
Genome-Resolved Metagenomic Analyses Reveal the Presence of a Putative Bacterial Endosymbiont in an Avian Nasal Mite (Rhinonyssidae; Mesostigmata)


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
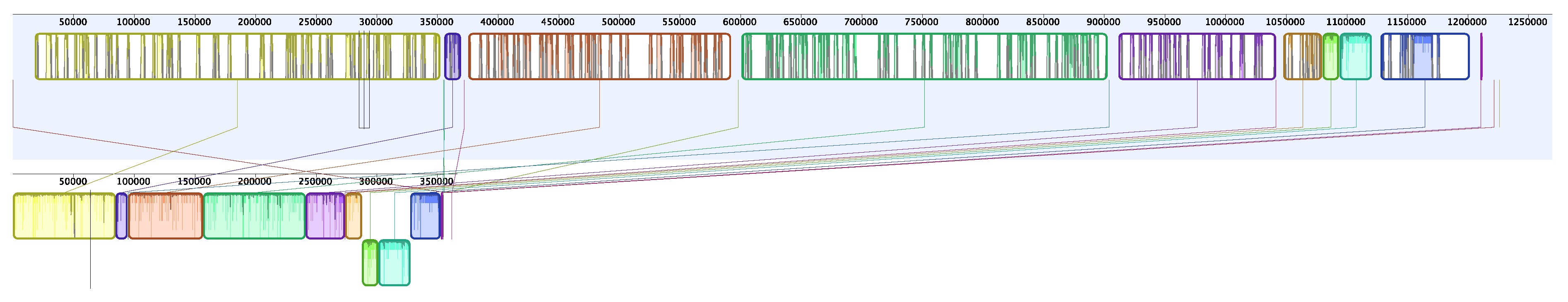
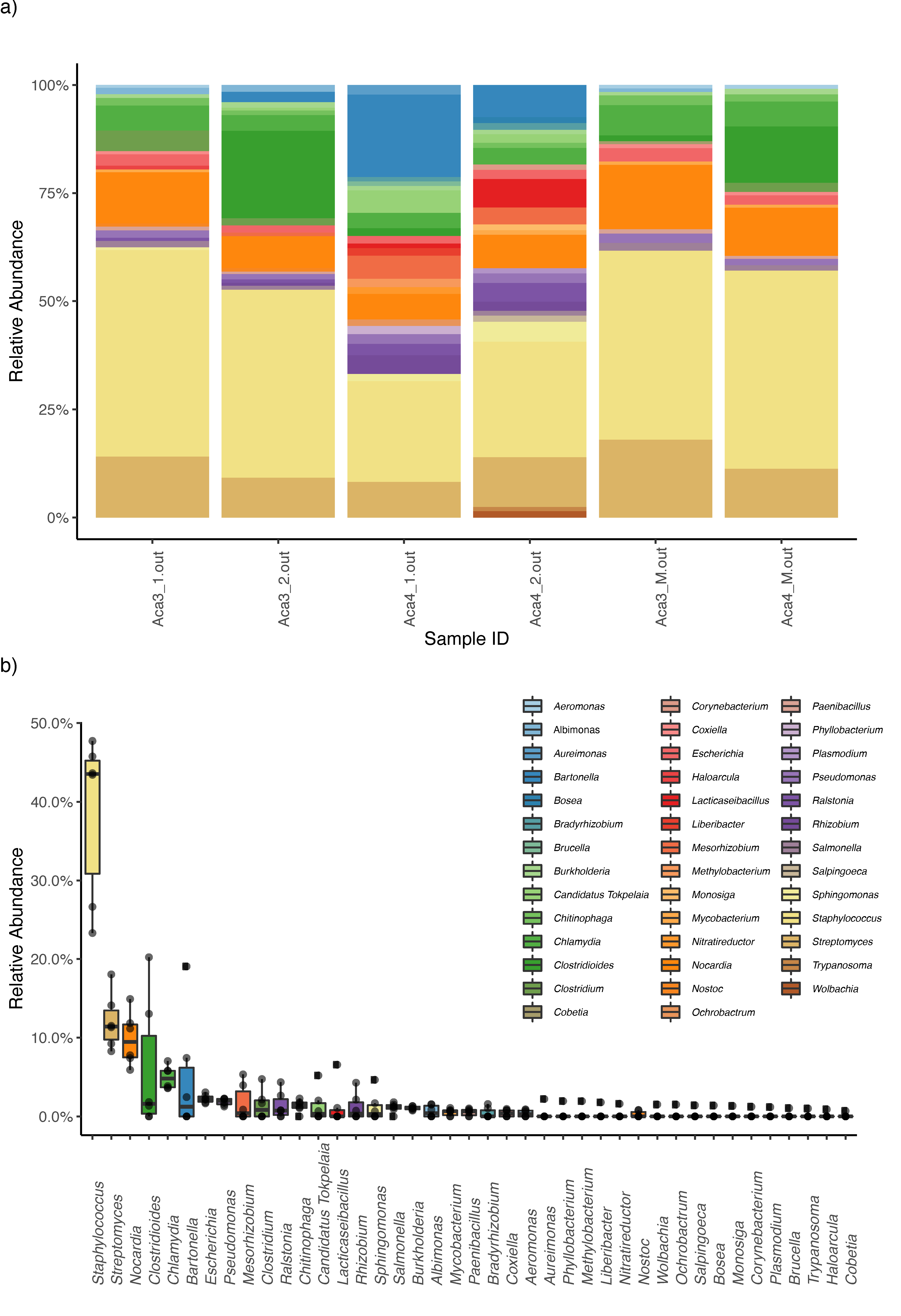
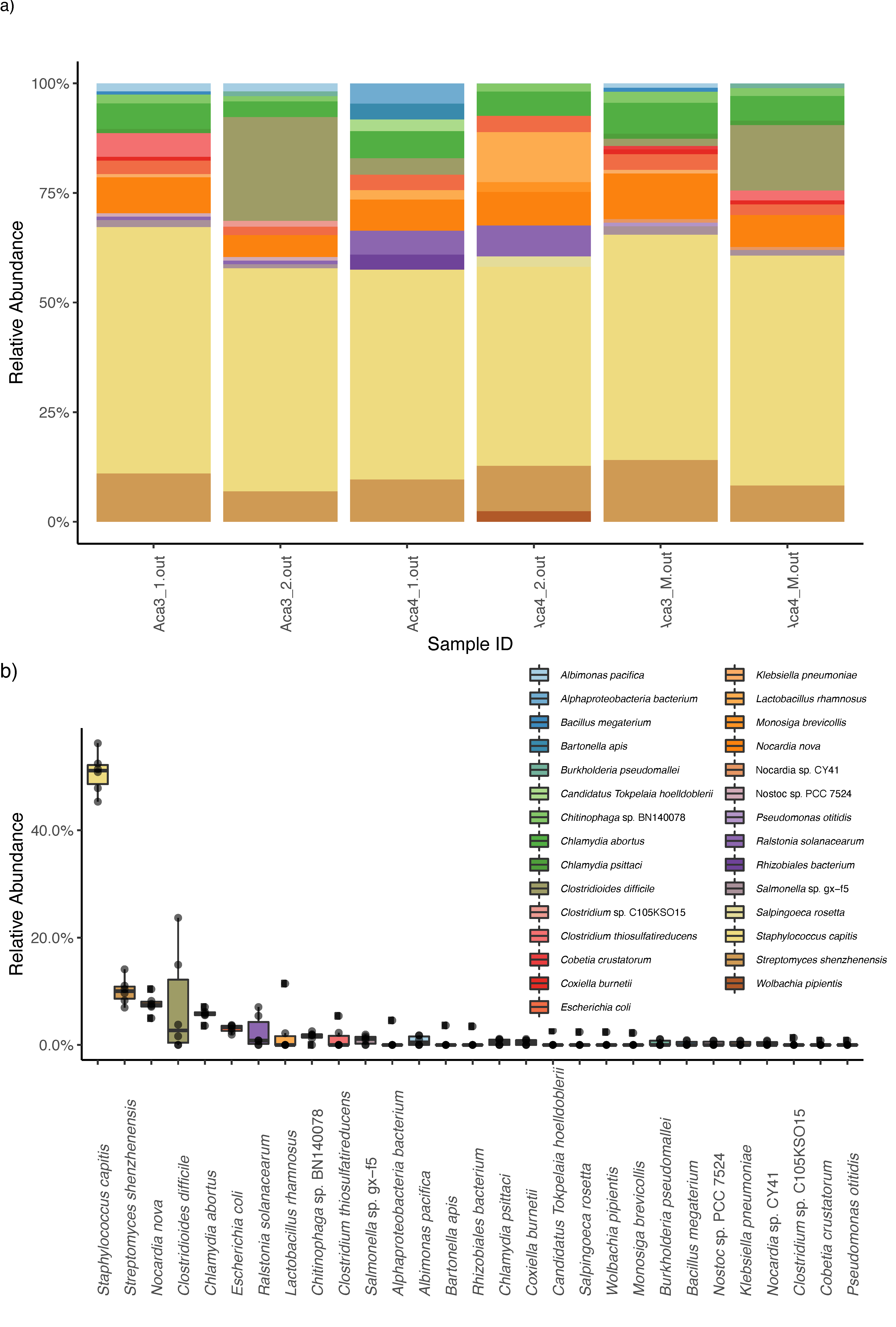
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walter, D.E.; Proctor, H.C. Mites: Ecology, Evolution, and Behaviour; Springer: Dordrecht, The Netherlands, 1999; pp. 1–9. [Google Scholar]
- Proctor, H.; Pritchard, G. Neglected predators: Water mites (Acari: Parasitengona: Hydrachnellae) in freshwater communities. J. N. Am. Benthol. Soc. 1989, 8, 100–111. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Gerecke, R.; Martin, P. The biology and ecology of lotic water mites (Hydrachnidia). Freshw. Biol. 2000, 44, 47–62. [Google Scholar] [CrossRef]
- Solhøy, T. Oribatid mites. In Tracking Environmental Change Using Lake Sediments; Springer: Dordrecht, The Netherlands, 2001; pp. 81–104. [Google Scholar]
- Proctor, H.C. Aquatic Mites from Genes to Communities; Springer: Dordrecht, The Netherlands, 2004; pp. 1–2. [Google Scholar]
- Gergócs, V.; Hufnagel, L. Application of oribatid mites as indicators. Appl. Ecol. Env. Res. 2009, 7, 79–98. [Google Scholar] [CrossRef]
- Lindo, Z.; Winchester, N.N. Spatial and environmental factors contributing to patterns in arboreal and terrestrial oribatid mite diversity across spatial scales. Oecologia 2009, 160, 817–825. [Google Scholar] [CrossRef]
- Maraun, M.; Erdmann, G.; Schulz, G.; Norton, R.A.; Scheu, S.; Domes, K. Multiple convergent evolution of arboreal life in oribatid mites indicates the primacy of ecology. Proc. R. Soc. B. 2009, 276, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.E.; Proctor, H.C. Animals as Habitats. Mites: Ecology, Evolution & Behaviour; Springer: Dordrecht, The Netherlands, 2013; pp. 341–422. [Google Scholar]
- Doña, J.; Proctor, H.; Mironov, S.; Serrano, D.; Jovani, R. Global associations between birds and vane-dwelling feather mites. Ecology 2016, 97, 3242. [Google Scholar] [CrossRef]
- De Rojas, M.; Doña, J.; Dimov, I. A comprehensive survey of rhinonyssid mites (Mesostigmata: Rhinonyssidae) in Northwest Russia: New mite-host associations and prevalence data. Biodivers. Data J. 2020, 8, e49535. [Google Scholar] [CrossRef] [PubMed]
- Sastre, N.; Calvete, O.; Martínez-Vargas, J.; Medarde, N.; Casellas, J.; Altet, L.; Sánchez, A.; Ventura, J. Skin mites in mice (Mus musculus): High prevalence of Myobia sp. (Acari, Arachnida) in Robertsonian mice. Parasitol. Res. 2018, 117, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Doña, J.; Proctor, H.; Serrano, D.; Johnson, K.P.; Oploo, A.O.V.; Huguet-Tapia, J.C.; Ascunce, M.; Jovani, R. Feather mites play a role in cleaning host feathers: New insights from DNA metabarcoding and microscopy. Mol. Ecol. 2019, 28, 203–218. [Google Scholar] [CrossRef]
- Kim, K.C. Coevolution of Parasitic Arthropods and Mammals; Wiley: New York, NY, USA, 1985; pp. 1–800. [Google Scholar]
- Pesapane, R.; Dodd, E.; Javeed, N.; Miller, M.; Foley, J. Molecular characterization and prevalence of Halarachne halichoeri in threatened southern sea otters (Enhydra lutris nereis). Int. J. Parasitol. Parasites. Wildl. 2018, 386–390. [Google Scholar] [CrossRef]
- González-Candela, M.; Léon-Vizcaíno, L.; Cubero-Pablo, M.J. Population effects of sarcoptic mange in Barbary sheep (Ammotragus lervia) from Sierra Espuña Regional Park, Spain. J. Wildl. Dis. 2004, 40, 456–465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hartley, M.; English, A. Sarcoptes scabei var. wombati infection in the common wombat (Vombatus ursinus). Eur. J. Wildl. Res. 2005, 51, 117–121. [Google Scholar] [CrossRef]
- Soulsbury, C.D.; Iossa, G.; Baker, P.J.; Cole, N.C.; Funk, S.M.; Harris, S. The impact of sarcoptic mange Sarcoptes scabiei on the British fox Vulpes vulpes population. Mammal Rev. 2007, 37, 278–296. [Google Scholar]
- Arlian, L.G.; Morgan, M.S. A review of Sarcoptes scabiei: Past, present and future. Parasites. Vectors 2007, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, P.; Aumeier, P.; Ziegelmann, B. Biology and control of Varroa destructor. J. Invertebr. Pathol. 2010, 103, S96–S119. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Takahashi, M.; Matsutani, M.; Takada, N.; Noda, S.; Saijo, M. Obligate intracellular bacteria diversity in unfed Leptotrombidium scutellare larvae highlights novel bacterial endosymbionts of mites. Microbiol. Immunol. 2020, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ahantarig, A.; Trinachartvanit, W.; Baimai, V.; Grubhoffer, L. Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia. Microbiol. 2013, 58, 419–428. [Google Scholar] [CrossRef]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The tick microbiome: Why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent insights into the tick microbiome gained through next-generation sequencing. Parasites. Vectors 2018, 11, 1–14. [Google Scholar] [CrossRef]
- George, J.E. The nasal mites of the genus Ptilonyssus (Acarina: Rhinonyssidae) occurring in some North American passeriform birds. J. Kans. Entomol. Soc. 1961, 34, 105–132. [Google Scholar]
- Fain, A. Adaptation, specificity and host-parasite coevolution in mites (Acari). Int. J. Parasitol. 1994, 24, 1273–1283. [Google Scholar] [CrossRef]
- De Rojas, M.; Úbeda, J.M.; Cutillas, C.; Mora, M.D.; Ariza, C.; Guevara, D. Utility of ITS1–5.8 S–ITS2 and 16S mitochondrial DNA sequences for species identification and phylogenetic inference within the Rhinonyssus coniventris species complex (Acari: Rhinonyssidae). Parasitol. Res. 2007, 100, 1041–1046. [Google Scholar] [CrossRef]
- Dimov, I.; de Rojas, M. One new species of nasal mites of the genus Vitznyssus (Rhinonyssidae) from the Leningrad province, Russia. J. Acarol. Soc. Jap. 2012, 21, 125–130. [Google Scholar] [CrossRef]
- Proctor, H.; Owens, I. Mites and birds: Diversity, parasitism and coevolution. Trends. Ecol. Evol. 2000, 15, 358–364. [Google Scholar] [CrossRef]
- Vitzthum, H.G. Milben aus der Nasenhöhle von Vögeln. J. Ornithol. 1935, 83, 563–587. [Google Scholar] [CrossRef]
- Reeves, W.K.; Dowling, A.P.; Dasch, G.A. Rickettsial agents from parasitic Dermanyssoidea (Acari: Mesostigmata). Exp. Appl. Acarol. 2006, 38, 181–188. [Google Scholar] [CrossRef]
- Osuna-Mascaró, C.; Doña, J.; Johnson, K.P.; Esteban, R.; De Rojas, M. Complete mitochondrial genomes and bacterial metagenomic data from two species of parasitic avian nasal-mites (Rhinonyssidae: Mesostigmata). Front. Ecol. Evol. 2020, 8, 142. [Google Scholar] [CrossRef]
- Fain, A. Les Rhinonyssides parasites des Pigeons. Rev. Zool. Bot. Afr. 1962, 65, 305–324. [Google Scholar]
- Fain, A. Diagnoses d’acariens parasites nouveaux. Rev. Zool. Bot. Afr. 1965, 72, 152–160. [Google Scholar]
- Bonnefoy, X.; Kampen, H.; Sweeney, K. Public Health Significance of Urban Pests; World Health Organization: Geneva, Switzerland, 2008; pp. 53–84. [Google Scholar]
- Knee, W.; Proctor, H.; Galloway, T. Survey of nasal mites (Rhinonyssidae, Ereynetidae, and Turbinoptidae) associated with birds in Alberta and Manitoba, Canada. Can. Entomol. 2008, 140, 364–379. [Google Scholar] [CrossRef]
- Dimov, I.; Mironov, S. Two new species of nasal mites of the genus Ptilonyssus (Rhinonyssidae) from sparrows from the Leningrad province, Russia. J. Hell. Vet. Med. Soc. 2012, 63, 167–176. [Google Scholar] [CrossRef][Green Version]
- Dimov, I.D. Rhinonyssid Mites of Birds from Northwest of Russia; LLC Zhigulin: Saint Petersburg, Russia, 2018; pp. 1–232. [Google Scholar]
- Dimov, I.D. Kleshchi-Rinonissidy ptic Severo-Zapada Ross; LLC Zhigulin: Saint Petersburg, Russia, 2018; pp. 1–232. [Google Scholar]
- Vizcaíno, A.; Doña, J.; Vierna, J.; Marí-Mena, N.; Esteban, R.; Mironov, S.; Urien, C.; Serrano, D.; Jovani, R. Enabling large- scale feather mite studies: An Illumina DNA metabarcoding pipeline. Exp. Appl. Acarol. 2018, 76, 81–97. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBMap Short Read Aligner, and Other Bioinformatic Tools. 2014. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 10 August 2021).
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 113. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome. Res. 2015, 25, 1043–1055. [Google Scholar]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic. Acids. Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commu. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wernegreen, J.J. Endosymbiont evolution: Predictions from theory and surprises from genomes. Ann. N. Y. Acad. Sci. 2015, 1360, 16. [Google Scholar] [CrossRef] [PubMed]
- Doña, J.; Virrueta Herrera, S.; Nyman, T.; Kunnasranta, M.; Johnson, K.P. Patterns of microbiome variation among infrapopulations of permanent bloodsucking parasites. Front. Microbiol. 2021, 12, 884. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome. Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Moran, N.A.; Baumann, P. Bacterial endosymbionts in animals. Curr Opin Microbiol. 2000, 3, 270–275. [Google Scholar] [CrossRef]
- Enigl, M.; Schausberger, P. Incidence of the endosymbionts Wolbachia, Cardinium and Spiroplasma in phytoseiid mites and associated prey. Exp. Appl. Acarol. 2007, 42, 75–85. [Google Scholar] [CrossRef]
- Derbala, A.; Ghazi, Y. Some investigations on brucella and psoroptes mites infections among Barki sheep flocks. Mansoura. Vet. Med. J. 2001, 3, 173–183. [Google Scholar] [CrossRef]
- Hosseini-Chegeni, A.; Tavakoli, M.; Telmadarraiy, Z.; Sedaghat, M.M.; Faghihi, F. Detection of a brucella-like (Alphaproteobacteria) bacterium in boophilus spp. (Acari: Ixodidae) from Iran. J. Med. Microbiol. Infect. Dis. 2017, 5, 66–68. [Google Scholar] [CrossRef]
- Dröge, J.; Gregor, I.; McHardy, A.C. Taxator-tk: Precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics 2015, 31, 817–824. [Google Scholar] [CrossRef]
- Kim, C.-M.; Kim, J.-Y.; Yi, Y.-H.; Lee, M.-J.; Cho, M.-R.; Shah, D.H.; Klein, T.A.; Kim, H.-C.; Song, J.-W.; Chong, S.-T.; et al. Detection of Bartonella species from ticks, mites and small mammals in Korea. J. Vet. Sci. 2005, 6, 327–334. [Google Scholar] [CrossRef]
- Hubert, J.; Erban, T.; Kopecky, J.; Sopko, B.; Nesvorna, M.; Lichovnikova, M. Comparison of microbiomes between red poultry mite populations (Dermanyssus gallinae): Predominance of Bartonella-like bacteria. Microb. Ecol. 2017, 74, 947–960. [Google Scholar] [CrossRef]
- Walter, D.E.; Proctor, H.C. Mites that Cause and Transmit Disease. In Mites: Ecology, Evolution & Behaviour; Springer: Dordrecht, The Netherlans, 2013. [Google Scholar]
- Tang, V.H.; Chang, B.J.; Srinivasan, A.; Mathaba, L.T.; Harnett, G.B.; Stewart, G.A. Skin-associated Bacillus, staphylococcal and micrococcal species from the house dust mite, Dermatophagoides pteronyssinus and bacteriolytic enzymes. Exp. Appl. Acarol. 2013, 61, 431–447. [Google Scholar] [CrossRef]
- Kopecký, J.; Nesvorná, M.; Hubert, J. Bartonella-like bacteria carried by domestic mite species. Exp. Appl. Acarol. 2014, 64, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Kopecky, J.; Sagova-Mareckova, M.; Nesvorna, M.; Zurek, L.; Erban, T. Assessment of bacterial communities in thirteen species of laboratory-cultured domestic mites (Acari: Acaridida). J. Econ. Entomol. 2016, 109, 1887–1896. [Google Scholar]
- Hubert, J.; Kopecký, J.; Perotti, M.A.; Nesvorná, M.; Braig, H.R.; Ságová-Marečková, M.; Macovei, L.; Zurek, L. Detection and identification of species-specific bacteria associated with synanthropic mites. Microb. Ecol. 2012, 63, 919–928. [Google Scholar] [CrossRef]
- Dzoro, S.; Mittermann, I.; Resch-Marat, Y.; Vrtala, S.; Nehr, M.; Hirschl, A.M.; Wikberg, G.; Lundeberg, L.; Johansson, C.; Scheynius, A.; et al. House dust mites as potential carriers for IgE sensitization to bacterial antigens. Allergy 2018, 73, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Molva, V.; Nesvorna, M.; Hubert, J. Feeding interactions between microorganisms and the house dust mites Dermatophagoides pteronyssinus and Dermatophagoides farinae (Astigmata: Pyroglyphidae). J. Med. Entomol. 2019, 56, 1669–1677. [Google Scholar] [CrossRef]
- Erban, T.; Klimov, P.; Molva, V.; Hubert, J. Whole genomic sequencing and sex-dependent abundance estimation of Cardinium sp., a common and hyperabundant bacterial endosymbiont of the American house dust mite, Dermatophagoides farinae. Exp. Appl. Acarol. 2020, 80, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.P.; Solonenko, N.E.; Gazitúa, M.C.; Kenny, D.V.; Mosley-Thompson, E.; Rich, V.I.; Sullivan, M.B. Clean low-biomass procedures and their application to ancient ice core microorganisms. Front. Microbiol. 2018, 9, 1094. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends. Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Swe, P.M.; Zakrzewski, M.; Waddell, R.; Sriprakash, K.S.; Fischer, K. High-throughput metagenome analysis of the Sarcoptes scabiei internal microbiota and in-situ identification of intestinal Streptomyces sp. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Guyomar, C.; Delage, W.; Legeai, F.; Mougel, C.; Simon, J.C.; Lemaitre, C. MinYS: Mine your symbiont by targeted genome assembly in symbiotic communities. NAR. Genom. Bioinform. 2020, 2, lqaa047. [Google Scholar] [CrossRef] [PubMed]
- Wales, A.D.; Carrique-Mas, J.J.; Rankin, M.; Bell, B.; Thind, B.B.; Davies, R.H. Review of the carriage of zoonotic bacteria by arthropods, with special reference to Salmonella in mites, flies and litter beetles. Zoonoses Public Health 2010, 57, 299–314. [Google Scholar] [PubMed]
- Veiga, J.; Dimov, I.; de Rojas, M. Endoparasitic Mites (Rhinonyssidae) on Urban Pigeons and Doves: Updating Morphological and Epidemiological Information. Divers 2021, 13, 11. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osuna-Mascaró, C.; Doña, J.; Johnson, K.P.; de Rojas, M. Genome-Resolved Metagenomic Analyses Reveal the Presence of a Putative Bacterial Endosymbiont in an Avian Nasal Mite (Rhinonyssidae; Mesostigmata). Microorganisms 2021, 9, 1734. https://doi.org/10.3390/microorganisms9081734
Osuna-Mascaró C, Doña J, Johnson KP, de Rojas M. Genome-Resolved Metagenomic Analyses Reveal the Presence of a Putative Bacterial Endosymbiont in an Avian Nasal Mite (Rhinonyssidae; Mesostigmata). Microorganisms. 2021; 9(8):1734. https://doi.org/10.3390/microorganisms9081734
Chicago/Turabian StyleOsuna-Mascaró, Carolina, Jorge Doña, Kevin P. Johnson, and Manuel de Rojas. 2021. "Genome-Resolved Metagenomic Analyses Reveal the Presence of a Putative Bacterial Endosymbiont in an Avian Nasal Mite (Rhinonyssidae; Mesostigmata)" Microorganisms 9, no. 8: 1734. https://doi.org/10.3390/microorganisms9081734
APA StyleOsuna-Mascaró, C., Doña, J., Johnson, K. P., & de Rojas, M. (2021). Genome-Resolved Metagenomic Analyses Reveal the Presence of a Putative Bacterial Endosymbiont in an Avian Nasal Mite (Rhinonyssidae; Mesostigmata). Microorganisms, 9(8), 1734. https://doi.org/10.3390/microorganisms9081734