Genome-Wide Association Study of Body Weight Traits in Chinese Fine-Wool Sheep

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Phenotypic Measurements
2.3. DNA Resequencing and Data Preprocessing
2.4. SNP Identification and Annotation
2.5. Genome-wide Association Studies
2.6. Bioinformatics Analysis
2.7. Statistical Analysis
3. Results
3.1. Phenotypic Data Analysis of Body Weight Traits
3.2. Summary of Sequencing Data
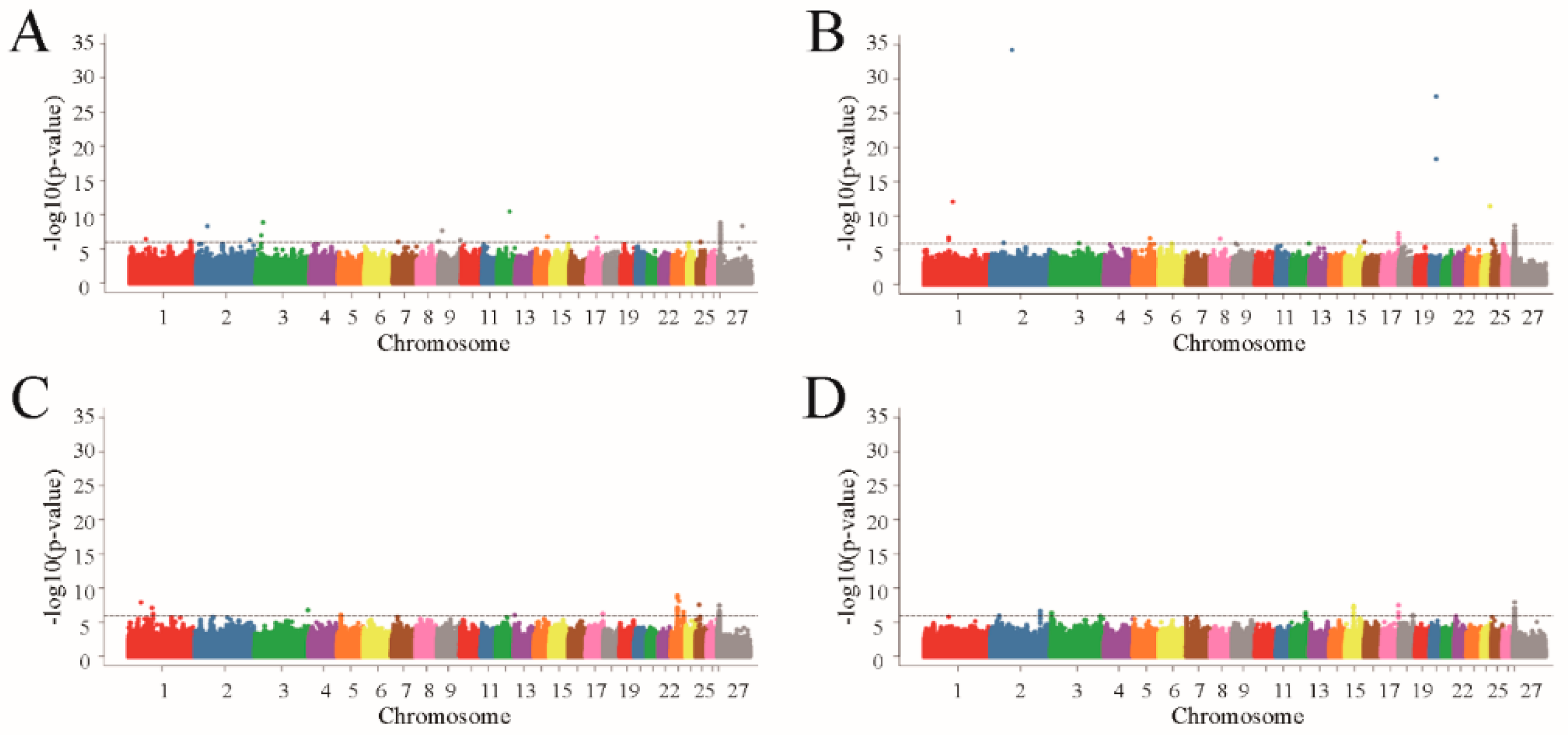
3.3. Genome-Wide Association Study
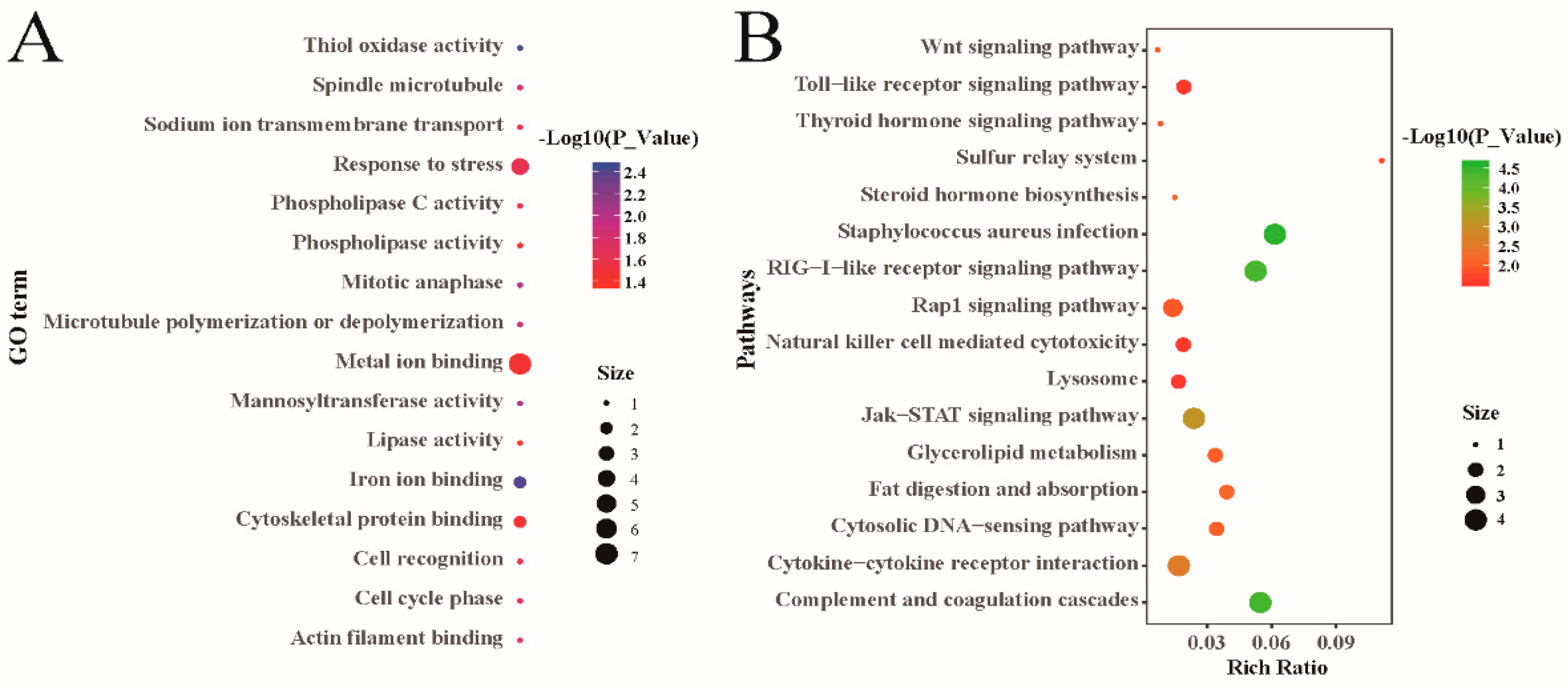
3.4. Bioinformatic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Chen, Y.L.; Liu, X.H.; Zhao, Y.X.; Zhu, L.; Liu, A.L.; Jiang, W.; Peng, X.; Zhang, C.L.; Tang, Z.S.; et al. Weighted single-step GWAS identified candidate genes associated with semen traits in a Duroc boar population. BMC Genomics 2019, 20, 797. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fang, L.; Null, D.J.; Hutchison, J.L.; Connor, E.E.; VanRaden, P.M.; VandeHaar, M.J.; Tempelman, R.J.; Weigel, K.A.; Cole, J.B. High-density genome-wide association study for residual feed intake in Holstein dairy cattle. J. Dairy Sci. 2019, 102, 11067–11080. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Zamani, P.; Vatankhah, M.; Abdoli, R. Genome-wide association study of birth weight in sheep. Animals 2019, 13, 1797–1803. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, N.; Yan, Y.Y.; Li, G.Q.; Liu, A.Q.; Wu, G.Q.; Sun, C.J. Genome-wide association analysis of egg production performance in chickens across the whole laying period. BMC Genet. 2019, 20, 67. [Google Scholar] [CrossRef]
- Wei, C.H.; Liu, C.S. Encyclopedia of Modern Sheep Production Technology; China Agriculture Press: Beijing, China, 2014; pp. 70–77. [Google Scholar]
- Al-Mamun, H.A.; Kwan, P.; Clark, S.A.; Ferdosi, M.H.; Tellam, R.; Gondro, C. Genome-wide association study of body weight in Australian Merino sheep reveals an orthologous region on OAR6 to human and bovine genomic regions affecting height and weight. Genet. Sel. Evol. 2015, 47, 66. [Google Scholar] [CrossRef]
- Matika, O.; Riggio, V.; Anselme-Moizan, M.; Law, A.S.; Pong-Wong, R.; Archibald, A.L.; Bishop, S.C. Genome-wide association reveals QTL for growth, bone and in vivo carcass traits as assessed by computed tomography in Scottish Blackface lambs. Genet. Sel. Evol. 2016, 48, 11. [Google Scholar] [CrossRef]
- Rupp, R.; Senin, P.; Sarry, J.; Allain, C.; Tasca, C.; Ligat, L.; Portes, D.; Woloszyn, F.; Bouchez, O.; Tabouret, G.; et al. A point mutation in suppressor of cytokine signalling 2 (Socs2) increases the susceptibility to inflammation of the mammary gland while associated with higher body weight and size and higher milk production in a sheep model. PLoS Genet. 2015, 11, e1005629. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, J.S.; Zhao, F.P.; Ren, H.X.; Xu, L.Y.; Lu, J.; Zhang, S.F.; Zhang, X.N.; Wei, C.H.; Lu, G.B.; et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS ONE 2013, 8, e66569. [Google Scholar] [CrossRef]
- Riggio, V.; Matika, O.; Pong-Wong, R.; Stear, M.J.; Bishop, S.C. Genome-wide association and regional heritability mapping to identify loci underlying variation in nematode resistance and body weight in Scottish Blackface lambs. Heredity 2013, 110, 420–429. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 2nd ed.; SAGE Press: Thousand Oaks, CA, USA, 2011; pp. 285–287. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 15 October 2019).
- Listed, N. References to molecular biology techniques. Curr. Protoc. Hum. Genet. 2001, 19, A.3A.1–A.3A.2. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.Y.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Nicodemus, K.K.; Liu, W.; Chase, G.A.; Tsai, Y.Y.; Fallin, M.D. Comparison of type I error for multiple test corrections in large single-nucleotide polymorphism studies using principal components versus haplotype blocking algorithms. BMC Genet. 2005, 6, S78. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Price, A.L.; Zaitlen, N.A.; Reich, D.; Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Z. Sheep Production; China Agriculture Press: Beijing, China, 2013; pp. 594–595. [Google Scholar]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.Y.; Peng, D.Z.; Gu, X.R.; Gong, Y.Z.; Sheng, Z.Y.; Hu, X.X. Polygenic basis and variable genetic architectures contribute to the complex nature of body weight -a genome-wide study in four Chinese indigenous chicken breeds. Front. Genet. 2018, 9, 229. [Google Scholar] [CrossRef]
- Dervishi, E.; Joy, M.; Alvarez-Rodriguez, J.; Serrano, M.; Calvo, J.H. The forage type (grazing versus hay pasture) fed to ewes and the lamb sex affect fatty acid profile and lipogenic gene expression in the longissimus muscle of suckling lambs. J. Anim. Sci. 2012, 90, 54–66. [Google Scholar] [CrossRef]
- Metzler-Zebeli, B.U.; Hollmann, M.; Sabitzer, S.; Podstatzky-Lichtenstein, L.; Klein, D.; Zebeli, Q. Epithelial response to high-grain diets involves alteration in nutrient transporters and Na+/K+-ATPase mRNA expression in rumen and colon of goats. J. Anim. Sci. 2013, 91, 4256–4266. [Google Scholar] [CrossRef]
- Lu, Z.K.; Chu, M.X.; Li, Q.; Jin, M.L.; Fei, X.J.; Ma, L.; Zhang, L.P.; Wei, C.H. Transcriptomic analysis provides novel insights into heat stress responses in sheep. Animals 2019, 9, 387. [Google Scholar] [CrossRef]
- Jing, X.P.; Peng, Q.H.; Hu, R.; Zou, H.W.; Wang, H.Z.; Yu, X.Q.; Zhou, J.W.; Degen, A.; Wang, Z.S. Dietary supplements during the cold season increase rumen microbial abundance and improve rumen epithelium development in Tibetan sheep. J. Anim. Sci. 2018, 96, 293–305. [Google Scholar] [CrossRef]
- Gholizadeh, M.; Rahimi-Mianji, G.; Nejati-Javaremi, A. Genomewide association study of body weight traits in Baluchi sheep. J. Genet. 2015, 94, 143–146. [Google Scholar] [CrossRef]
- Zhang, W.C.; Bin, Y.; Zhang, J.J.; Cui, L.L.; Ma, J.W.; Chen, C.Y.; Ai, H.S.; Xiao, S.J.; Ren, J.; Huang, L.S. Genome-wide association studies for fatty acid metabolic traits in five divergent pig populations. Sci. Rep. 2016, 6, 24718. [Google Scholar] [CrossRef]
- Altshuler, D.; Hirschhorn, J.N. Upsetting the balance: VGF and the regulation of body weight. Neuron 1999, 23, 415–417. [Google Scholar] [CrossRef][Green Version]
- Jiang, C.; Lin, W.J.; Sadahiro, M.; Shin, A.C.; Buettner, C.; Salton, S.R. Embryonic ablation of neuronal VGF increases energy expenditure and reduces body weight. Neuropeptides 2017, 64, 75–83. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lewis, J.E.; Brameld, J.M.; Hill, P.; Cocco, C.; Noli, B.; Ferri, G.L.; Barrett, P.; Ebling, F.J.; Jethwa, P.H. Hypothalamic over-expression of VGF in the Siberian hamster increases energy expenditure and reduces body weight gain. PLoS ONE 2017, 12, e0172724. [Google Scholar]
- Foglesong, G.D.; Huang, W.; Liu, X.L.; Slater, A.M.; Siu, J.; Yildiz, V.; Salton, S.R.; Cao, L. Role of hypothalamic VGF in energy balance and metabolic adaption to environmental enrichment in mice. Endocrinology 2016, 157, 983–996. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fargali, S.; Scherer, T.; Shin, A.C.; Sadahiro, M.; Buettner, C.; Salton, S.R. Germline ablation of VGF increases lipolysis in white adipose tissue. J. Endocrinol. 2012, 215, 313–322. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watson, E.; Hahm, S.; Mizuno, T.M.; Windsor, J.; Montgomery, C.; Scherer, P.E.; Mobbs, C.V.; Salton, S.R. VGF ablation blocks the development of hyperinsulinemia and hyperglycemia in several mouse models of obesity. Endocrinology 2005, 146, 5151–5163. [Google Scholar] [CrossRef]
- Jelinek, D.; Heidenreich, R.A.; Erickson, R.P.; Garver, W.S. Decreased Npc1 gene dosage in mice is associated with weight gain. Obesity 2010, 18, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, D.; Millward, V.; Birdi, A.; Trouard, T.P.; Heidenreich, R.A.; Garver, W.S. Npc1 haploinsufficiency promotes weight gain and metabolic features associated with insulin resistance. Hum. Mol. Genet. 2011, 20, 312–321. [Google Scholar] [CrossRef][Green Version]
- Bambace, C.; Dahlman, I.; Arner, P.; Kulyte, A. NPC1 in human white adipose tissue and obesity. BMC Endocr. Disord. 2013, 13, 5. [Google Scholar] [CrossRef]
- Gajewska, J.; Kurylowicz, A.; Mierzejewska, E.; Ambroszkiewicz, J.; Chelchowska, M.; Weker, H.; Pietrzykowska, M.; Puzianowska-Kuznicka, M. Are omentin rs2274907 and vaspin rs2236242 gene polymorphisms related to body composition, lipid profile and other adipokines in prepubertal healthy children? Endocr. Res. 2019, 15, 1–8. [Google Scholar] [CrossRef]
- Breitfeld, J.; Wiele, N.; Gutsmann, B.; Stumvoll, M.; Bluher, M.; Scholz, M.; Kovacs, P.; Tonjes, A. Circulating adipokine VASPIN is associated with serum lipid profiles in Humans. Lipids 2019, 54, 203–210. [Google Scholar] [CrossRef]
- Wigmore, P.M.; Stickland, N.C. Muscle development in large and small pig fetuses. J. Anat. 1983, 137, 235–245. [Google Scholar] [PubMed]
- Picard, B.; Berri, C.; Lefaucheur, L.; Molette, C.; Sayd, T.; Terlouw, C. Skeletal muscle proteomics in livestock production. Brief. Funct. Genom. 2010, 9, 259–278. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Kaur, P.; Bunnag, N.; Suresh, J.; Sung, I.C.H.; Tan, Q.H.; Gruber, J.; Tolwinski, N.S. WNT signaling in disease. Cells 2019, 8, 826. [Google Scholar] [CrossRef]
- Abu-Elmagd, M.; Robson, L.; Sweetman, D.; Hadley, J.; Francis-West, P.; Munsterberg, A. Wnt/Lef1 signaling acts via Pitx2 to regulate somite myogenesis. Dev. Biol. 2010, 337, 211–219. [Google Scholar] [CrossRef]
- Jones, A.E.; Price, F.D.; Le Grand, F.; Soleimani, V.D.; Dick, S.A.; Megeney, L.A.; Rudnicki, M.A. Wnt/beta-catenin controls follistatin signalling to regulate satellite cell myogenic potential. Skelet. Muscle 2015, 5, 14. [Google Scholar] [CrossRef]
- Murphy, M.M.; Keefe, A.C.; Lawson, J.A.; Flygare, S.D.; Yandell, M.; Kardon, G. Transiently active Wnt/beta-catenin signaling is not required but must be silenced for stem cell function during muscle regeneration. Stem Cell Rep. 2014, 3, 475–488. [Google Scholar] [CrossRef]
- Rudnicki, M.A.; Williams, B.O. Wnt signaling in bone and muscle. Bone 2015, 80, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.J.; Chen, B.C.; Tsai, F.C.; Anastassiadis, K.; Meyer, T.; Betzig, E.; Nusse, R. A localized Wnt signal orients asymmetric stem cell division in vitro. Science 2013, 339, 1445–1448. [Google Scholar] [CrossRef]
- Karner, C.M.; Long, F. Wnt signaling and cellular metabolism in osteoblasts. Cell. Mol. Life Sci. 2017, 74, 1649–1657. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The regulation of bone metabolism and disorders by wnt signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef]
- Trenerry, M.K.; Della Gatta, P.A.; Cameron-Smith, D. JAK/STAT signaling and human in vitro myogenesis. BMC Physiol. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Traits | Chr | Position (bp) | MAF | −log10 (p-Value) | Distance (bp) | Gene | Variant Effect |
|---|---|---|---|---|---|---|---|
| birth weight | ch1 | 253212365 | 0.10 | 6.111011551 | −19101 | SLCO2A1 | intergenic |
| ch9 | 14382214 | 0.49 | 7.664224268 | −2084 | LY6K | intergenic | |
| ch9 | 90030866 | 0.15 | 6.323004957 | −26215 | RALYL | intergenic | |
| ch12 | 52839429 | 0.50 | 10.45675318 | 0 | AADACL3 | intronic | |
| ch17 | 39807635 | 0.06 | 6.65587487 | 0 | C17H4orf45 | intronic | |
| ch25 | 13791395 | 0.48 | 6.006388368 | −17811 | BICC1 | intergenic | |
| ch27 | 6893599 | 0.16 | 8.797304666 | 10577 | GPR143 | intergenic | |
| ch27 | 7049963 | 0.15 | 8.355957873 | 0 | SHROOM2 | intronic | |
| weaning weight | ch1 | 101447761 | 0.21 | 6.814810126 | 7370 | C1H1orf68 | intergenic |
| ch1 | 118783826 | 0.50 | 12.06478576 | −16239 | CLIC6 | intergenic | |
| ch3 | 118519704 | 0.07 | 6.007008521 | 0 | TMTC2 | intronic | |
| ch20 | 26410829 | 0.50 | 27.43388012 | −16968 | STK19 | intergenic | |
| ch20 | 26410829 | 0.50 | 27.43388012 | −25619 | DXO | intergenic | |
| ch20 | 26410829 | 0.50 | 27.43388012 | −27940 | SKIV2L | intergenic | |
| ch24 | 35415932 | 0.49 | 11.41651058 | −19192 | NAT16 | intergenic | |
| ch24 | 35415932 | 0.49 | 11.41651058 | −28975 | VGF | intergenic | |
| ch27 | 6892015 | 0.21 | 8.574556919 | 8993 | GPR143 | intergenic | |
| ch27 | 6892015 | 0.21 | 8.574556919 | −87231 | SHROOM2 | intergenic | |
| yearling weight | ch1 | 51280492 | 0.50 | 7.90882916 | −29110 | RABGGTB | intergenic |
| ch1 | 96929132 | 0.50 | 7.121270413 | 29522 | TRNAQ-CUG | intronic | |
| ch1 | 96929132 | 0.50 | 7.121270413 | 27565 | TRNAN-GUU | intronic | |
| ch1 | 101447761 | 0.21 | 6.206583675 | 7370 | C1H1orf68 | intergenic | |
| ch5 | 13636592 | 0.11 | 6.080914687 | 17967 | ZNF557 | intergenic | |
| ch13 | 1884594 | 0.35 | 6.049430267 | 0 | PLCB4 | intronic | |
| ch23 | 33324859 | 0.50 | 8.093591468 | 0 | NPC1 | intronic | |
| ch23 | 33324859 | 0.50 | 8.093591468 | −16422 | C23H18orf8 | intronic | |
| ch23 | 52260021 | 0.12 | 6.009205597 | 0 | DCC | intronic | |
| ch25 | 12802212 | 0.50 | 7.554054255 | −22550 | ZNF25 | intergenic | |
| ch27 | 7053187 | 0.19 | 7.464649666 | 170165 | GPR143 | intronic | |
| ch27 | 7053187 | 0.19 | 7.464649666 | 0 | SHROOM2 | intronic | |
| adult weight | ch2 | 206466290 | 0.06 | 6.642091631 | 0 | PARD3B | intronic |
| ch3 | 4796131 | 0.09 | 6.34382137 | 0 | MED27 | intronic | |
| ch18 | 57956306 | 0.11 | 6.049410169 | 24508 | SERPINA14 | intergenic | |
| ch18 | 57956306 | 0.11 | 6.049410169 | −4231 | SERPINA12 | intergenic | |
| ch27 | 6743376 | 0.05 | 7.929222061 | 0 | TBL1X | intronic | |
| ch27 | 6997506 | 0.19 | 6.124895773 | 0 | SHROOM2 | intronic |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Z.; Yue, Y.; Yuan, C.; Liu, J.; Chen, Z.; Niu, C.; Sun, X.; Zhu, S.; Zhao, H.; Guo, T.; et al. Genome-Wide Association Study of Body Weight Traits in Chinese Fine-Wool Sheep. Animals 2020, 10, 170. https://doi.org/10.3390/ani10010170
Lu Z, Yue Y, Yuan C, Liu J, Chen Z, Niu C, Sun X, Zhu S, Zhao H, Guo T, et al. Genome-Wide Association Study of Body Weight Traits in Chinese Fine-Wool Sheep. Animals. 2020; 10(1):170. https://doi.org/10.3390/ani10010170
Chicago/Turabian StyleLu, Zengkui, Yaojing Yue, Chao Yuan, Jianbin Liu, Zhiqiang Chen, Chune Niu, Xiaoping Sun, Shaohua Zhu, Hongchang Zhao, Tingting Guo, and et al. 2020. "Genome-Wide Association Study of Body Weight Traits in Chinese Fine-Wool Sheep" Animals 10, no. 1: 170. https://doi.org/10.3390/ani10010170
APA StyleLu, Z., Yue, Y., Yuan, C., Liu, J., Chen, Z., Niu, C., Sun, X., Zhu, S., Zhao, H., Guo, T., & Yang, B. (2020). Genome-Wide Association Study of Body Weight Traits in Chinese Fine-Wool Sheep. Animals, 10(1), 170. https://doi.org/10.3390/ani10010170