
Assessment of Carbon Substrate Catabolism Pattern and Functional Metabolic Pathway for Microbiota of Limestone Caves

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection Information
2.2. Carbon Substrate Utilization Pattern
2.3. Microbial Genomic DNA Extraction
2.4. 16S rRNA Amplicon Library Preparation and Functional Prediction
2.5. Data Visualization and Statistical Analysis
3. Results
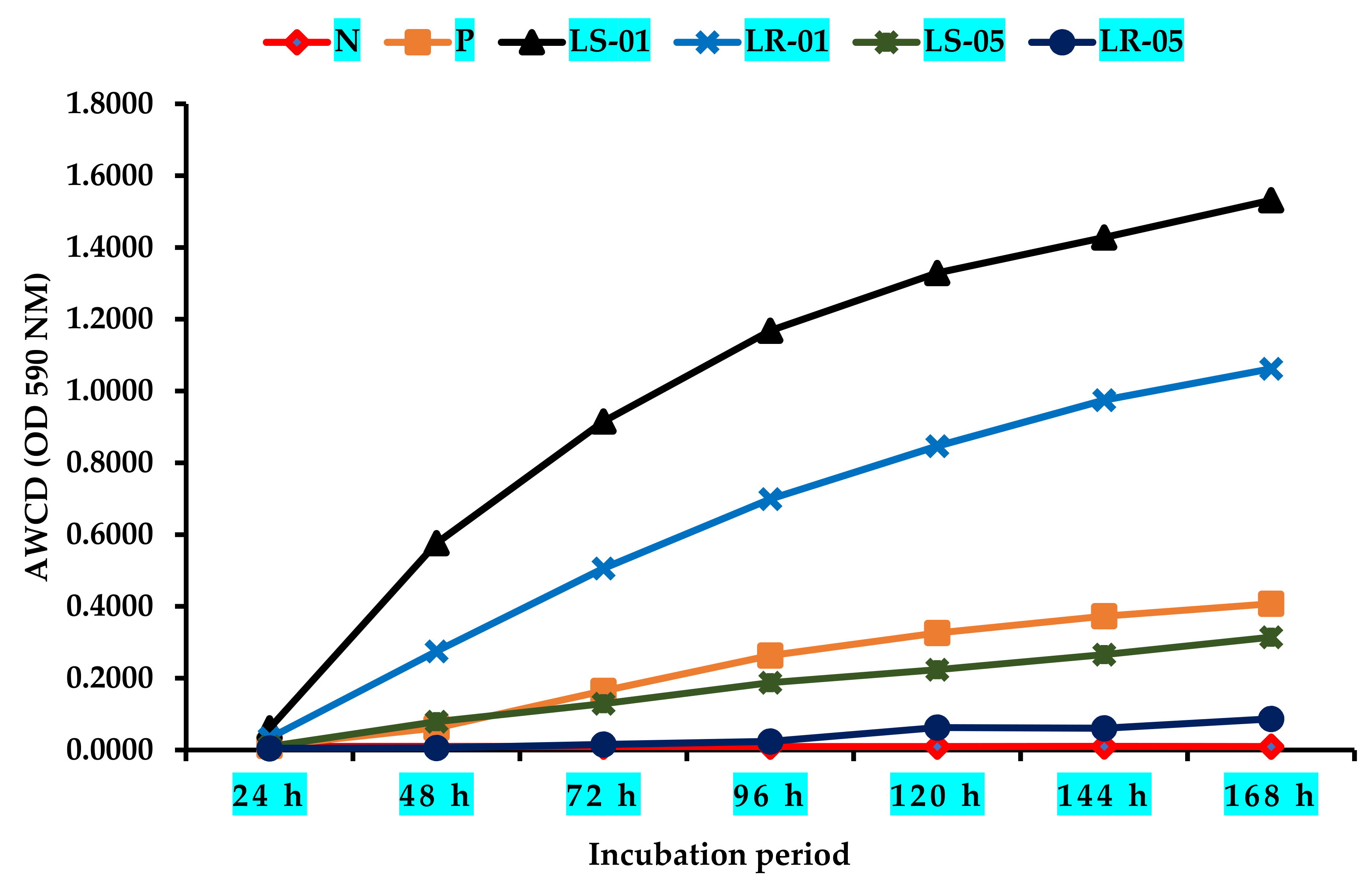
3.1. Variation in Average Well Color Development (AWCD) during Incubation Period
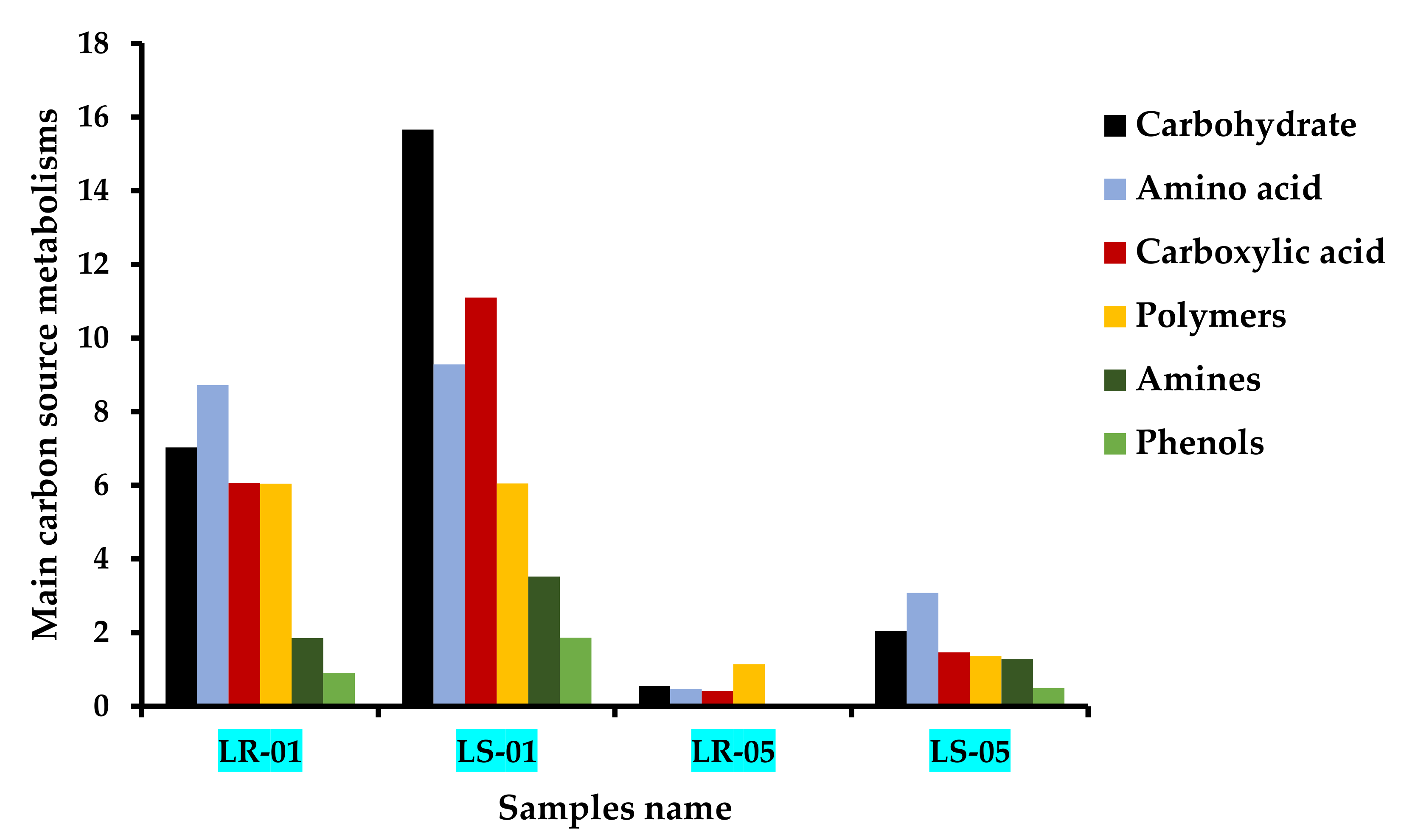
3.2. Major Carbon Source Assimilation Pattern at End Point (168 h)
3.3. Analysis of Bacterial Community Compositions and Distribution Patterns Using 16S rRNA Amplicon-Based Techniques
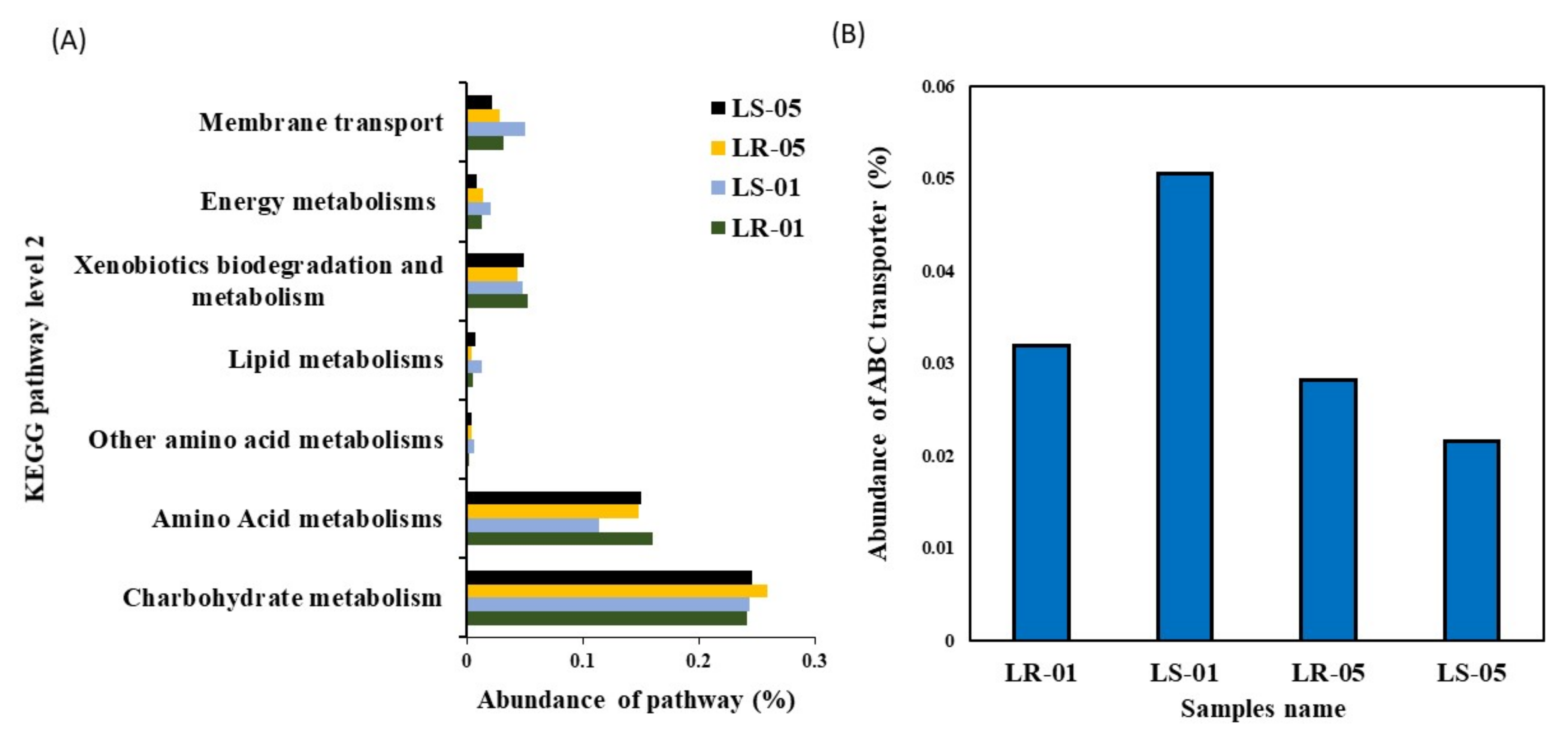
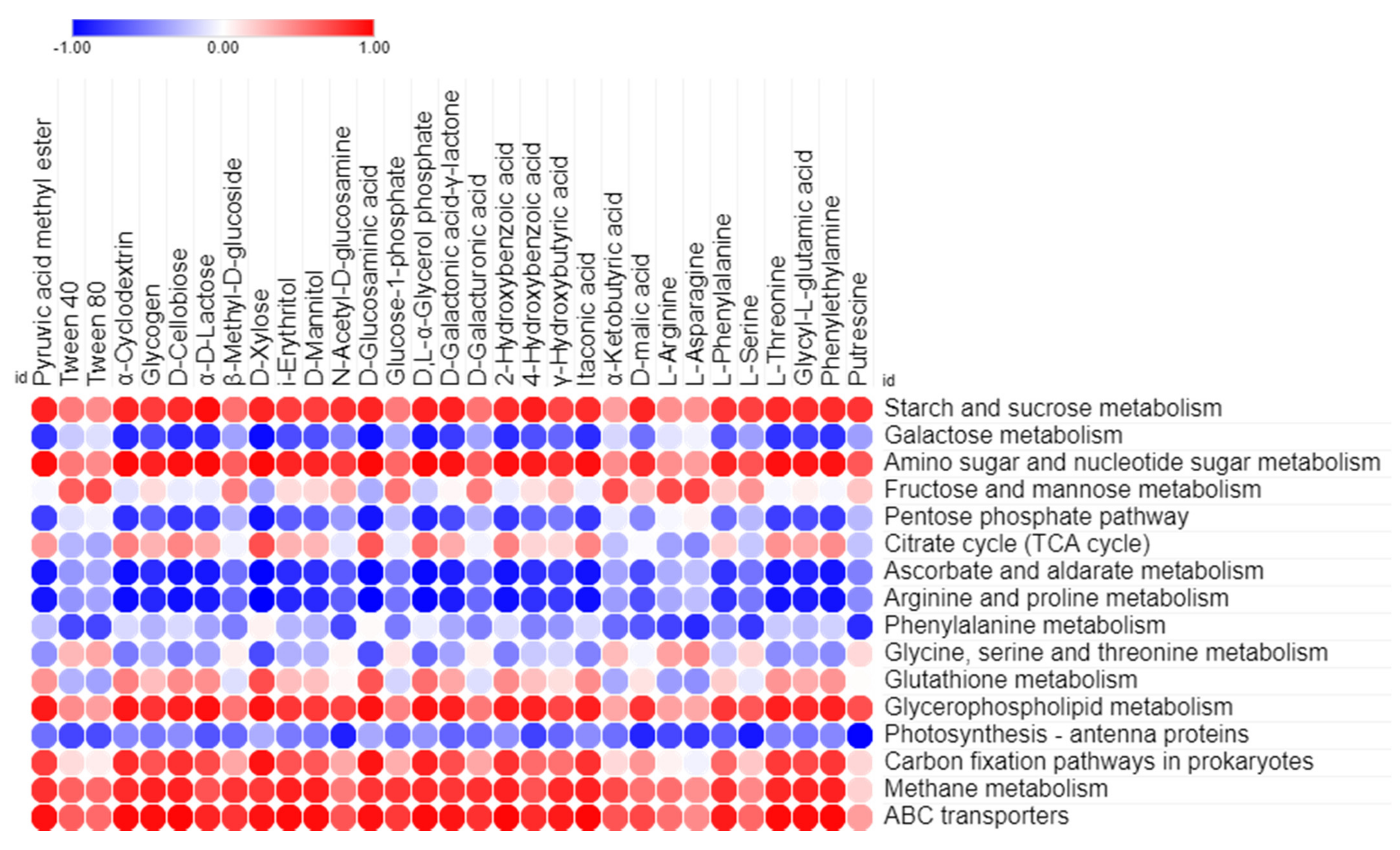
3.4. Correlation between Predicted Functional Pathways and Carbon Utilization Pattern
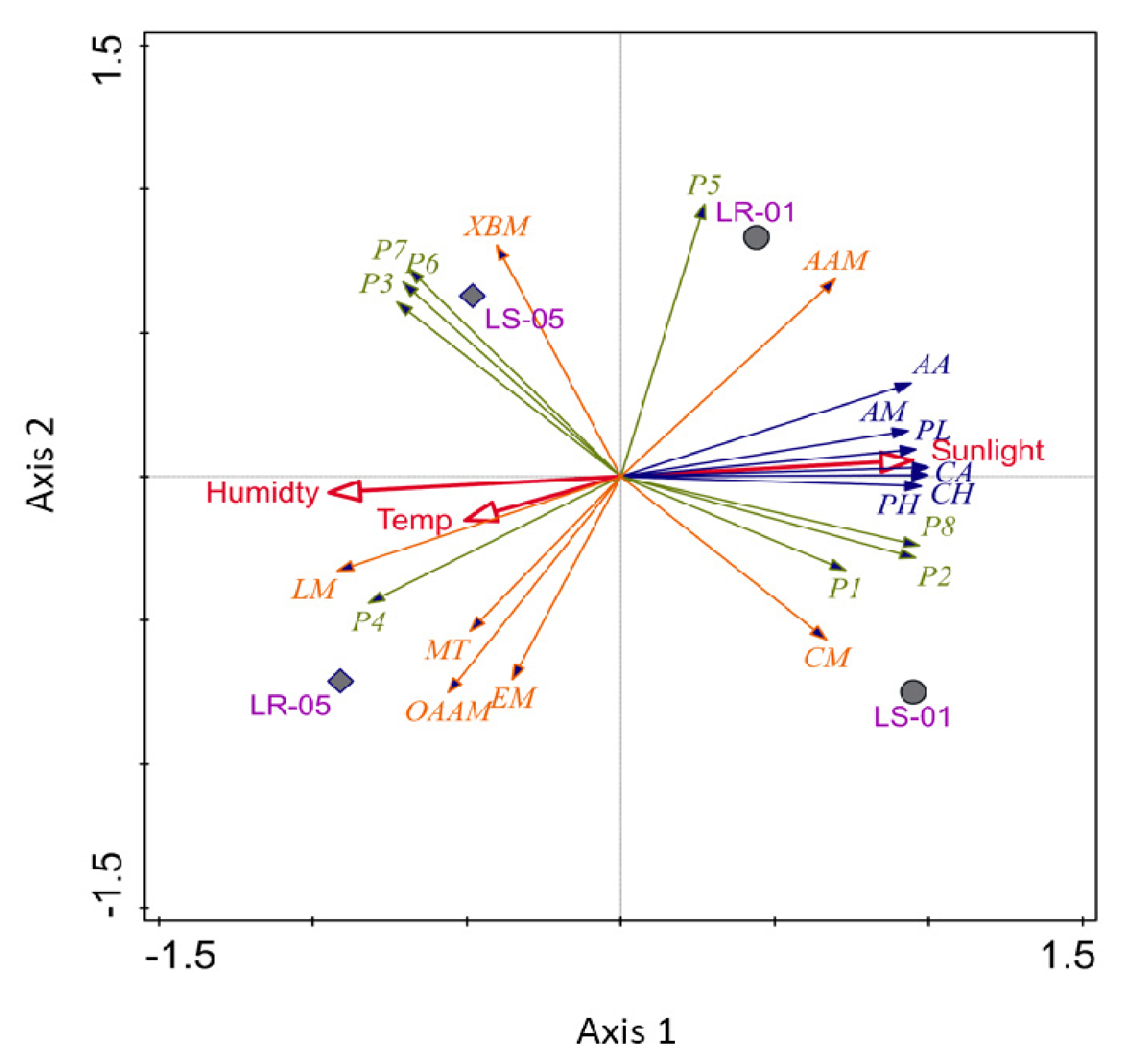
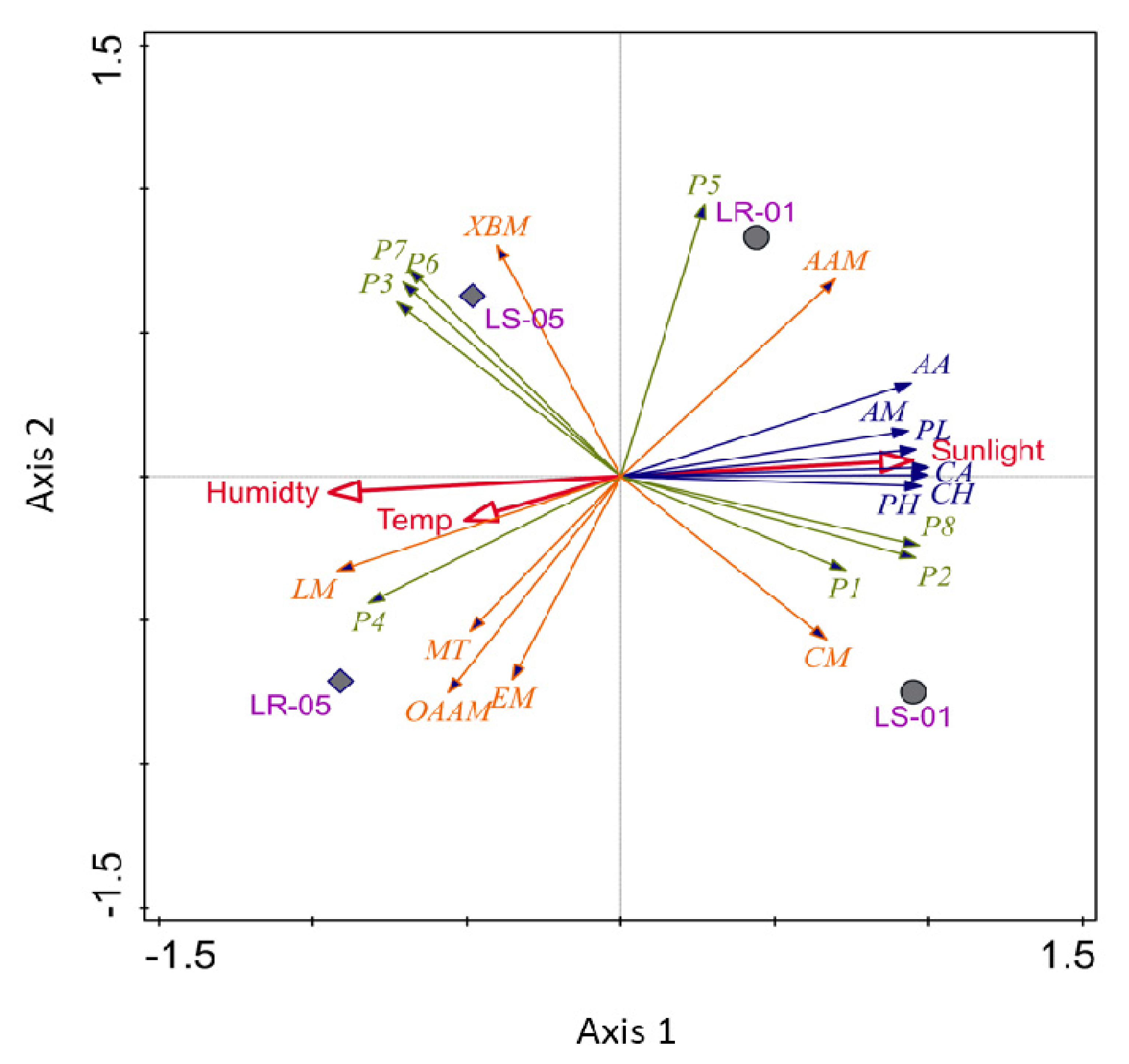
3.5. Visualization of Multivariate Analysis of Eco Plate and 16S rRNA Amplicon Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seifan, M.; Berenjian, A. Microbially induced calcium carbonate precipitation: A widespread phenomenon in the biological world. Appl. Microbiol. Biotechnol. 2019, 103, 4693–4708. [Google Scholar] [CrossRef]
- Reddy, M.S. Biomineralization of calcium carbonates and their engineered applications: A review. Front. Microbiol. 2013, 4, 314. [Google Scholar]
- Kim, Y.; Roh, Y. Microbially Induced Carbonate Precipitation Using Microorganisms Enriched from Calcareous Materials in Marine Environments and Their Metabolites. Minerals 2019, 9, 722. [Google Scholar] [CrossRef] [Green Version]
- Anbu, P.; Kang, C.-H.; Shin, Y.-J.; So, J.-S. Formations of calcium carbonate minerals by bacteria and its multiple applications. SpringerPlus 2016, 5, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Haiming, T.; Xiaoping, X.; Chao, L.; Xiaochen, P.; Kaikai, C.; Weiyan, L.; Ke, W. Microbial carbon source utilization in rice rhizosphere and nonrhizosphere soils with short-term manure N input rate in paddy field. Sci. Rep. 2020, 10, 6487–6489. [Google Scholar] [CrossRef]
- Waschina, S.; D’Souza, G.G.; Kost, C.; Kaleta, C. Metabolic network architecture and carbon source determine metabolite production costs. FEBS J. 2016, 283, 2149–2163. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Qiu, T.; Sun, Y.; Song, Y.; Wang, X.; Gao, M. Diversity of tetracycline- and erythromycin-resistant bacteria in aerosols and manures from four types of animal farms in China. Environ. Sci. Pollut. Res. 2019, 26, 24213–24222. [Google Scholar] [CrossRef]
- Sutfin, N.A.; Wohl, E.E.; Dwire, K.A. Banking carbon: A review of organic carbon storage and physical factors influencing retention in floodplains and riparian ecosystems. Earth Surf. Process. Landf. 2015, 41, 38–60. [Google Scholar] [CrossRef]
- Babur, E.; Dindaroglu, T. Seasonal Changes of Soil Organic Carbon and Microbial Biomass Carbon in Different Forest Ecosystems. In Environmental Factors Affecting Human Health; IntechOpen: London, UK, 2020. [Google Scholar]
- DeJong, J.T.; Soga, K.; Banwart, S.A.; Whalley, W.R.; Ginn, T.R.; Nelson, D.C.; Mortensen, B.M.; Martinez, B.C.; Barkouki, T. Soil engineering in vivo: Harnessing natural biogeochemical systems for sustainable, multi-functional engineering solutions. J. R. Soc. Interface 2011, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, J.; Zhang, W.; Wang, L.; Putnis, C.V. Direct Observations of the Occlusion of Soil Organic Matter within Calcite. Environ. Sci. Technol. 2019, 53, 8097–8104. [Google Scholar] [CrossRef]
- Ondrasek, G.; Begić, H.B.; Zovko, M.; Filipović, L.; Meriño-Gergichevich, C.; Savic, R.; Rengel, Z. Biogeochemistry of soil organic matter in agroecosystems & environmental implications. Sci. Total Environ. 2018, 658, 1559–1573. [Google Scholar] [CrossRef]
- Van Hees, P.A.; Jones, D.L.; Finlay, R.; Godbold, D.L.; Lundström, U.S. The carbon we do not see—The impact of low molecular weight compounds on carbon dynamics and respiration in forest soils: A review. Soil Biol. Biochem. 2005, 37, 1–13. [Google Scholar] [CrossRef]
- Cotrufo, M.F.; Soong, J.L.; Horton, A.J.; Campbell, E.E.; Haddix, M.; Wall, D.H.; Parton, W.J. Formation of soil organic matter via biochemical and physical pathways of litter mass loss. Nat. Geosci. 2015, 8, 776–779. [Google Scholar] [CrossRef]
- Wiseschart, A.; Mhuantong, W.; Tangphatsornruang, S.; Chantasingh, D.; Pootanakit, K. Shotgun metagenomic sequencing from Manao-Pee cave, Thailand, reveals insight into the microbial community structure and its metabolic potential. BMC Microbiol. 2019, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, L.; Boden, R.; Hillebrand, A.; Kumaresan, D.; Moussard, H.; Baciu, M.; Lu, Y.; Murrell, J.C. Life without light: Microbial diversity and evidence of sulfur- and ammonium-based chemolithotrophy in Movile Cave. ISME J. 2009, 3, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ersan, Y.C.; Boon, N.; De Belie, N. Application of microorganisms in concrete: A promising sustainable strategy to improve concrete durability. Appl. Microbiol. Biotechnol. 2016, 100, 2993–3007. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Dittrich, M. Carbonate precipitation through microbial activities in natural environment, and their potential in biotechnology: A review. Front. Bioeng. Biotechnol. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanier, S.; Le Métayer-Levrel, G.; Perthuisot, J.-P. Bacterial Roles in the Precipitation of Carbonate Minerals. In Microbial Sediments; Springer: Berlin/Heidelberg, Germany, 2000; pp. 32–39. [Google Scholar] [CrossRef]
- Ortiz, M.; Legatzki, A.; Neilson, J.W.; Fryslie, B.; Nelson, W.M.; Wing, R.; Soderlund, C.A.; Pryor, B.M.; Maier, R. Making a living while starving in the dark: Metagenomic insights into the energy dynamics of a carbonate cave. ISME J. 2013, 8, 478–491. [Google Scholar] [CrossRef]
- Feigl, V.; Ujaczki, E.; Vaszita, E.; Molnár, M. Influence of red mud on soil microbial communities: Application and comprehensive evaluation of the Biolog EcoPlate approach as a tool in soil microbiological studies. Sci. Total Environ. 2017, 595, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Salomo, S.; Münch, C.; Röske, I. Evaluation of the metabolic diversity of microbial communities in four different filter layers of a constructed wetland with vertical flow by Biolog™ analysis. Water Res. 2009, 43, 4569–4578. [Google Scholar] [CrossRef]
- Grządziel, J.; Furtak, K.; Gałązka, A. Community-Level Physiological Profiles of Microorganisms from Different Types of Soil That are Characteristic to Poland—A Long-Term Microplot Experiment. Sustainability 2018, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Gryta, A.; Frąc, M.; Oszust, K. The Application of the Biolog EcoPlate Approach in Ecotoxicological Evaluation of Dairy Sewage Sludge. Appl. Biochem. Biotechnol. 2014, 174, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Chan, K.-G.; Tay, Y.-L.; Chua, Y.-H.; Goh, K.M. Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing. Front. Microbiol. 2015, 6, 177. [Google Scholar] [CrossRef] [Green Version]
- D’Auria, G.; Artacho, A.; Rojas, R.A.; Bautista, J.S.; Méndez, R.; Gamboa, M.T.; Gamboa, J.R.; Gómez-Cruz, R. Metagenomics of Bacterial Diversity in Villa Luz Caves with Sulfur Water Springs. Genes 2018, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- De Mandal, S.; Chatterjee, R.; Kumar, N.S. Dominant bacterial phyla in caves and their predicted functional roles in C and N cycle. BMC Microbiol. 2017, 17, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, E.; Carey, M.; Meharg, A.A.; Meharg, C. Microbiome and ecotypic adaption of Holcus lanatus (L.) to extremes of its soil pH range, investigated through transcriptome sequencing. Microbiome 2018, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenarova, A.; Radeva, G.; Traykov, I.; Boteva, S. Community level physiological profiles of bacterial communities inhabiting uranium mining impacted sites. Ecotoxicol. Environ. Saf. 2014, 100, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Grządziel, J.; Gałązka, A. Microplot long-term experiment reveals strong soil type influence on bacteria composition and its functional diversity. Appl. Soil Ecol. 2018, 124, 117–123. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- DouglasGM, M.; ZaneveldJ, Y.; BrownJR, T.; HuttenhowerC, L. PICRUSt2: An improved and customizable approach for metagenome inference. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kurten, G.L.; Barkoh, A. Evaluation of Community-Level Physiological Profiling for Monitoring Microbial Community Function in Aquaculture Ponds. N. Am. J. Aquac. 2015, 78, 34–44. [Google Scholar] [CrossRef]
- Bais, A.F.; Lucas, R.M.; Bornman, J.F.; Williamson, C.E.; Sulzberger, B.; Austin, A.; Wilson, S.R.; Andrady, A.L.; Bernhard, G.; McKenzie, R.L.; et al. Environmental effects of ozone depletion, UV radiation and interactions with climate change: UNEP Environmental Effects Assessment Panel, update 2017. Photochem. Photobiol. Sci. 2018, 17, 127–179. [Google Scholar] [CrossRef]
- Pieristè, M.; Forey, E.; Sahraoui, A.L.-H.; Meglouli, H.; Laruelle, F.; Delporte, P.; Robson, T.M.; Chauvat, M. Spectral Composition of Sunlight Affects the Microbial Functional Structure of Beech Leaf Litter During the Initial Phase of Decomposition. Plant Soil 2020, 451, 515–530. [Google Scholar] [CrossRef]
- Ward, C.P.; Nalven, S.G.; Crump, B.C.; Kling, G.W.; Cory, R.M. Photochemical alteration of organic carbon draining permafrost soils shifts microbial metabolic pathways and stimulates respiration. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Zhu, H.-Z.; Zhang, Z.-F.; Zhou, N.; Jiang, C.-Y.; Wang, B.-J.; Cai, L.; Liu, S.-J. Diversity, Distribution and Co-occurrence Patterns of Bacterial Communities in a Karst Cave System. Front. Microbiol. 2019, 10, 1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochner, B.R. Global phenotypic characterization of bacteria. FEMS Microbiol. Rev. 2008, 33, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Gunina, A.; Smith, A.R.; Kuzyakov, Y.; Jones, D. Microbial uptake and utilization of low molecular weight organic substrates in soil depend on carbon oxidation state. Biogeochemistry 2017, 133, 89–100. [Google Scholar] [CrossRef]
- Ravn, N.R.; Michelsen, A.; Reboleira, A.S. Decomposition of Organic Matter in Caves. Front. Ecol. Evol. 2020, 8. [Google Scholar] [CrossRef]
- Wu, Y.; Tan, L.; Liu, W.; Wang, B.; Wang, J.; Cai, Y.; Lin, X. Profiling bacterial diversity in a limestone cave of the western Loess Plateau of China. Front. Microbiol. 2015, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Singh, M.; Kastner, A.; Merten, D.; Büchel, G.; Kothe, E. Microbial communities in carbonate rocks-from soil via groundwater to rocks. J. Basic Microbiol. 2017, 57, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk-Żak, K.; Zielenkiewicz, U. Microbial Diversity in Caves. Geomicrobiol. J. 2015, 33, 20–38. [Google Scholar] [CrossRef]
- Kumaresan, D.; Wischer, D.; Stephenson, J.; Hillebrand-Voiculescu, A.; Murrell, J.C. Microbiology of Movile Cave—A Chemolithoautotrophic Ecosystem. Geomicrobiol. J. 2014, 31, 186–193. [Google Scholar] [CrossRef]
- Cui, J.; Davidson, A.L. ABC solute importers in bacteria. Essays Biochem. 2011, 50, 85–99. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Lu, J.; Wang, Y.; Jiao, N. Genomic reconstructions and potential metabolic strategies of generalist and specialist heterotrophic bacteria associated with an estuary Synechococcus culture. FEMS Microbiol. Ecol. 2019, 95, fiz017. [Google Scholar] [CrossRef] [Green Version]
- Bergauer, K.; Fernandez-Guerra, A.; Garcia, J.A.L.; Sprenger, R.; Stepanauskas, R.; Pachiadaki, M.; Jensen, O.N.; Herndl, G.J. Organic matter processing by microbial communities throughout the Atlantic water column as revealed by metaproteomics. Proc. Natl. Acad. Sci. USA 2017, 115, E400–E408. [Google Scholar] [CrossRef] [Green Version]
- Ren, Q.; Paulsen, I.T. Large-Scale Comparative Genomic Analyses of Cytoplasmic Membrane Transport Systems in Prokaryotes. J. Mol. Microbiol. Biotechnol. 2007, 12, 165–179. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | LR-01 | LS-01 | LR-05 | LS-05 | p Value |
|---|---|---|---|---|---|
| AWCD | 1.06 | 1.53 | 0.09 | 0.31 | <0.05 |
| Shannon Index H’ | 3.00 | 3.38 | 2.18 | 2.65 | <0.05 |
| Simpson Index D | 0.98 | 0.98 | 1.00 | 0.98 | >0.05 |
| Shannon evenness | 0.87 | 0.98 | 0.63 | 0.77 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koner, S.; Chen, J.-S.; Hsu, B.-M.; Tan, C.-W.; Fan, C.-W.; Chen, T.-H.; Hussain, B.; Nagarajan, V. Assessment of Carbon Substrate Catabolism Pattern and Functional Metabolic Pathway for Microbiota of Limestone Caves. Microorganisms 2021, 9, 1789. https://doi.org/10.3390/microorganisms9081789
Koner S, Chen J-S, Hsu B-M, Tan C-W, Fan C-W, Chen T-H, Hussain B, Nagarajan V. Assessment of Carbon Substrate Catabolism Pattern and Functional Metabolic Pathway for Microbiota of Limestone Caves. Microorganisms. 2021; 9(8):1789. https://doi.org/10.3390/microorganisms9081789
Chicago/Turabian StyleKoner, Suprokash, Jung-Sheng Chen, Bing-Mu Hsu, Chao-Wen Tan, Cheng-Wei Fan, Tsung-Hsien Chen, Bashir Hussain, and Viji Nagarajan. 2021. "Assessment of Carbon Substrate Catabolism Pattern and Functional Metabolic Pathway for Microbiota of Limestone Caves" Microorganisms 9, no. 8: 1789. https://doi.org/10.3390/microorganisms9081789
APA StyleKoner, S., Chen, J.-S., Hsu, B.-M., Tan, C.-W., Fan, C.-W., Chen, T.-H., Hussain, B., & Nagarajan, V. (2021). Assessment of Carbon Substrate Catabolism Pattern and Functional Metabolic Pathway for Microbiota of Limestone Caves. Microorganisms, 9(8), 1789. https://doi.org/10.3390/microorganisms9081789