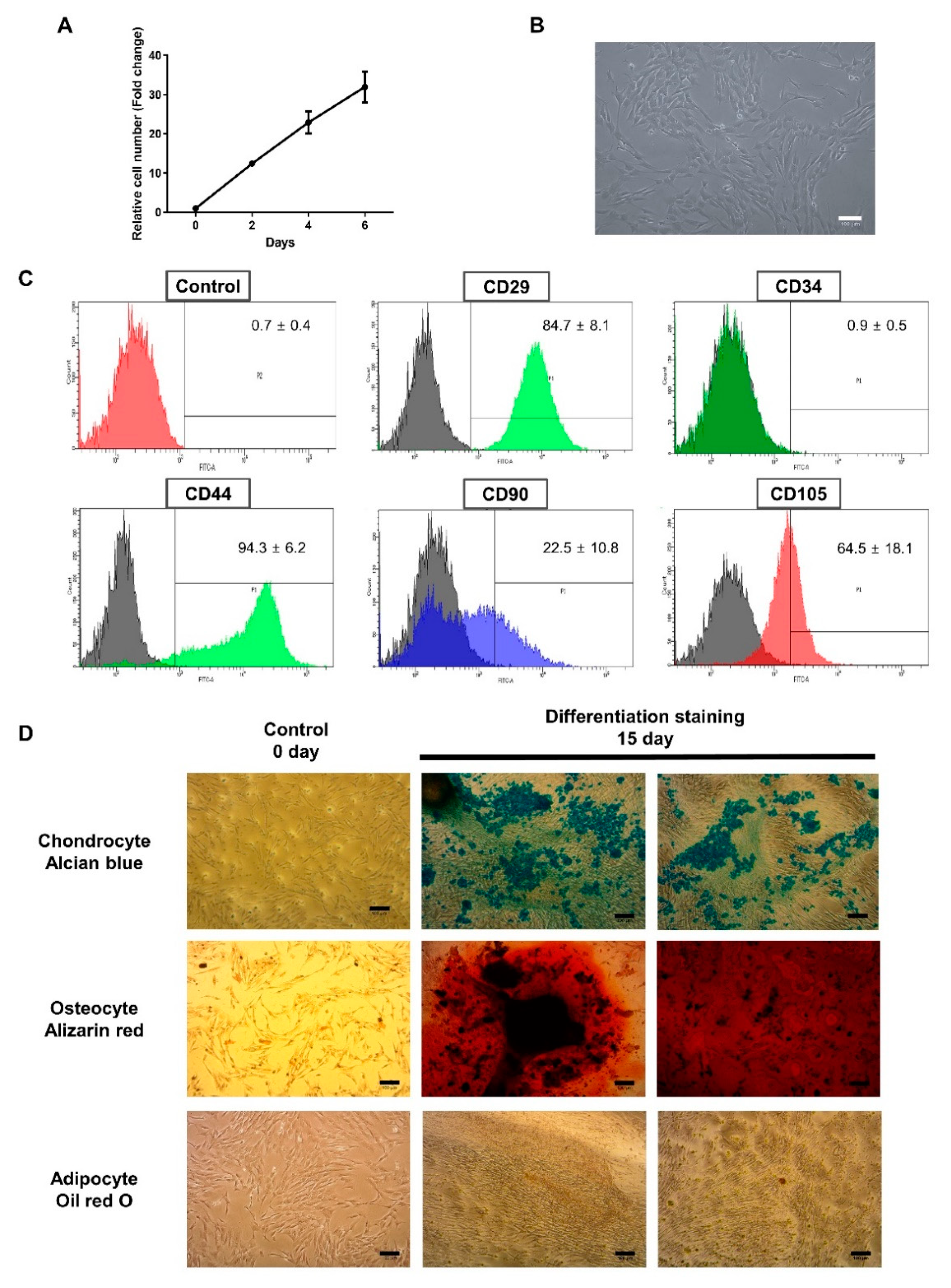
3.1. Characterization of BMCs
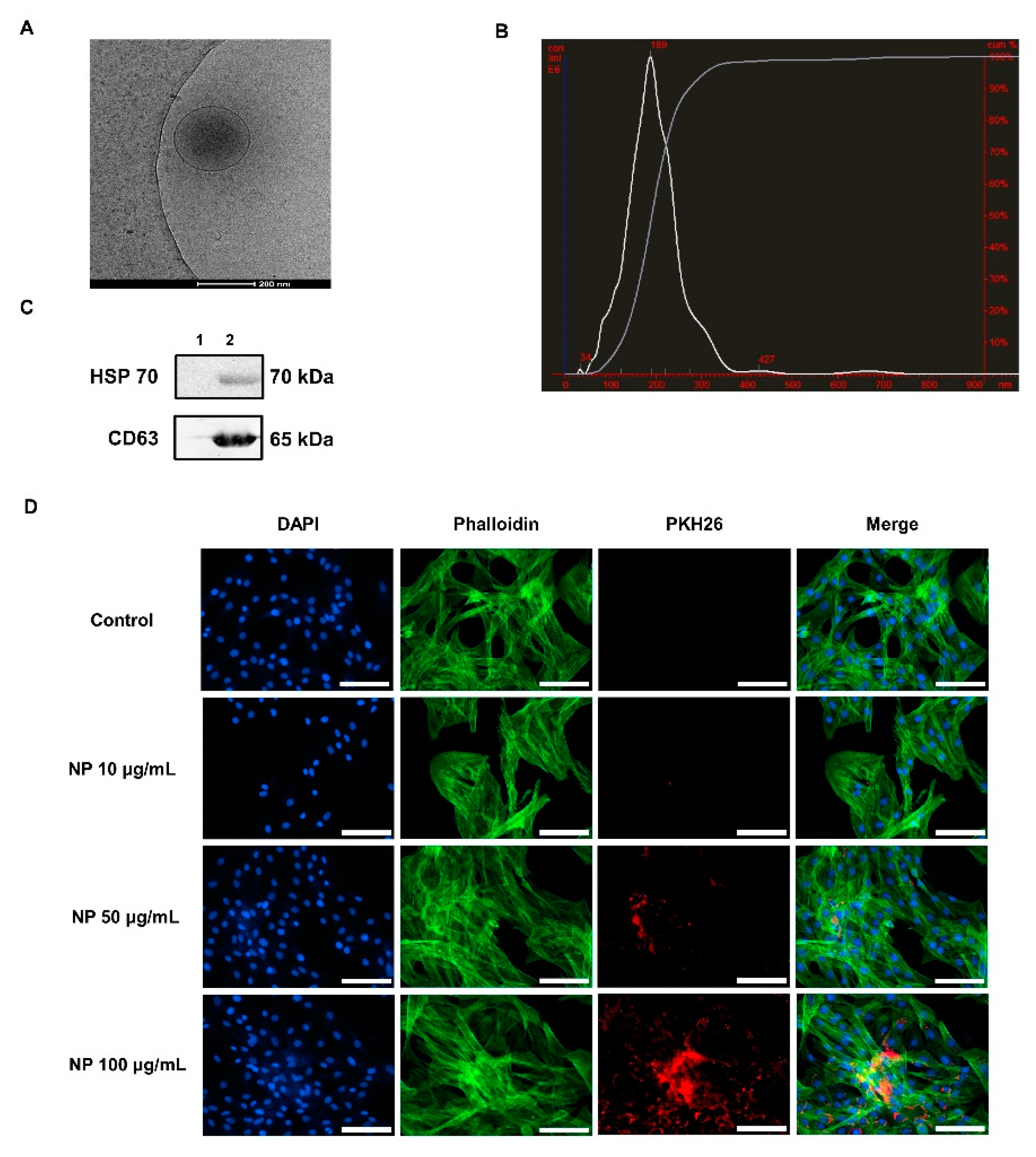
3.2. Characterization of BMC-Derived NPs
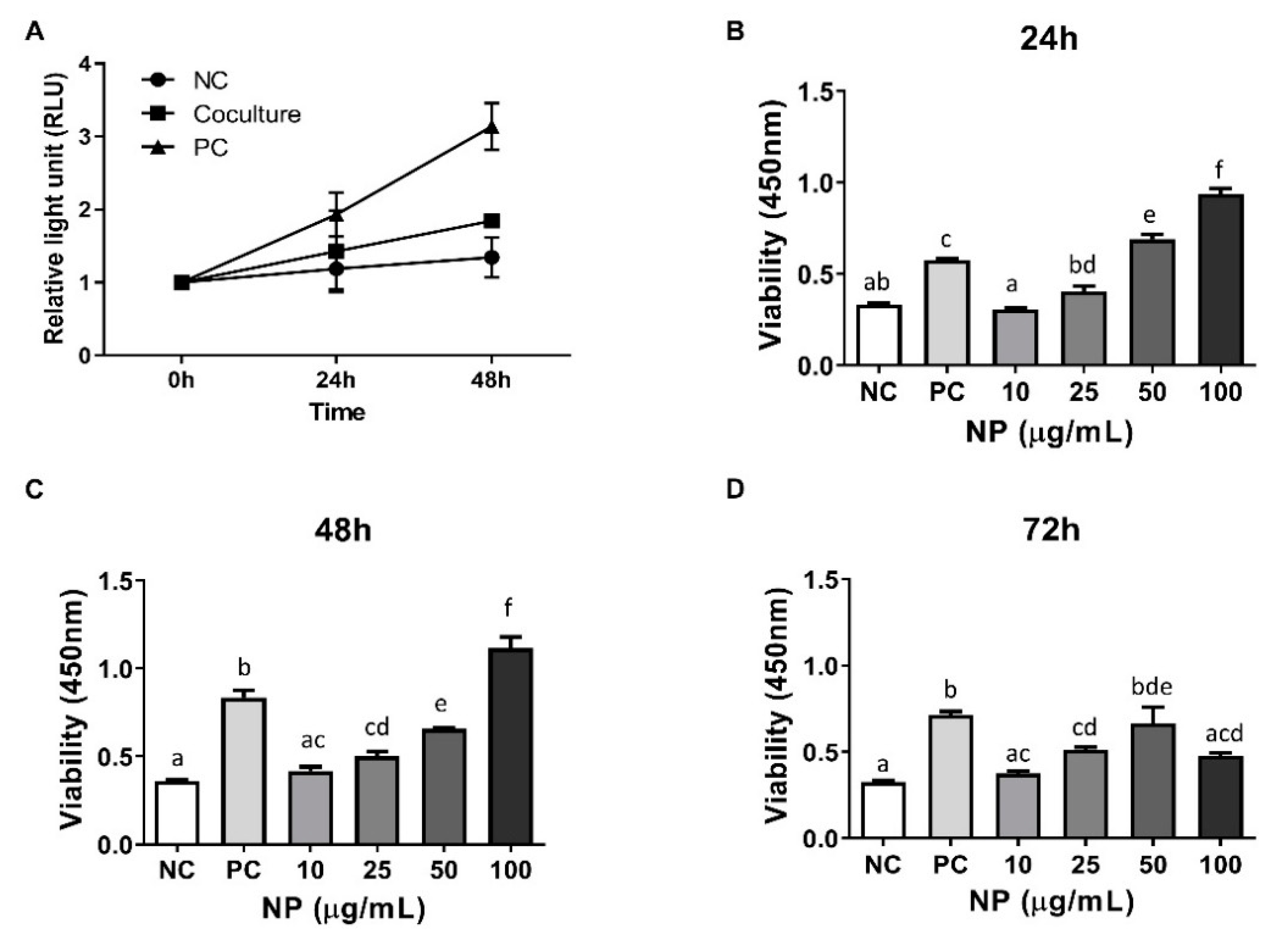
3.3. Assessment of the Role of BMCs and BMC-Derived NPs in the Viability of Equine Chondrocytes
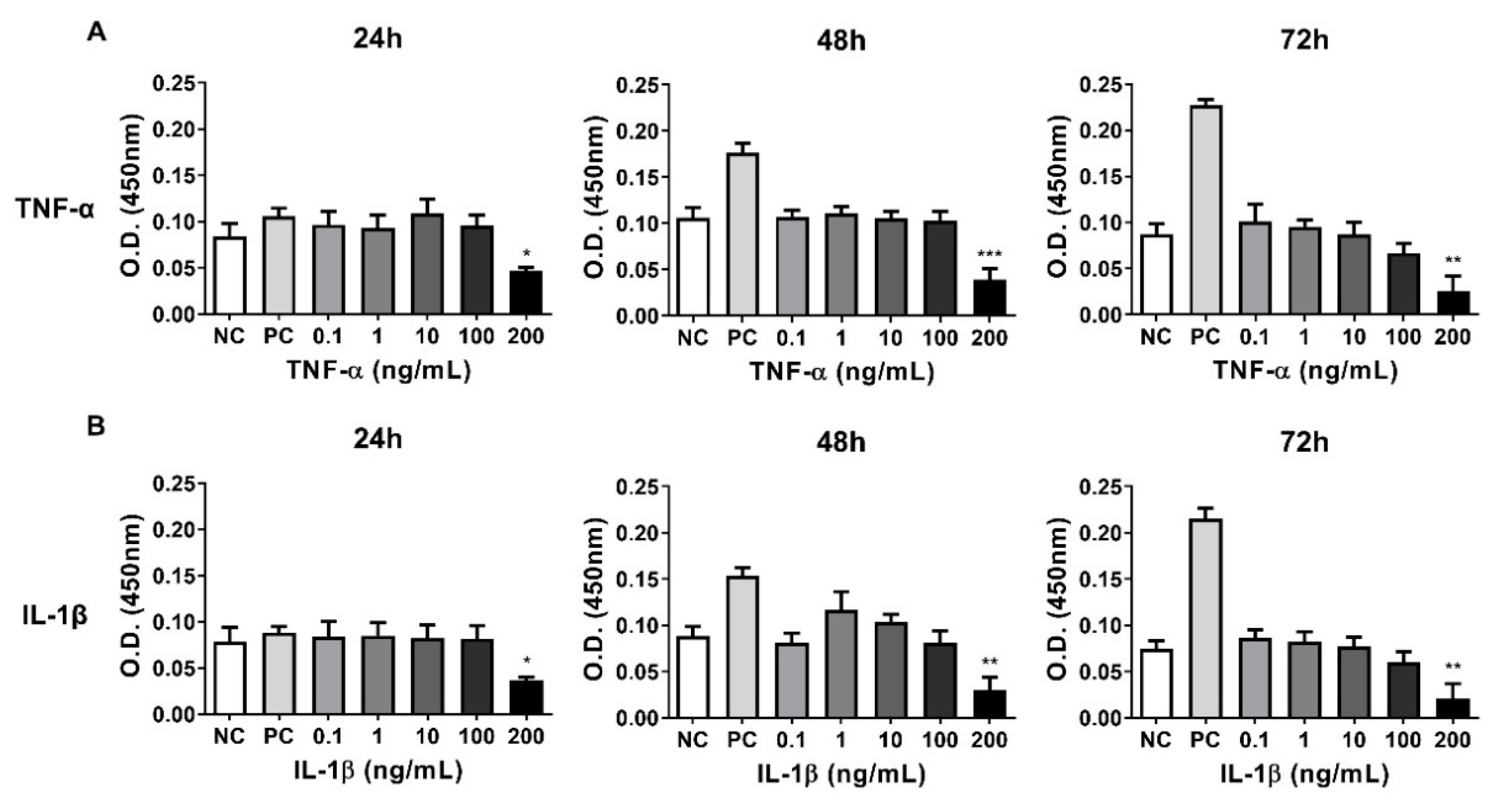
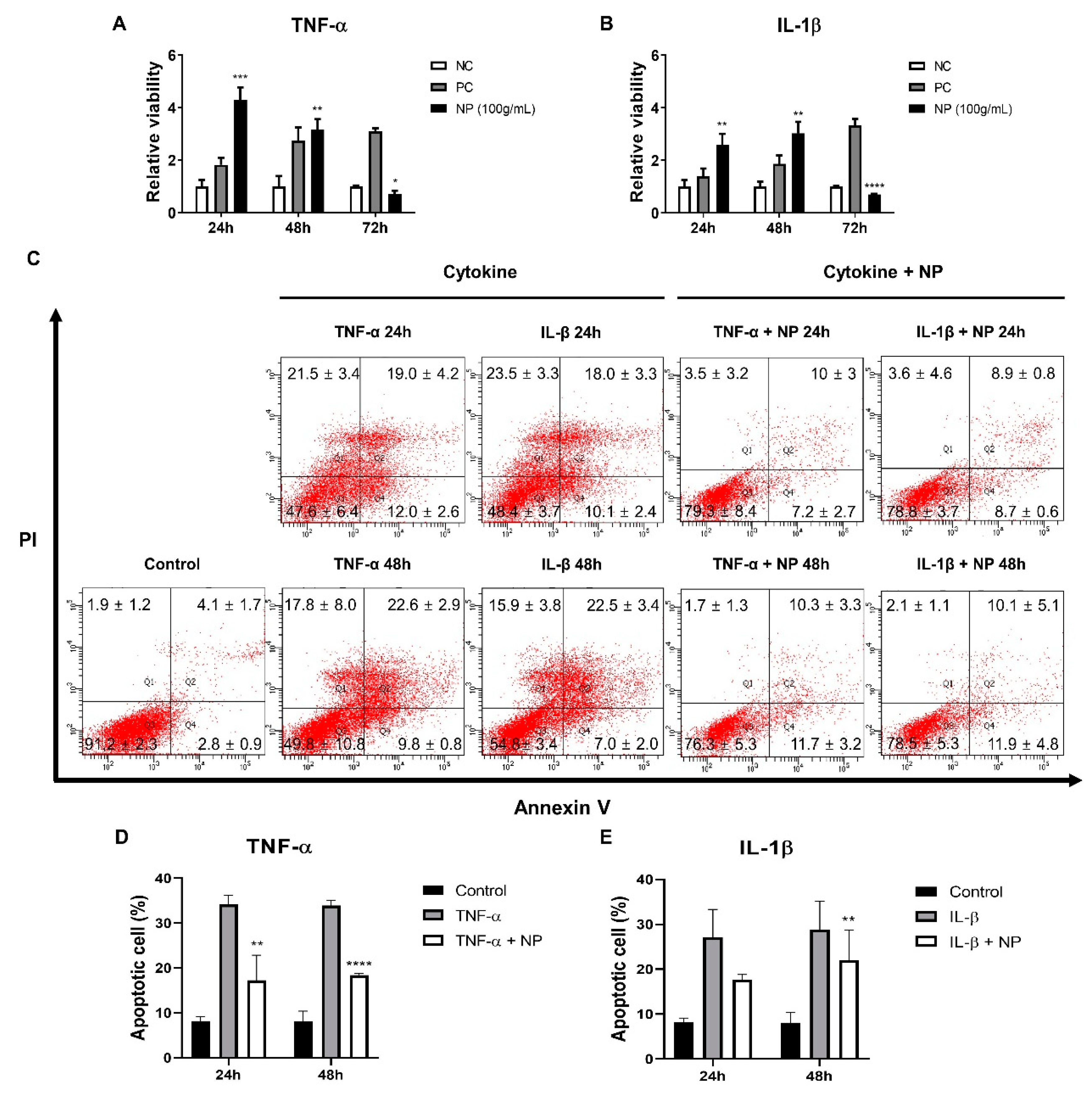
3.4. Establishing a Inflammatory Injury Model in Equine Chondrocytes
3.5. The Effect of the BMC-NPs on the Survival of Injured Chondrocytes
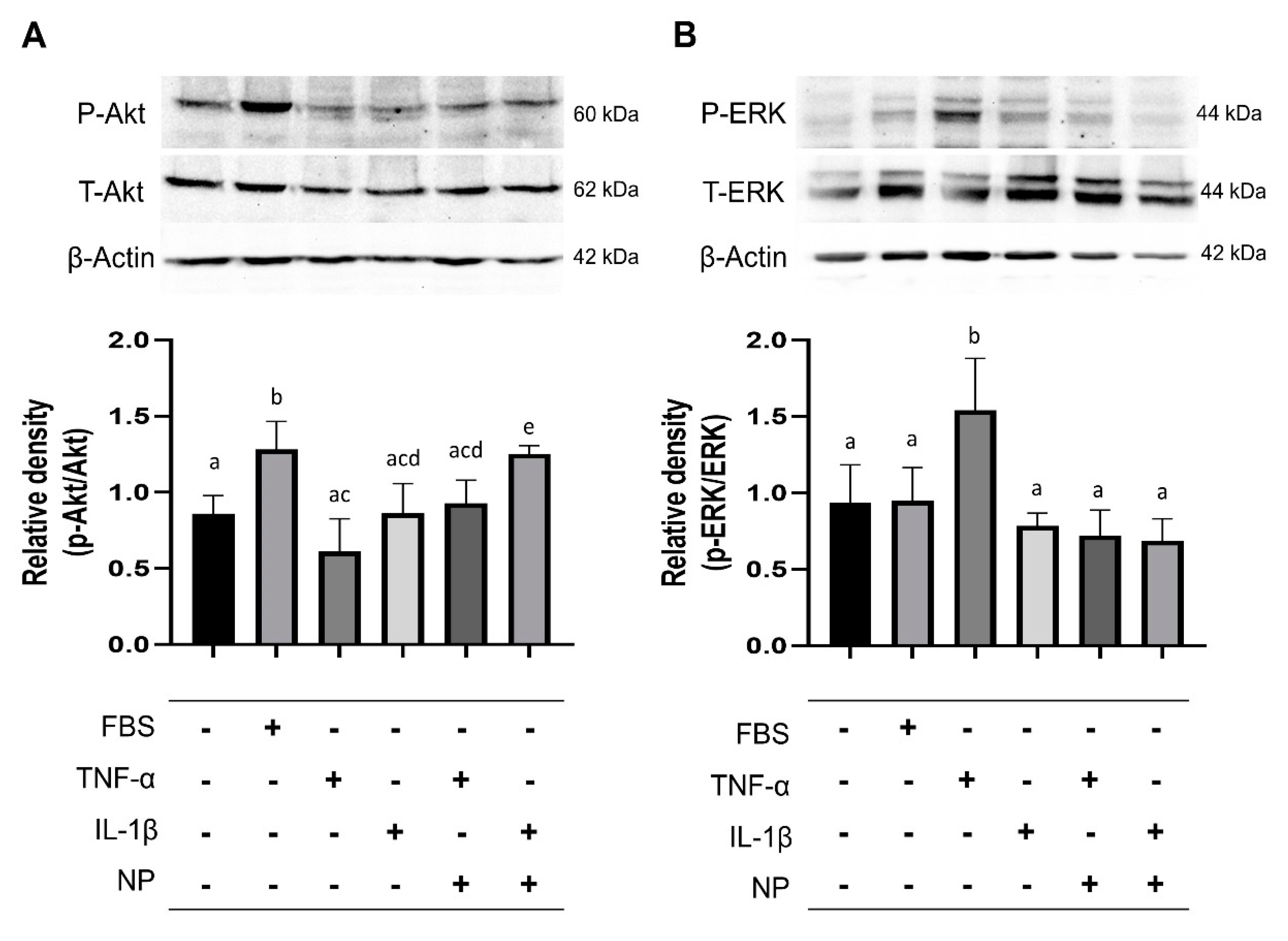
3.6. Immunoblotting of Signaling Pathway Molecules
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ekstrand, C.; Bondesson, U.; Giving, E.; Hedeland, M.; Ingvast-Larsson, C.; Jacobsen, S.; Lofgren, M.; Moen, L.; Rhodin, M.; Saetra, T.; et al. Disposition and effect of intra-articularly administered dexamethasone on lipopolysaccharide induced equine synovitis. Acta Vet. Scand. 2019, 61, 28. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, T.E. Fell-Muir lecture: Cartilage 2010—the known unknowns. Int. J. Exp. Pathol. 2010, 91, 203–209. [Google Scholar] [CrossRef]
- Neundorf, R.H.; Lowerison, M.B.; Cruz, A.M.; Thomason, J.J.; McEwen, B.J.; Hurtig, M.B. Determination of the prevalence and severity of metacarpophalangeal joint osteoarthritis in Thoroughbred racehorses via quantitative macroscopic evaluation. Am. J. Vet. Res. 2010, 71, 1284–1293. [Google Scholar] [CrossRef]
- Reed, S.R.; Jackson, B.F.; Mc Ilwraith, C.W.; Wright, I.M.; Pilsworth, R.; Knapp, S.; Wood, J.L.; Price, J.S.; Verheyen, K.L. Descriptive epidemiology of joint injuries in Thoroughbred racehorses in training. Equine Vet. J. 2012, 44, 13–19. [Google Scholar] [CrossRef]
- Broeckx, S.Y.; Seys, B.; Suls, M.; Vandenberghe, A.; Marien, T.; Adriaensen, E.; Declercq, J.; Van Hecke, L.; Braun, G.; Hellmann, K.; et al. Equine Allogeneic Chondrogenic Induced Mesenchymal Stem Cells Are an Effective Treatment for Degenerative Joint Disease in Horses. Stem Cells Dev. 2019, 28, 410–422. [Google Scholar] [CrossRef]
- Colbath, A.C.; Dow, S.W.; McIlwraith, C.W.; Goodrich, L.R. Mesenchymal stem cells for treatment of musculoskeletal disease in horses: Relative merits of allogeneic versus autologous stem cells. Equine Vet. J. 2020, 52, 654–663. [Google Scholar] [CrossRef]
- Marinas-Pardo, L.; Garcia-Castro, J.; Rodriguez-Hurtado, I.; Rodriguez-Garcia, M.I.; Nunez-Naveira, L.; Hermida-Prieto, M. Allogeneic Adipose-Derived Mesenchymal Stem Cells (Horse Allo 20) for the Treatment of Osteoarthritis-Associated Lameness in Horses: Characterization, Safety, and Efficacy of Intra-Articular Treatment. Stem Cells Dev. 2018, 27, 1147–1160. [Google Scholar] [CrossRef]
- Broeckx, S.Y.; Martens, A.M.; Bertone, A.L.; Van Brantegem, L.; Duchateau, L.; Van Hecke, L.; Dumoulin, M.; Oosterlinck, M.; Chiers, K.; Hussein, H.; et al. The use of equine chondrogenic-induced mesenchymal stem cells as a treatment for osteoarthritis: A randomised, double-blinded, placebo-controlled proof-of-concept study. Equine Vet. J. 2019, 51, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Ardanaz, N.; Vazquez, F.J.; Romero, A.; Remacha, A.R.; Barrachina, L.; Sanz, A.; Ranera, B.; Vitoria, A.; Albareda, J.; Prades, M.; et al. Inflammatory response to the administration of mesenchymal stem cells in an equine experimental model: Effect of autologous, and single and repeat doses of pooled allogeneic cells in healthy joints. BMC Vet. Res. 2016, 12, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zayed, M.; Adair, S.; Ursini, T.; Schumacher, J.; Misk, N.; Dhar, M. Concepts and challenges in the use of mesenchymal stem cells as a treatment for cartilage damage in the horse. Res. Vet. Sci. 2018, 118, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Kourembanas, S. Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol. 2015, 77, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, C.; Withrow, J.; Hunter, M.; Liu, Y.; Tang, Y.L.; Fulzele, S.; Hamrick, M.W. Emerging role of extracellular vesicles in musculoskeletal diseases. Mol. Asp. Med. 2018, 60, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.; Hunter, M.; Kolhe, R.; Isales, C.; Hamrick, M.; Fulzele, S. Therapeutic potential of mesenchymal stem cell based therapy for osteoarthritis. Clin. Transl. Med. 2016, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, H.S.; Suh, J.H.; Lee, H.J.; Bang, E.S.; Lee, J.M. Current Knowledge and Future Perspectives on Mesenchymal Stem Cell-Derived Exosomes as a New Therapeutic Agent. Int. J. Mol. Sci. 2020, 21, 727. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Hosseini-Beheshti, E.; Grau, G.E.; Zreiqat, H.; Little, C.B. Stem Cell-Derived Extracellular Vesicles for Treating Joint Injury and Osteoarthritis. Nanomaterials (Basel) 2019, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lin, L.; Zou, R.; Wen, C.; Wang, Z.; Lin, F. MSC-derived exosomes promote proliferation and inhibit apoptosis of chondrocytes via lncRNA-KLF3-AS1/miR-206/GIT1 axis in osteoarthritis. Cell Cycle 2018, 17, 2411–2422. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Teo, K.Y.W.; Chuah, S.J.; Lai, R.C.; Lim, S.K.; Toh, W.S. MSC exosomes alleviate temporomandibular joint osteoarthritis by attenuating inflammation and restoring matrix homeostasis. Biomaterials 2019, 200, 35–47. [Google Scholar] [CrossRef]
- Zhang, S.; Chuah, S.J.; Lai, R.C.; Hui, J.H.P.; Lim, S.K.; Toh, W.S. MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials 2018, 156, 16–27. [Google Scholar] [CrossRef]
- Mao, G.; Zhang, Z.; Hu, S.; Zhang, Z.; Chang, Z.; Huang, Z.; Liao, W.; Kang, Y. Exosomes derived from miR-92a-3p-overexpressing human mesenchymal stem cells enhance chondrogenesis and suppress cartilage degradation via targeting WNT5A. Stem Cell Res. 2018, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noel, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8, 1399–1410. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xie, H.; Chen, C.; Tao, Z.; Zhang, C.; Cai, L. Inhibiting the PI3K/AKT/NF-κB signal pathway with nobiletin for attenuating the development of osteoarthritis: In vitro and in vivo studies. Food Funct. 2019, 10, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-kappaB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Nejadnik, H.; Hui, J.H.; Feng Choong, E.P.; Tai, B.-C.; Lee, E.H. Autologous bone marrow–derived mesenchymal stem cells versus autologous chondrocyte implantation: An observational cohort study. Am. J. Sports Med. 2010, 38, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.T.; Ren, X.; Afizah, M.H.; Tarigan-Panjaitan, S.; Yang, Z.; Wu, Y.; Chian, K.S.; Mikos, A.G.; Hui, J.H. Repair of osteochondral defects with rehydrated freeze-dried oligo[poly(ethylene glycol) fumarate] hydrogels seeded with bone marrow mesenchymal stem cells in a porcine model. Tissue Eng. Part A 2013, 19, 1852–1861. [Google Scholar] [CrossRef] [Green Version]
- Bjorge, I.M.; Kim, S.Y.; Mano, J.F.; Kalionis, B.; Chrzanowski, W. Extracellular vesicles, exosomes and shedding vesicles in regenerative medicine—A new paradigm for tissue repair. Biomater. Sci. 2017, 6, 60–78. [Google Scholar] [CrossRef]
- Toh, W.S.; Lai, R.C.; Hui, J.H.P.; Lim, S.K. MSC exosome as a cell-free MSC therapy for cartilage regeneration: Implications for osteoarthritis treatment. Semin. Cell Dev. Biol. 2017, 67, 56–64. [Google Scholar] [CrossRef]
- Chen, K.; Man, C.; Zhang, B.; Hu, J.; Zhu, S.S. Effect of in vitro chondrogenic differentiation of autologous mesenchymal stem cells on cartilage and subchondral cancellous bone repair in osteoarthritis of temporomandibular joint. Int. J. Oral Maxillofac. Surg. 2013, 42, 240–248. [Google Scholar] [CrossRef]
- Toh, W.S.; Foldager, C.B.; Pei, M.; Hui, J.H. Advances in mesenchymal stem cell-based strategies for cartilage repair and regeneration. Stem Cell Rev. Rep. 2014, 10, 686–696. [Google Scholar] [CrossRef]
- Hofer, H.R.; Tuan, R.S. Secreted trophic factors of mesenchymal stem cells support neurovascular and musculoskeletal therapies. Stem Cell Res. 2016, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.C.; Yuan, T.; Zhang, Y.L.; Yin, W.J.; Guo, S.C.; Zhang, C.Q. Exosomes derived from miR-140-5p-overexpressing human synovial mesenchymal stem cells enhance cartilage tissue regeneration and prevent osteoarthritis of the knee in a rat model. Theranostics 2017, 7, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, D.; Liu, Z.; Zhou, F.; Dai, J.; Wu, B.; Zhou, J.; Heng, B.C.; Zou, X.H.; Ouyang, H.; et al. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res. 2017, 8, 189. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, Y.; Zhao, B.; Niu, X.; Hu, B.; Li, Q.; Zhang, J.; Ding, J.; Chen, Y.; Wang, Y. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res. 2017, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Chu, W.C.; Lai, R.C.; Lim, S.K.; Hui, J.H.; Toh, W.S. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr. Cartil. 2016, 24, 2135–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Li, Y.; Yang, Y.; Fang, Z.; Chen, X.; Wang, Y.; Kang, J.; Qu, X.; Yuan, W.; Dai, K.; et al. Osteoinductivity and Antibacterial Properties of Strontium Ranelate-Loaded Poly(Lactic-co-Glycolic Acid) Microspheres With Assembled Silver and Hydroxyapatite Nanoparticles. Front. Pharm. 2018, 9, 368. [Google Scholar] [CrossRef]
- Wu, J.; Kuang, L.; Chen, C.; Yang, J.; Zeng, W.-N.; Li, T.; Chen, H.; Huang, S.; Fu, Z.; Li, J. miR-100-5p-abundant exosomes derived from infrapatellar fat pad MSCs protect articular cartilage and ameliorate gait abnormalities via inhibition of mTOR in osteoarthritis. Biomaterials 2019, 206, 87–100. [Google Scholar] [CrossRef]
- Zavatti, M.; Beretti, F.; Casciaro, F.; Bertucci, E.; Maraldi, T. Comparison of the therapeutic effect of amniotic fluid stem cells and their exosomes on monoiodoacetate-induced animal model of osteoarthritis. Biofactors 2020, 46, 106–117. [Google Scholar] [CrossRef]
- Guillot, P.V.; Gotherstrom, C.; Chan, J.; Kurata, H.; Fisk, N.M. Human first-trimester fetal MSC express pluripotency markers and grow faster and have longer telomeres than adult MSC. Stem Cells 2007, 25, 646–654. [Google Scholar] [CrossRef]
- Stenderup, K.; Justesen, J.; Clausen, C.; Kassem, M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003, 33, 919–926. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Teoh, S.H.; Chong, M.S.; Schantz, J.T.; Fisk, N.M.; Choolani, M.A.; Chan, J. Superior osteogenic capacity for bone tissue engineering of fetal compared with perinatal and adult mesenchymal stem cells. Stem Cells 2009, 27, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Campagnoli, C.; Roberts, I.A.; Kumar, S.; Bennett, P.R.; Bellantuono, I.; Fisk, N.M. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood 2001, 98, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. The mesengenic process. Clin. Plast. Surg. 1994, 21, 429–435. [Google Scholar] [CrossRef]
- Mattar, P.; Bieback, K. Comparing the immunomodulatory properties of bone marrow, adipose tissue, and birth-associated tissue mesenchymal stromal cells. Front. Immunol. 2015, 6, 560. [Google Scholar] [CrossRef] [Green Version]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells-current trends and future prospective. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Gotherstrom, C.; Ringden, O.; Westgren, M.; Tammik, C.; Le Blanc, K. Immunomodulatory effects of human foetal liver-derived mesenchymal stem cells. Bone Marrow Transpl. 2003, 32, 265–272. [Google Scholar] [CrossRef]
- Götherström, C.; Lundqvist, A.; Duprez, I.R.; Childs, R.; Berg, L.; le Blanc, K.J.C. Fetal and adult multipotent mesenchymal stromal cells are killed by different pathways. Cytotherapy 2011, 13, 269–278. [Google Scholar] [CrossRef]
- Gotherstrom, C.; Ringden, O.; Tammik, C.; Zetterberg, E.; Westgren, M.; Le Blanc, K. Immunologic properties of human fetal mesenchymal stem cells. Am. J. Obs. Gynecol. 2004, 190, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, C.; Choi, Y.S.; Kim, M.; Park, C.; Suh, Y. Age-related alterations in mesenchymal stem cells related to shift in differentiation from osteogenic to adipogenic potential: Implication to age-associated bone diseases and defects. Mech. Ageing Dev. 2012, 133, 215–225. [Google Scholar] [CrossRef]
- Guillot, P.V.; De Bari, C.; Dell’Accio, F.; Kurata, H.; Polak, J.; Fisk, N.M. Comparative osteogenic transcription profiling of various fetal and adult mesenchymal stem cell sources. Differentiation 2008, 76, 946–957. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.K.; Kim, H.; Kim, T.M. Exosomes Secreted from Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Accelerate Skin Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, X.; Li, H.; Chen, C.; Hu, B.; Niu, X.; Li, Q.; Zhao, B.; Xie, Z.; Wang, Y. Exosomes/tricalcium phosphate combination scaffolds can enhance bone regeneration by activating the PI3K/Akt signaling pathway. Stem Cell Res. 2016, 7, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barberini, D.J.; Freitas, N.P.P.; Magnoni, M.S.; Maia, L.; Listoni, A.J.; Heckler, M.C.; Sudano, M.J.; Golim, M.A.; da Cruz Landim-Alvarenga, F.; Amorim, R.M. Equine mesenchymal stem cells from bone marrow, adipose tissue and umbilical cord: Immunophenotypic characterization and differentiation potential. Stem Cell Res. 2014, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranera, B.; Lyahyai, J.; Romero, A.; Vazquez, F.J.; Remacha, A.R.; Bernal, M.L.; Zaragoza, P.; Rodellar, C.; Martin-Burriel, I. Immunophenotype and gene expression profiles of cell surface markers of mesenchymal stem cells derived from equine bone marrow and adipose tissue. Vet. Immunol. Immunopathol. 2011, 144, 147–154. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, N.; Marsano, A.; Vunjak-Novakovic, G.; Zhang, Y.; Lopez, M.J. In vitro mesenchymal trilineage differentiation and extracellular matrix production by adipose and bone marrow derived adult equine multipotent stromal cells on a collagen scaffold. Stem Cell Rev. Rep. 2013, 9, 858–872. [Google Scholar] [CrossRef] [Green Version]
- Hillmann, A.; Ahrberg, A.B.; Brehm, W.; Heller, S.; Josten, C.; Paebst, F.; Burk, J. Comparative Characterization of Human and Equine Mesenchymal Stromal Cells: A Basis for Translational Studies in the Equine Model. Cell Transpl. 2016, 25, 109–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capomaccio, S.; Cappelli, K.; Bazzucchi, C.; Coletti, M.; Gialletti, R.; Moriconi, F.; Passamonti, F.; Pepe, M.; Petrini, S.; Mecocci, S.; et al. Equine Adipose-Derived Mesenchymal Stromal Cells Release Extracellular Vesicles Enclosing Different Subsets of Small RNAs. Stem Cells Int. 2019, 2019, 4957806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascucci, L.; Alessandri, G.; Dall’Aglio, C.; Mercati, F.; Coliolo, P.; Bazzucchi, C.; Dante, S.; Petrini, S.; Curina, G.; Ceccarelli, P. Membrane vesicles mediate pro-angiogenic activity of equine adipose-derived mesenchymal stromal cells. Vet. J. 2014, 202, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Klymiuk, M.C.; Balz, N.; Elashry, M.I.; Heimann, M.; Wenisch, S.; Arnhold, S. Exosomes isolation and identification from equine mesenchymal stem cells. BMC Vet. Res. 2019, 15, 42. [Google Scholar] [CrossRef]
- Kojima, M.; Gimenes-Junior, J.A.; Langness, S.; Morishita, K.; Lavoie-Gagne, O.; Eliceiri, B.; Costantini, T.W.; Coimbra, R. Exosomes, not protein or lipids, in mesenteric lymph activate inflammation: Unlocking the mystery of post-shock multiple organ failure. J. Trauma Acute Care Surg. 2017, 82, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Chai, Y.; Lv, S.; Ye, M.; Wu, M.; Xie, L.; Fan, Y.; Zhu, X.; Gao, Z. Endothelial cell-derived exosomes protect SH-SY5Y nerve cells against ischemia/reperfusion injury. Int. J. Mol. Med. 2017, 40, 1201–1209. [Google Scholar] [CrossRef]
- Schey, K.L.; Luther, J.M.; Rose, K.L. Proteomics characterization of exosome cargo. Methods 2015, 87, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Shin, H.; Kim, J.; Kim, J.; Park, J. Isolation of High-Purity Extracellular Vesicles by Extracting Proteins Using Aqueous Two-Phase System. PLoS ONE 2015, 10, e0129760. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Park, Y.H.; Kim, Y.G.; Lee, J.Y.; Park, J. Aqueous two-phase system to isolate extracellular vesicles from urine for prostate cancer diagnosis. PLoS ONE 2018, 13, e0194818. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Sagini, K.; Sandvig, K.; Llorente, A. An emerging focus on lipids in extracellular vesicles. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.H.; Park, T.S.; Cho, B.-W.; Kim, T.M. Nanoparticles from Equine Fetal Bone Marrow-Derived Cells Enhance the Survival of Injured Chondrocytes. Animals 2020, 10, 1723. https://doi.org/10.3390/ani10101723
Kim KH, Park TS, Cho B-W, Kim TM. Nanoparticles from Equine Fetal Bone Marrow-Derived Cells Enhance the Survival of Injured Chondrocytes. Animals. 2020; 10(10):1723. https://doi.org/10.3390/ani10101723
Chicago/Turabian StyleKim, Ki Hoon, Tae Sub Park, Byung-Wook Cho, and Tae Min Kim. 2020. "Nanoparticles from Equine Fetal Bone Marrow-Derived Cells Enhance the Survival of Injured Chondrocytes" Animals 10, no. 10: 1723. https://doi.org/10.3390/ani10101723
APA StyleKim, K. H., Park, T. S., Cho, B.-W., & Kim, T. M. (2020). Nanoparticles from Equine Fetal Bone Marrow-Derived Cells Enhance the Survival of Injured Chondrocytes. Animals, 10(10), 1723. https://doi.org/10.3390/ani10101723