
Adaptations for Pressure and Temperature in Dihydrofolate Reductases


Abstract
:
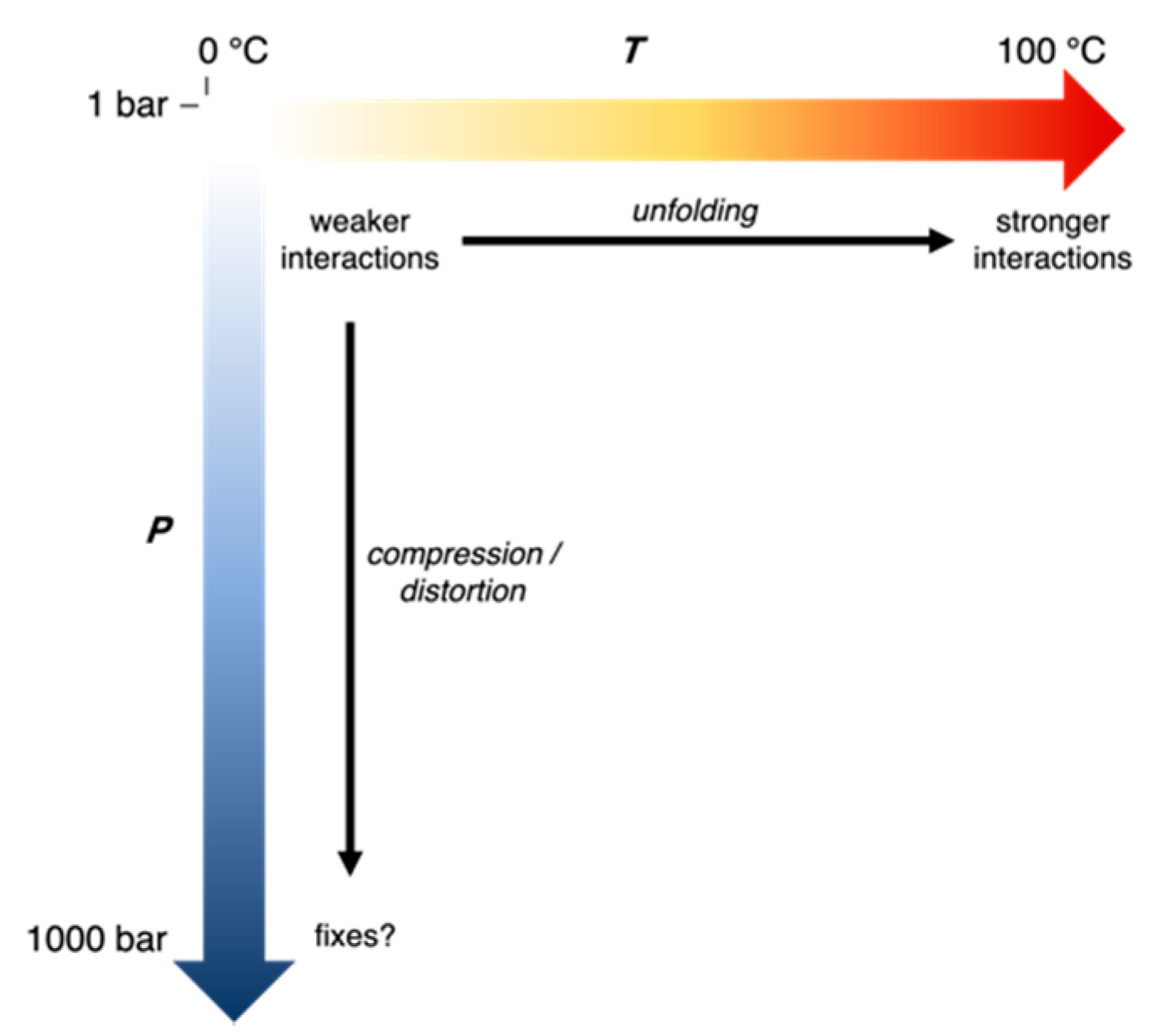
1. Introduction
2. Methods
3. Analysis
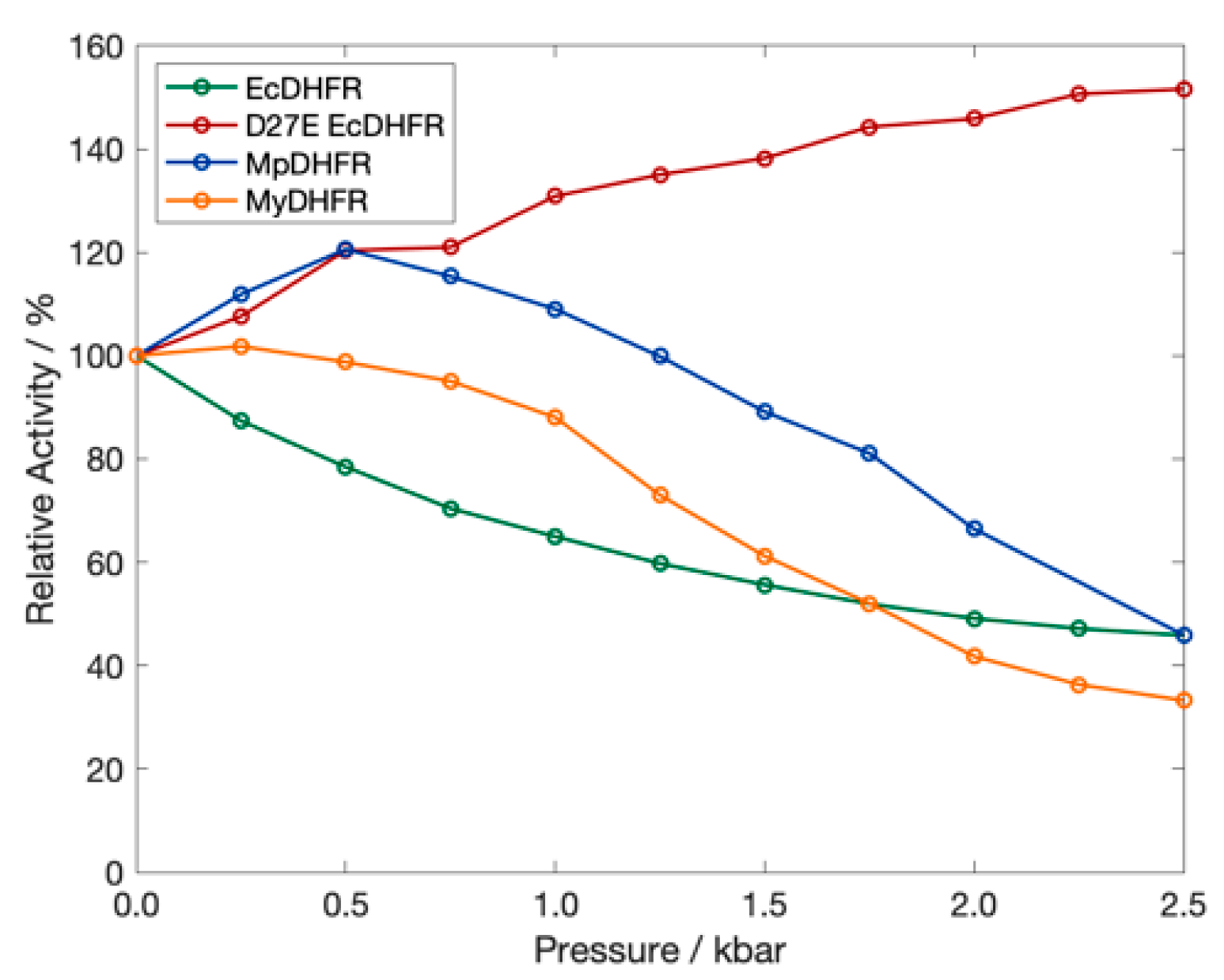
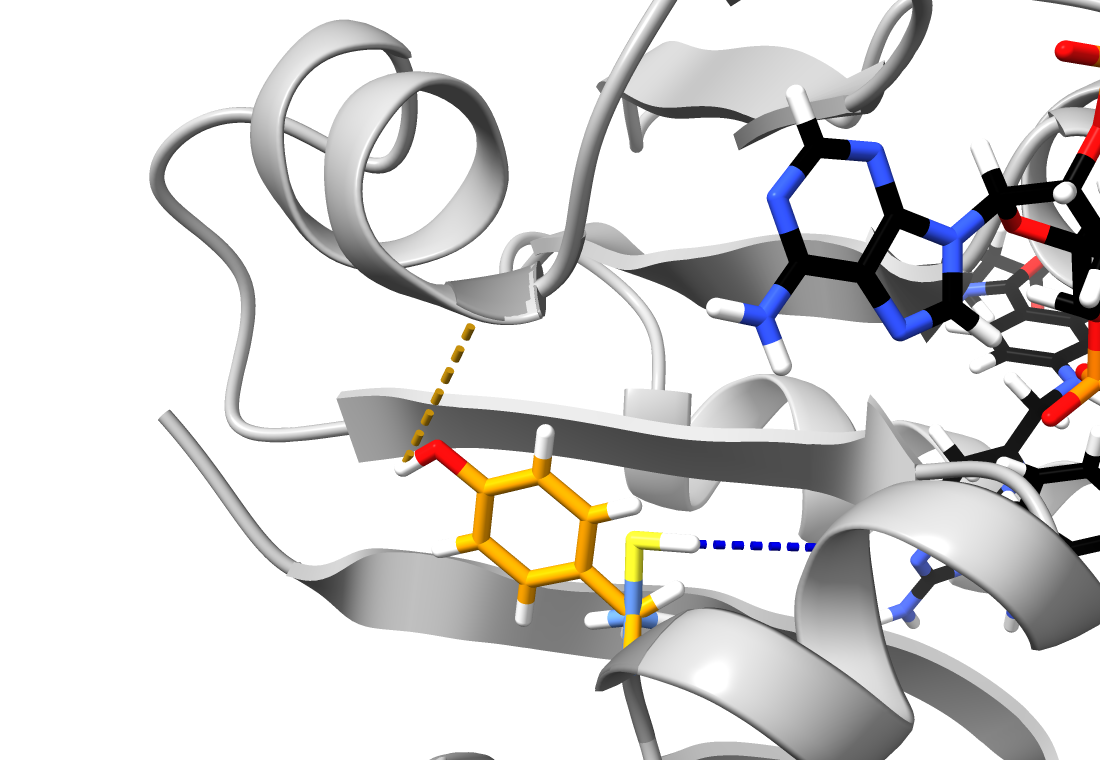
4. Results
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrison, J.P.; Gheeraert, N.; Tsigelnitskiy, D.; Cockell, C.S. The limits for life under multiple extremes. Trends Microbiol. 2013, 21, 204–212. [Google Scholar] [CrossRef]
- Ichiye, T. What makes proteins work: Exploring life in P-T-X. Phys. Biol. 2016, 13, 063001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junge, K.; Eicken, H.; Deming, J.W. Bacterial Activity at −2 to −20 degrees C in Arctic wintertime sea ice. Appl. Environ. Microbiol. 2004, 70, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, K.; Nakamura, K.; Toki, T.; Tsunogai, U.; Miyazaki, M.; Miyazaki, J.; Hirayama, H.; Nakagawa, S.; Nunoura, T.; Horikoshi, K. Cell proliferation at 122 °C and isotopically heavy CH4 production by a hyperthermophilic methanogen under high-pressure cultivation. Proc. Natl. Acad. Sci. USA 2008, 105, 10949–10954. [Google Scholar] [CrossRef] [Green Version]
- Somero, G.N. Proteins and temperature. Ann. Rev. Physiol. 1995, 57, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Jaenicke, R. Protein stability and molecular adaptation to extreme conditions. Eur. J. Biochem. 1991, 1991, 291–304. [Google Scholar] [CrossRef]
- Feller, G. Psychrophilic Enzymes: From Folding to Function and Biotechnology. Scientifica 2013, 2013, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.; Phillips, G.N. Structures and Analysis of Highly Homologous Psychrophilic, Mesophilic, and Thermophilic Adenylate Kinases. J. Biol. Chem. 2004, 279, 28202–28208. [Google Scholar] [CrossRef] [Green Version]
- Meinhold, L.; Clement, D.; Tehei, M.; Daniel, R.; Finney, J.L.; Smith, J.C. Protein Dynamics and Stability: The Distribution of Atomic Fluctuations in Thermophilic and Mesophilic Dihydrofolate Reductase Derived Using Elastic Incoherent Neutron Scattering. Biophys. J. 2008, 94, 4812–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.; Wilson, C.; Hoemberger, M.; Stiller, J.B.; Agafonov, R.V.; Kutter, S.; English, J.; Theobald, D.L.; Kern, D. Evolutionary drivers of thermoadaptation in enzyme catalysis. Science 2016, 355, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Arcus, V.L.; Prentice, E.J.; Hobbs, J.; Mulholland, A.; van der Kamp, M.; Pudney, C.; Parker, E.; Schipper, L.A. On the Temperature Dependence of Enzyme-Catalyzed Rates. Biochemistry 2016, 55, 1681–1688. [Google Scholar] [CrossRef]
- Pinney, M.M.; Mokhtari, D.A.; Akiva, E.; Yabukarski, F.; Sanchez, D.M.; Liang, R.; Doukov, T.; Martinez, T.J.; Babbitt, P.C.; Herschlag, D. Parallel molecular mechanisms for enzyme temperature adaptation. Science 2021, 371, eaay2784. [Google Scholar] [CrossRef]
- Berezovsky, I.N.; Shakhnovich, E.I. Physics and evolution of thermophilic adaptation. Proc. Natl. Acad. Sci. USA 2005, 102, 12742–12747. [Google Scholar] [CrossRef] [Green Version]
- Peterson, R.W.; Wand, J.A. Self-contained high-pressure cell, apparatus, and procedure for the preparation of encapsulated proteins dissolved in low viscosity fluids for nuclear magnetic resonance spectroscopy. Rev. Sci. Instrum. 2005, 76, 094101. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.D.; Kim, C.U.; Gruner, S.M. High-Pressure Protein Crystallography and NMR to Explore Protein Conformations. Annu. Rev. Biophys. 2011, 40, 81–98. [Google Scholar] [CrossRef] [Green Version]
- Ando, N.; Barstow, B.; Baase, W.A.; Fields, A.; Matthews, B.W.; Gruner, S.M. Structural and Thermodynamic Characterization of T4 Lysozyme Mutants and the Contributions of Internal Cavities to Pressure Denaturation. Biochemistry 2008, 47, 11097–11109. [Google Scholar] [CrossRef] [PubMed]
- Ortore, M.G.; Spinozzi, F.; Mariani, P.; Paciaroni, A.; Barbosa, L.; Amenitsch, H.; Steinhart, M.; Ollivier, J.; Russo, D. Combining structure and dynamics: Non-denaturing high-pressure effect on lysozyme in solution. J. R. Soc. Interface 2009, 6, S619–S634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filabozzi, A.; Deriu, A.; Di Bari, M.; Russo, D.; Croci, S.; Di Venere, A. Elastic incoherent neutron scattering as a probe of high pressure induced changes in protein flexibility. Biochim. Biophys. Acta 2010, 1804, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Scott, J.H.; Cody, G.D.; Fogel, M.L.; Hazen, R.M.; Hemley, R.J.; Huntress, W.T. Microbial Activity at Gigapascal Pressures. Science 2002, 295, 1514–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.; Martinez, N.; Michoud, G.; Cario, A.; Franzetti, B.; Oger, P.; Jebbar, M. Deep Sea Microbes Probed by Incoherent Neutron Scattering Under High Hydrostatic Pressure. Z. Phys. Chem. 2014, 228, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Deep Life Leadership. Available online: https://deepcarbon.net/content/deep-life-leadership (accessed on 30 July 2021).
- Gross, M.; Jaenicke, R. Review: Proteins under pressure: The influence of high hydrostatic pressure on structure, function and assembly of proteins and protein complexes. Eur. J. Biochem. 1994, 221, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Vanlint, D.; Mitchell, R.; Bailey, E.; Meersman, F.; McMillan, P.F.; Michiels, C.W.; Aertsen, A. Rapid aquisition of gigapascal-high-pressure resistance by Escherichia coli. mBio 2011, 2, e00130-00110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazael, R.; Foglia, F.; Kardzhaliyska, L.; Daniel, I.; Meersmen, F.; McMillan, P. Laboratory investigation of high pressure survival in Shewanella oneidensis MR-1 in the gigapascal pressure range. Front. Microbiol. 2014, 5, 612. [Google Scholar] [CrossRef] [Green Version]
- Nogi, Y.; Kato, C. Taxonomic studies of extremely barophilic bacteria isolated from the Mariana Trench and description of Moritella yayanosii sp. nov., a new barophilic bacterial isolate. Extremophiles 1999, 3, 71–77. [Google Scholar] [CrossRef]
- Sinha, N.; Nepal, S.; Kral, T.; Kumar, P. Survivability and growth kinetics of methanogenic archaea at various pHs and pressures: Implications for deep subsurface life on Mars. Planet. Space Sci. 2017, 136, 15–24. [Google Scholar] [CrossRef]
- Schnell, J.R.; Dyson, H.J.; Wright, P. Structure, Dynamics, and Catalytic Function of Dihydrofolate Reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, M.; Kraut, J. Loop and Subdomain Movements in the Mechanism of Escherichia coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef]
- Osborne, M.J.; Schnell, J.; Benkovic, S.J.; Dyson, H.J.; Wright, P.E. Backbone Dynamics in Dihydrofolate Reductase Complexes: Role of Loop Flexibility in the Catalytic Mechanism. Biochemistry 2001, 40, 9846–9859. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, R.; Sareth, S.; Yamada, H.; Ohmae, E.; Gekko, K.; Akasaka, K. High Pressure NMR Reveals Active-Site Hinge Motion of Folate-Bound Escherichia coli Dihydrofolate Reductase. Biochemistry 2000, 39, 12789–12795. [Google Scholar] [CrossRef]
- Nagae, T.; Yamada, H.; Watanabe, N. High-pressure protein crystal structure analysis of Escherichia coli dihydrofolate reductase complexed with folate and NADP+. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Ohmae, E.; Murakami, C.; Tate, S.-I.; Gekko, K.; Hata, K.; Akasaka, K.; Kato, C. Pressure dependence of activity and stability of dihydrofolate reductases of the deep-sea bacterium Moritella profunda and Escherichia coli. Biochim. Biophys. Acta 2012, 1824, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Nogi, Y.; Kato, C.; Liang, Z.; Rüger, H.-J.; De Kegel, D.; Glansdorff, N. Moritella profunda sp. nov. and Moritella abyssi sp. nov., two psychropiezophilic organisms isolated from deep Atlantic sediments. Int. J. Syst. Evol. Microbiol. 2003, 53, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Nagae, T.; Kato, C.; Watanabe, N. Structural analysis of 3-isopropylmalate dehydrogenase from the obligate piezophileShewanella benthicaDB21MT-2 and the nonpiezophileShewanella oneidensisMR-1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 265–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmae, E.; Miyashita, Y.; Kato, C. Thermodynamic and functional characteristics of deep-sea enzymes revealed by pressure effects. Extremophiles 2013, 17, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Ohmae, E.; Miyashita, Y.; Tate, S.-I.; Gekko, K.; Kitazawa, S.; Kitahara, R.; Kuwajima, K. Solvent environments significantly affect the enzymatic function of Escherichia coli dihydrofolate reductase: Comparison of wild-type protein and active-site mutant D27E. Biochim. Biophys. Acta 2013, 1834, 2782–2794. [Google Scholar] [CrossRef]
- Evans, R.M.; Behiry, E.M.; Tey, L.-H.; Guo, J.; Loveridge, E.J.; Allemann, R.K. Catalysis by Dihydrofolate Reductase from the Psychropiezophile Moritella profunda. ChemBioChem 2010, 11, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Stiller, J.B.; Kerns, S.J.; Hoemberger, M.; Cho, Y.-J.; Otten, R.; Hagan, M.F.; Kern, D. Probing the transition state in enzyme catalysis by high-pressure NMR dynamics. Nat. Catal. 2019, 2, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Delong, E.F.; Franks, D.G.; Yayanos, A. Evolutionary relationships of cultivated psychrophilic and barophilic deep-sea bacteria. Appl. Environ. Microbiol. 1997, 63, 2105–2108. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Extreme biophysics: Enzymes under pressure. J. Comput. Chem. 2017, 38, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Quasi-harmonic analysis of the energy landscapes of dihydrofolate reductase from piezophiles and mesophiles. J. Phys. Chem. B 2018, 122, 5527–5533. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Effects of Pressure and Temperature on the Atomic Fluctuations of Dihydrofolate Reductase from a Psychropiezophile and a Mesophile. Int. J. Mol. Sci. 2019, 20, 1452. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Rodgers, J.M.; Hemley, R.J.; Ichiye, T. Adaptations for pressure and temperature effects on loop motion in Escherichia coli and Moritella profunda dihydrofolate reductase. High Press. Res. 2019, 39, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Penhallurick, R.W.; Harold, A.; Durnal, M.D.; Ichiye, T. How adding a single methylene to dihydrofolate reductase can change its conformational dynamics. J. Chem. Phys. 2021, 154, 165103. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.L.; Johnson, L.A.; Behiry, E.M.; Loveridge, E.J.; Allemann, R.K. A Rapid Analysis of Variations in Conformational Behavior during Dihydrofolate Reductase Catalysis. Biochemistry 2017, 56, 2126–2133. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2016, 38, 1114–1124. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellot, M.; Dunbrack, R.L., Jr.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph, D.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain X1 and X2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.E.M.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Pavelites, J.J.; Gao, J.L.; Bash, P.A.; Mackerell, A.D. A molecular mechanics force field for NAD (+), NADH, and the pyrophosphate groups of nucleotides. J. Comput. Chem. 1997, 18, 221–239. [Google Scholar] [CrossRef]
- Coordinators, N.R.; Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; López, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Coutsias, E.A.; Seok, C.; Jacobson, M.; Dill, K.A. A kinematic view of loop closure. J. Comput. Chem. 2004, 25, 510–528. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Islam, M.S.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues. In Biomolecular Modelling and Simulations; Karabencheva-Christova, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 96, pp. 235–265. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Feller, G.; Gerday, C.; Glansdorff, N. Moritella cold-active dihydrofolate reductase: Are there natural limits to optimization of catalytic efficiency at low temperature? J. Bacteriol. 2003, 185, 5519–5526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, C.; Ohmae, E.; Tate, S.-I.; Gekko, K.; Nakasone, K.; Kato, C. Cloning and characterization of dihydrofolate reductases from deep-sea bacteria. J. Biochem. 2009, 147, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., III; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- York, D.M.; Pedersen, L.G.; Darden, T.A. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: A comparison of the Ewald and trucated list methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Roche, J.; Caro, J.A.; Norberto, D.R.; Barthe, P.; Roumestand, C.; Schlessman, J.L.; Garcia, A.E.; García-Moreno, E.B.; Royer, C.A. Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 6945–6950. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | P = 1 bar | Phi | ||
|---|---|---|---|---|
| NHB | 〈ΔrHA2〉 (Å2) | NHB | 〈ΔrHA2〉 (Å2) | |
| EcDHFR * | 105 ± 2 | 0.57 ± 0.07 | 104 ± 4 | 0.60 ± 0.08 |
| D27E EcDHFR * | 103 ± 3 | 0.60 ± 0.04 | 109 ± 1 | 0.50 ± 0.04 |
| MpDHFR | 104 ± 2 | 0.54 ± 0.04 | 106 ± 3 | 0.53 ± 0.03 |
| MyDHFR | 107 ± 2 | 0.54 ± 0.05 | 105 ± 1 | 0.54 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penhallurick, R.W.; Durnal, M.D.; Harold, A.; Ichiye, T. Adaptations for Pressure and Temperature in Dihydrofolate Reductases. Microorganisms 2021, 9, 1706. https://doi.org/10.3390/microorganisms9081706
Penhallurick RW, Durnal MD, Harold A, Ichiye T. Adaptations for Pressure and Temperature in Dihydrofolate Reductases. Microorganisms. 2021; 9(8):1706. https://doi.org/10.3390/microorganisms9081706
Chicago/Turabian StylePenhallurick, Ryan W., Maya D. Durnal, Alliyah Harold, and Toshiko Ichiye. 2021. "Adaptations for Pressure and Temperature in Dihydrofolate Reductases" Microorganisms 9, no. 8: 1706. https://doi.org/10.3390/microorganisms9081706
APA StylePenhallurick, R. W., Durnal, M. D., Harold, A., & Ichiye, T. (2021). Adaptations for Pressure and Temperature in Dihydrofolate Reductases. Microorganisms, 9(8), 1706. https://doi.org/10.3390/microorganisms9081706