
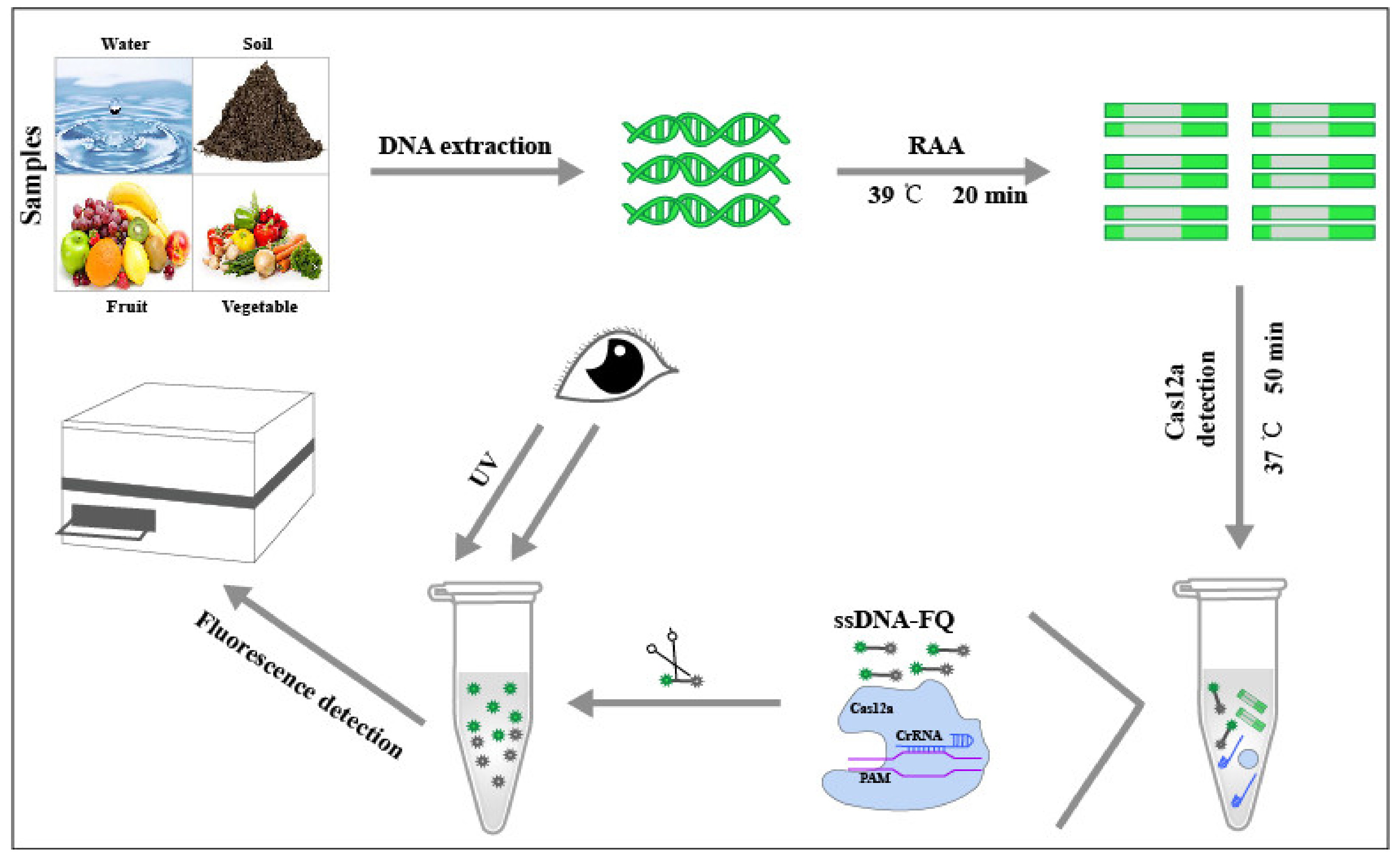
RAA-Cas12a-Tg: A Nucleic Acid Detection System for Toxoplasma gondii Based on CRISPR-Cas12a Combined with Recombinase-Aided Amplification (RAA)

 ,
,  ,
,
Abstract
:1. Introduction
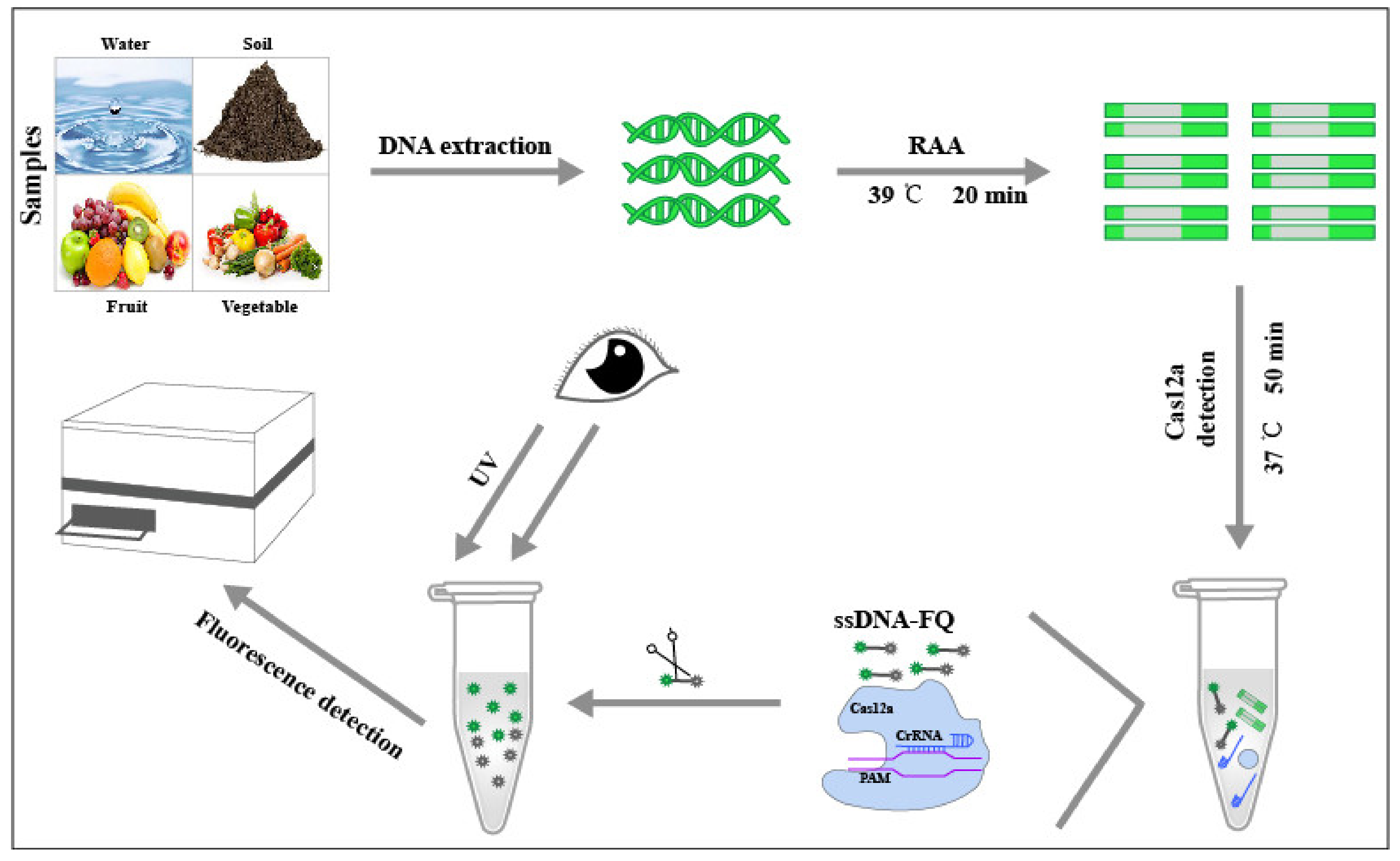
2. Materials and Methods
2.1. Materials
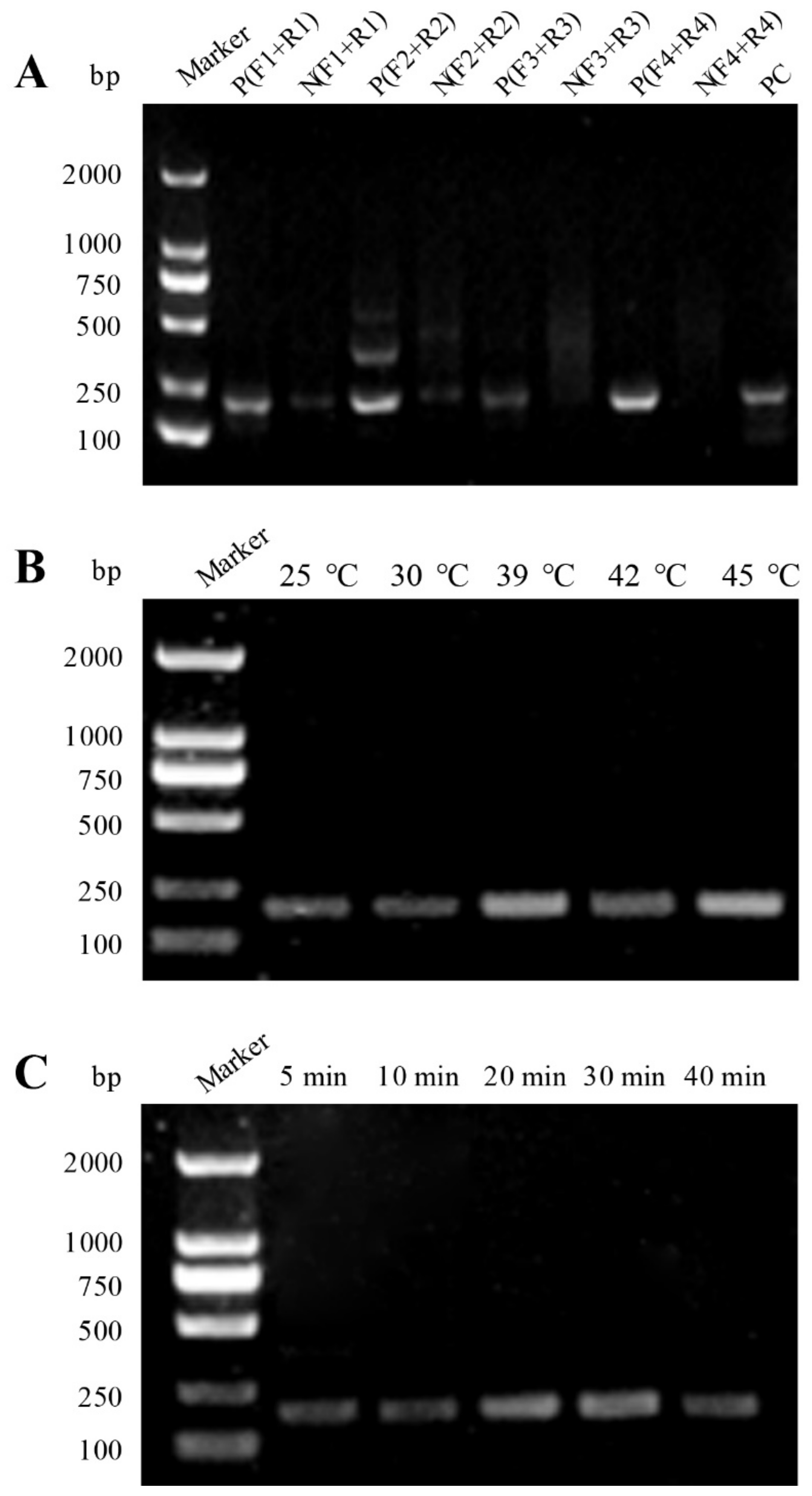
2.2. Establishment of 529 bp RE-Based RAA Assay
2.3. Construction of Positive Recombinant Plasmids pMD18-T-529 bp
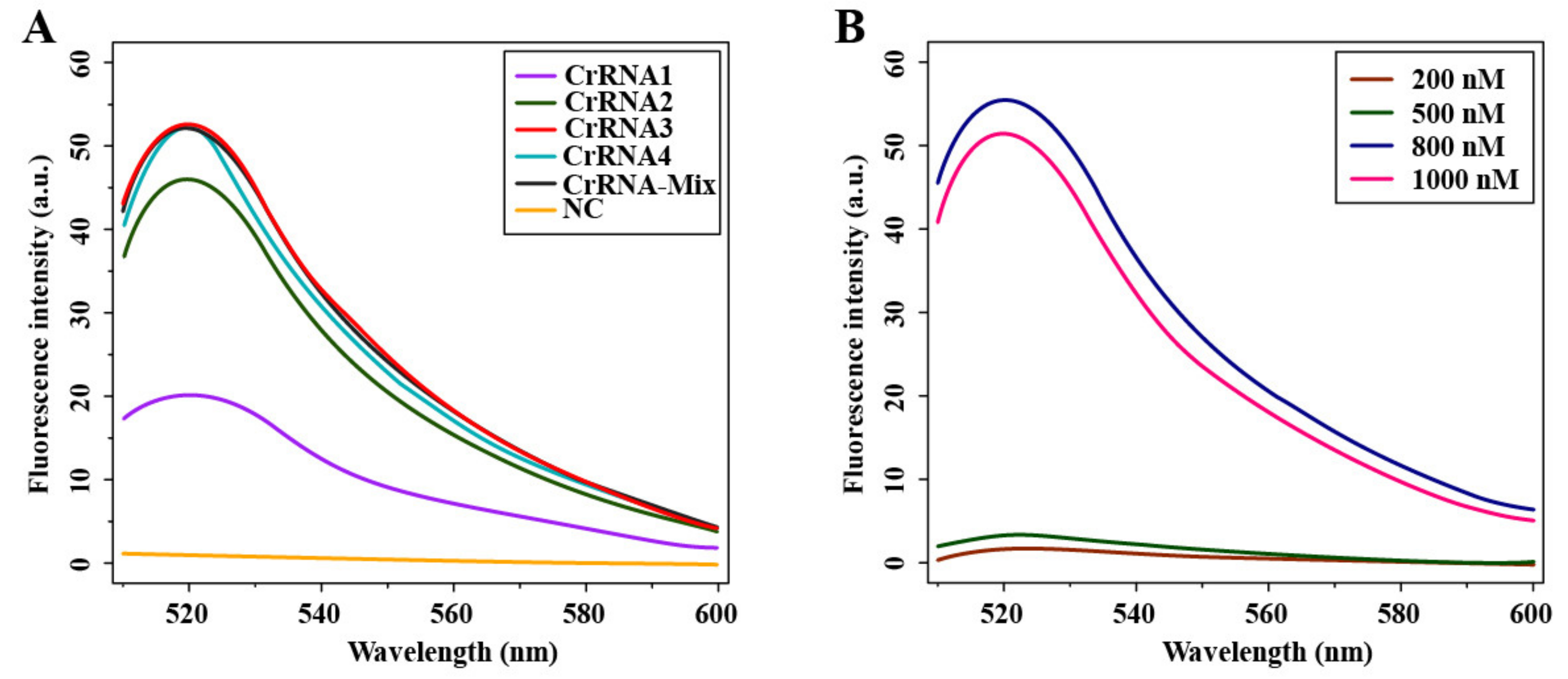
2.4. crRNA/ssDNA-FQ Preparation and Fluorescence Detection of Cas12a
2.5. Analysis of Specificity and Sensitivity of RAA-Cas12a System
2.6. Application of 529 bp RE-Based RAA-Cas12a System Detection to Soil Samples
3. Results
3.1. Optimization of the RAA System
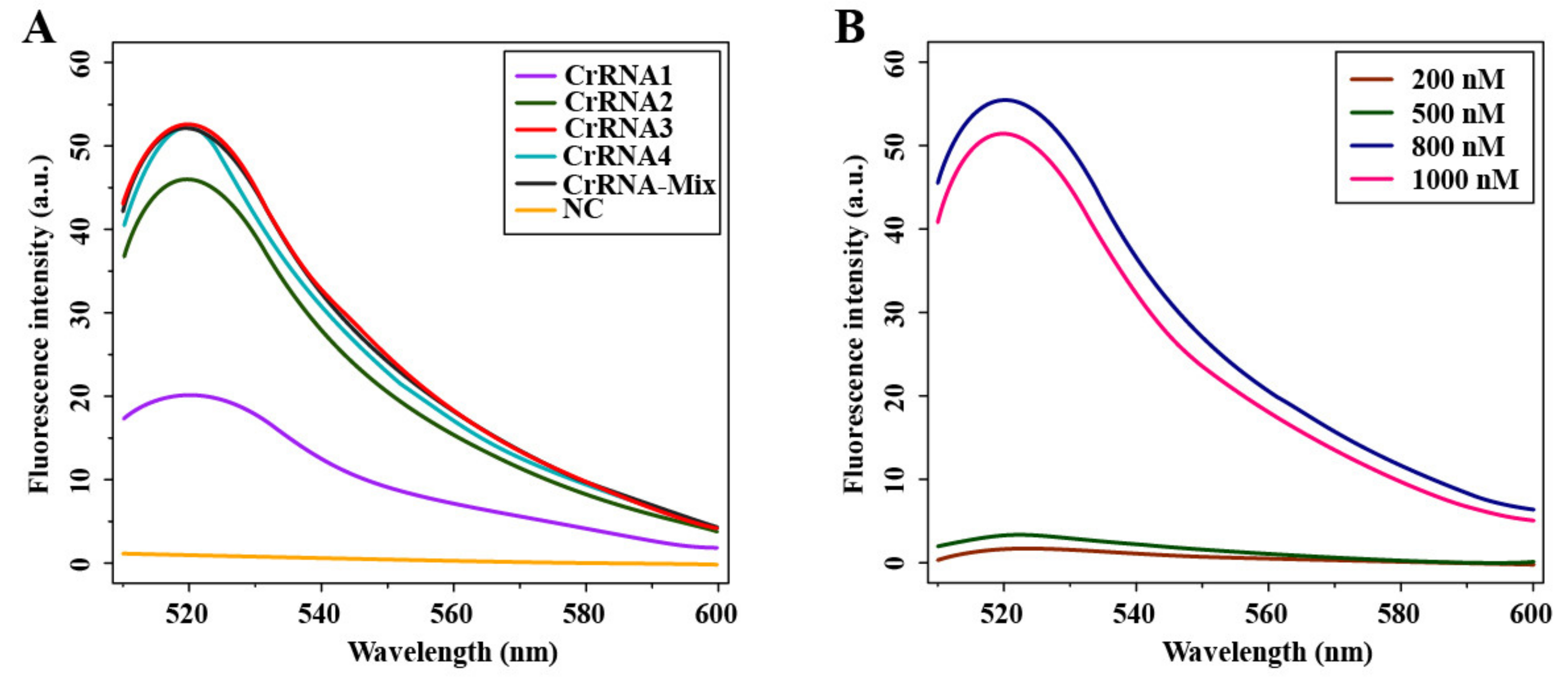
3.2. Optimization of Cas12a-Mediated Fluorescence Detection Assay
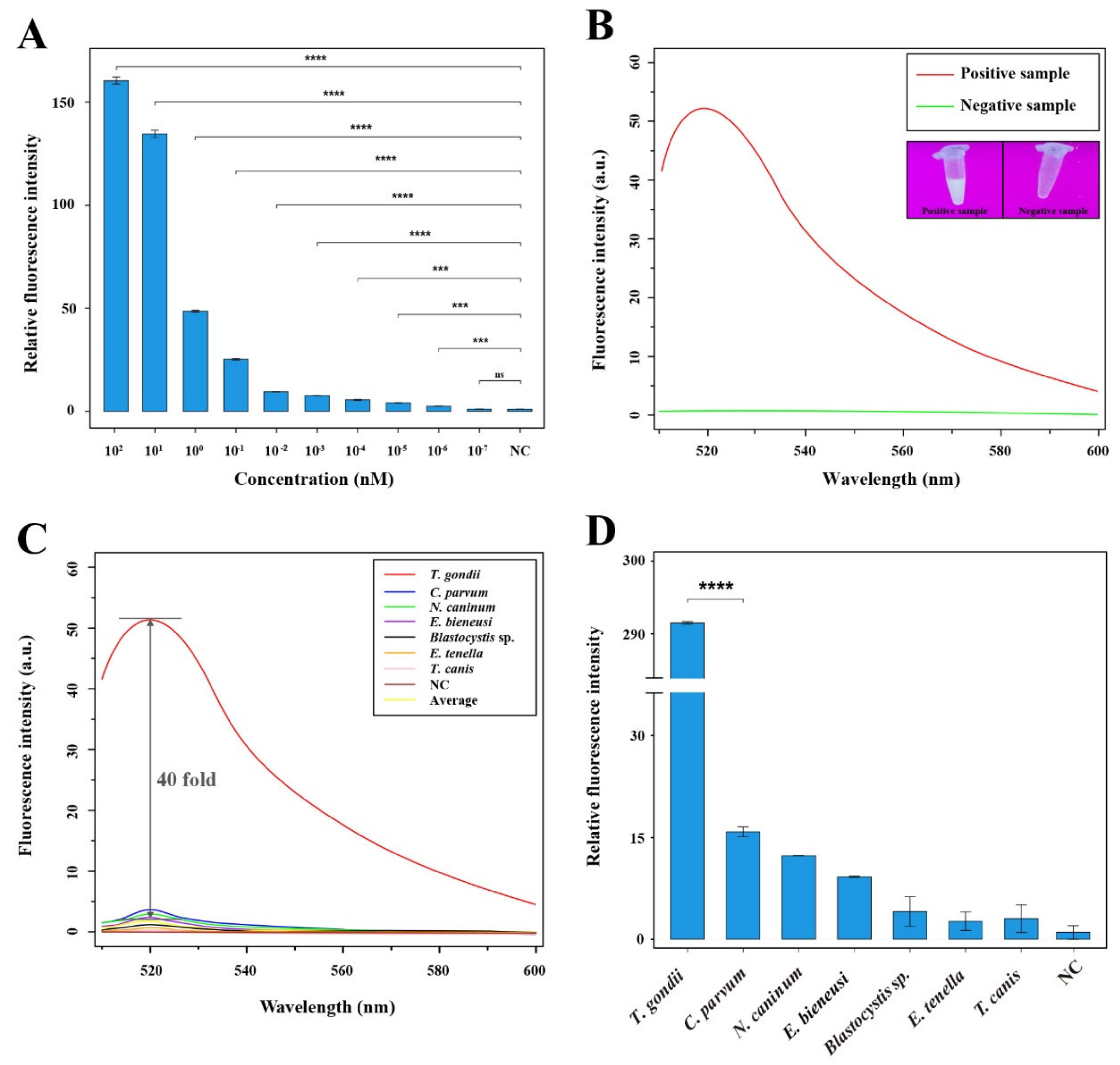
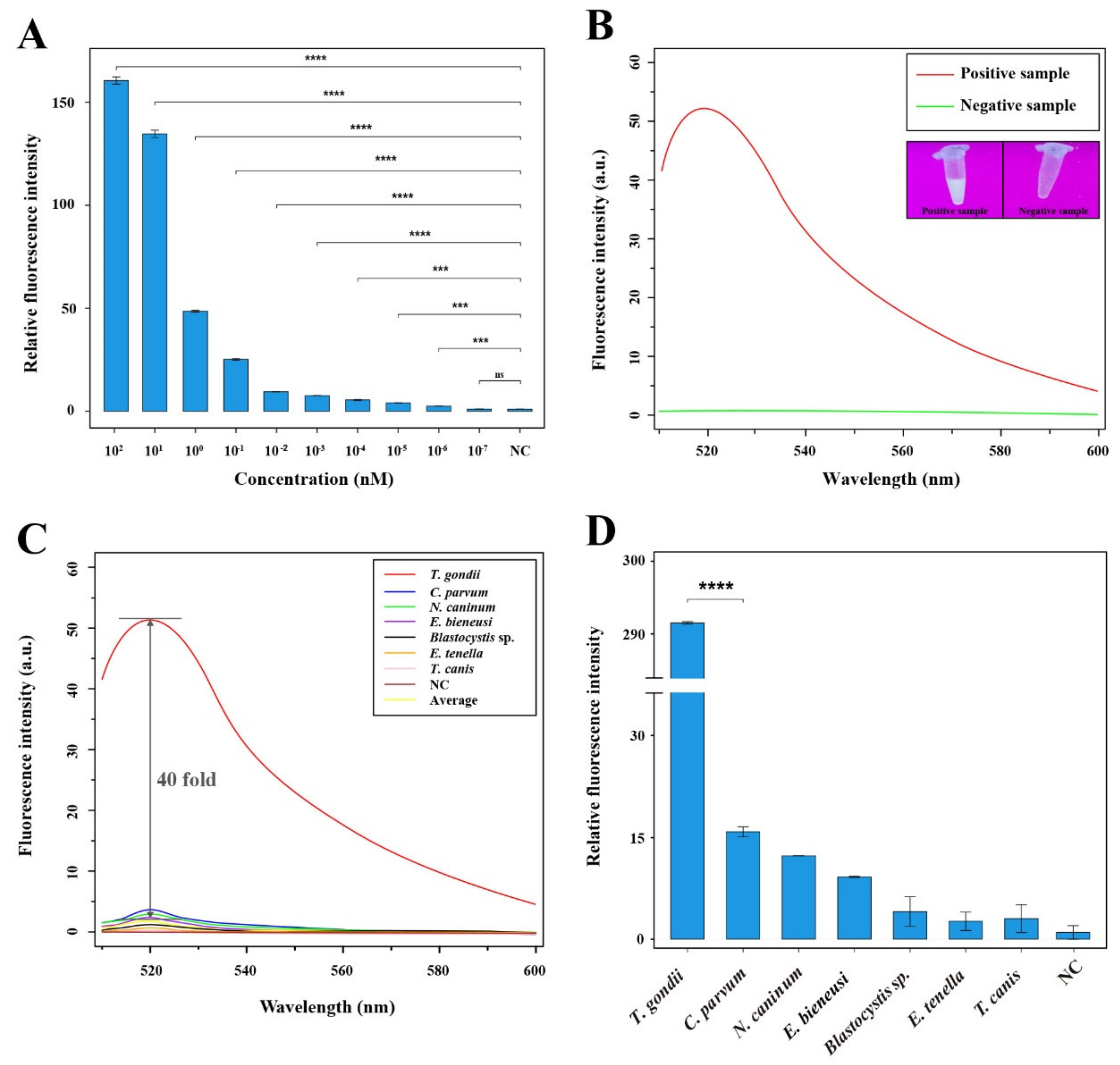
3.3. Evaluation of Sensitivity and Specificity of the RAA-Cas12a-Tg System
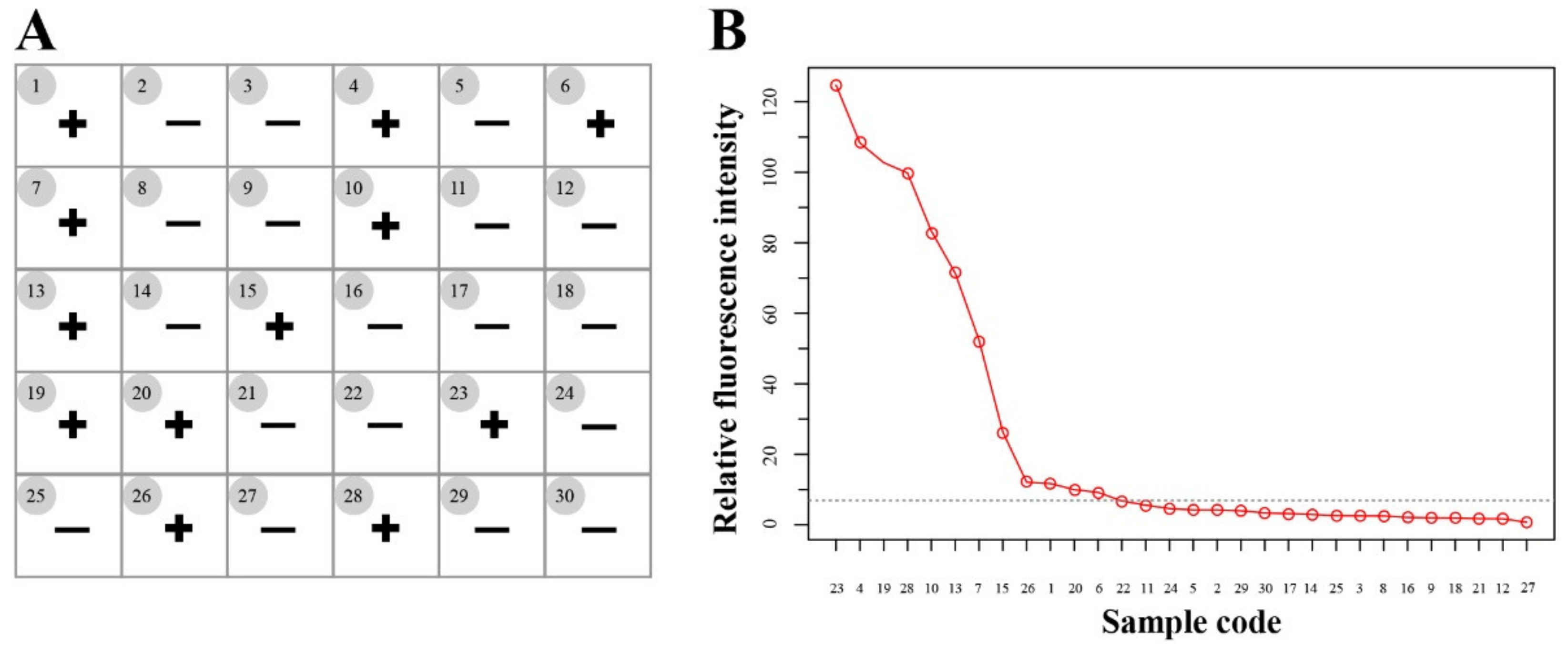
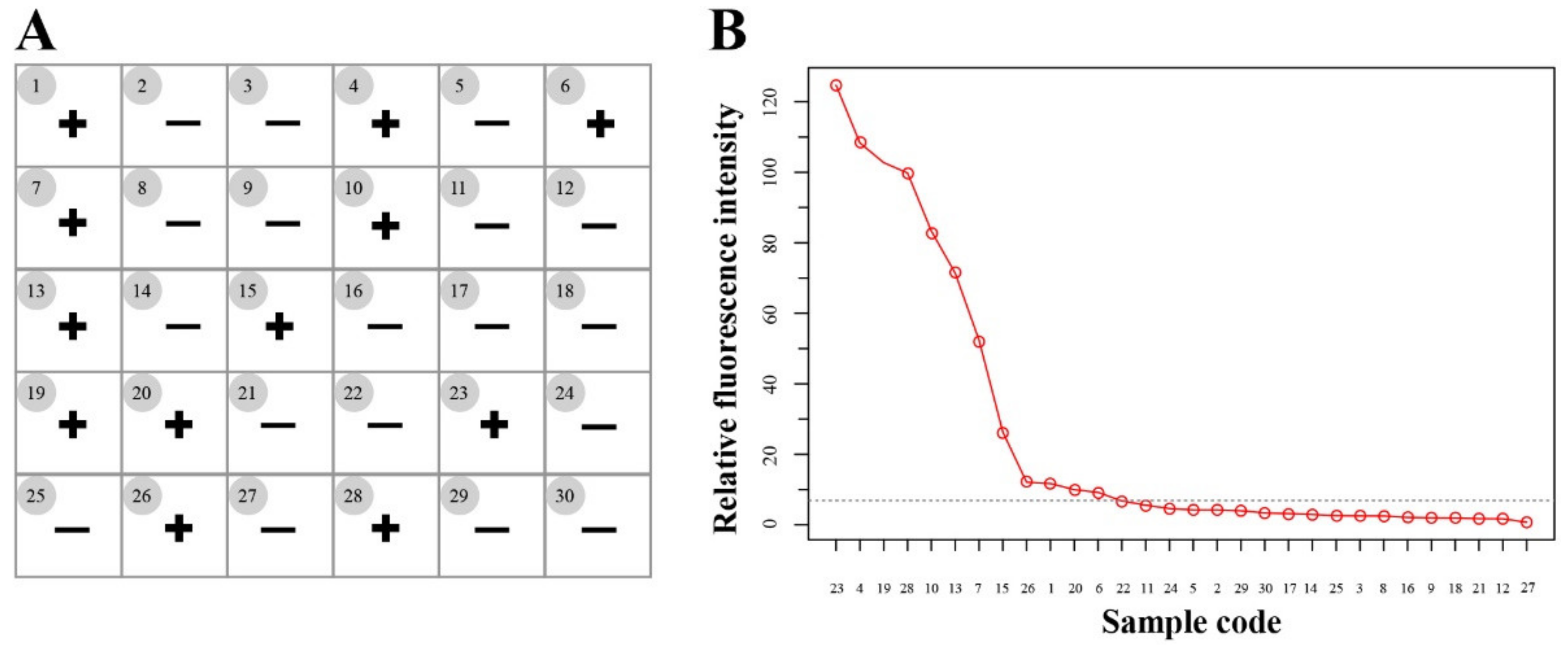
3.4. Application of the 529 bp RE-Based RAA-Cas12a-Tg System for T. gondii Detection in Soil Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dubey, J.P. Toxoplasmosis of Animals and Humans, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 1–313. [Google Scholar]
- Zhou, P.; Chen, Z.; Li, H.-L.; Zheng, H.; He, S.; Lin, R.-Q.; Zhu, X.-Q. Toxoplasma gondii infection in humans in China. Parasit. Vectors 2011, 4, 165. [Google Scholar] [CrossRef] [Green Version]
- Montoya, J.G.; Remington, J.S. Management of Toxoplasma gondii infection during pregnancy. Clin. Infect. Dis. 2008, 47, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Nissapatorn, V. Toxoplasma gondii and HIV: A never-ending story. Lancet HIV 2017, 4, e146–e147. [Google Scholar] [CrossRef]
- Nayeri, T.; Sarvi, S.; Moosazadeh, M.; Daryani, A. Global prevalence of Toxoplasma gondii infection in the aborted fetuses and ruminants that had an abortion: A systematic review and meta-analysis. Vet. Parasitol. 2021, 290, 109370. [Google Scholar] [CrossRef]
- Dawson, D. Foodborne protozoan parasites. Int. J. Food Microbiol. 2005, 103, 207–227. [Google Scholar] [CrossRef]
- Simon, J.; Kurdzielewicz, S.; Jeanniot, E.; Dupuis, E.; Marnef, F.; Aubert, D.; Villena, I.; Poulle, M.-L. Spatial distribution of soil contaminated with Toxoplasma gondii oocysts in relation to the distribution and use of domestic cat defecation sites on dairy farms. Int. J. Parasitol. 2017, 47, 357–367. [Google Scholar] [CrossRef]
- Lass, A.; Ma, L.; Kontogeorgos, I.; Zhang, X.; Li, X.; Karanis, P. First molecular detection of Toxoplasma gondii in vegetable samples in China using qualitative, quantitative real-time PCR and multilocus genotyping. Sci. Rep. 2019, 9, 1–11. [Google Scholar]
- Cong, W.; Zhang, N.Z.; Hu, R.S.; Zou, F.C.; Zou, Y.; Zhong, W.Y.; Wu, J.J.; Fallaize, C.J.; Zhu, X.Q.; Elsheikha, H.M. Prevalence, risk factors and genotype distribution of Toxoplasma gondii DNA in soil in China. Ecotoxicol. Environ. Saf. 2020, 189, 109999. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Karim, R.; Zhang, L. Detection of human intestinal protozoan parasites in vegetables and fruits: A review. Parasit. Vectors 2020, 13, 380. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Sousa, S.; Castro, A.; Da Costa, J.M.C. Detection of Toxoplasma gondii oocysts in fresh vegetables and berry fruits. Parasit. Vectors 2020, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.; Li, M.-Y.; Zou, Y.; Ma, J.-Y.; Wang, B.; Jiang, Z.-Y.; Elsheikha, H.M. Prevalence, genotypes and risk factors for Toxoplasma gondii contamination in marine bivalve shellfish in offshore waters in eastern China. Ecotoxicol. Environ. Saf. 2021, 213, 112048. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Exel, K.E.; Swart, A.; Marinović, A.A.B.; Dam-Deisz, C.; van der Giessen, J.W.; Opsteegh, M. Digging into Toxoplasma gondii infections via soil: A quantitative microbial risk assessment approach. Sci. Total Environ. 2021, 755, 143232. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; La, C.-S.; Dumètre, A.; Robertson, L.J.; Gargala, G.; Escotte-Binet, S.; Favennec, L.; Villena, I.; Gérard, C.; Aubert, D. Assessing viability and infectivity of foodborne and waterborne stages (cysts/oocysts) of Giardia duodenalis, Cryptosporidium spp., and Toxoplasma gondii: A review of methods. Parasite 2018, 25, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Wang, Z.-D.; Huang, S.-Y.; Zhu, X.-Q. Diagnosis of toxoplasmosis and typing of Toxoplasma gondii. Parasit. Vectors 2015, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Elsheikha, H.M.; Marra, C.M.; Zhu, X.Q. Epidemiology, pathophysiology, diagnosis, and management of cerebral toxoplasmosis. Clin. Microbiol. Rev. 2021, 34, e00115–e00119. [Google Scholar]
- Switaj, K.; Master, A.; Skrzypczak, M.; Zaborowski, P. Recent trends in molecular diagnostics for Toxoplasma gondii infections. Clin. Microbiol. Infect. 2005, 11, 170–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homan, W.; Vercammen, M.; De Braekeleer, J.; Verschueren, H. Identification of a 200- to 300-fold repetitive 529 bp DNA fragment in Toxoplasma gondii, and its use for diagnostic and quantitative PCR. Int. J. Parasitol. 2000, 30, 69–75. [Google Scholar] [CrossRef]
- Valian, H.K.; Mirhendi, H.; Mohebali, M.; Shojaee, S.; Fallahi, S.; Jafari, R.; Kheirandish, F.; Mousavi, P. Comparison of the RE-529 sequence and B1 gene for Toxoplasma gondii detection in blood samples of the at-risk seropositive cases using uracil DNA glycosylase supplemented loop-mediated isothermal amplification (UDG-LAMP) assay. Microb. Pathog. 2020, 140, 103938. [Google Scholar] [CrossRef]
- Calderaro, A.; Piccolo, G.; Gorrini, C.; Peruzzi, S.; Zerbini, L.; Bommezzadri, S.; Dettori, G.; Chezzi, C. Comparison between two real-time PCR assays and a nested-PCR for the detection of Toxoplasma gondii. Acta Bio-Medica Atenei Parm. 2006, 77, 75–80. [Google Scholar]
- Jauregui, L.H.; Higgins, J.; Zarlenga, D.; Dubey, J.P.; Lunney, J.K. Development of a real-time PCR assay for detection of Toxoplasma gondii in pig and mouse tissues. J. Clin. Microbiol. 2001, 39, 2065–2071. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Zhang, Y.; Zhang, H.; Zhou, Y.; Cao, J.; Zhou, J. Comparison of loop-mediated isothermal amplification (LAMP) and real-time PCR method targeting a 529-bp repeat element for diagnosis of toxoplasmosis. Vet. Parasitol. 2012, 185, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.; Zhang, D.; Yin, H.; Wang, M. Detection of acute toxoplasmosis in pigs using loop-mediated isothermal amplification and quantitative PCR. Korean J. Parasitol. 2013, 51, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Sashital, D.G. Pathogen detection in the CRISPR–Cas era. Genome Med. 2018, 10, 32. [Google Scholar] [CrossRef]
- Strich, J.R.; Chertow, D.S. CRISPR-Cas biology and its application to infectious diseases. J. Clin. Microbiol. 2019, 57, 01307–01318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Lin, H.; Li, H.; Zhou, Y.; Liu, J.; Zhong, G.; Wu, L.; Jiang, W.; Du, H.; Yang, J.; et al. Cas12a-based on-site and rapid nucleic acid detection of African swine fever. Front. Microbiol. 2019, 10, 2830. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Mei, Y.; Zhao, X.; Jiang, X. Reagents-loaded, automated assay that integrates recombinase-aided amplification and Cas12a nucleic acid detection for a point-of-care test. Anal. Chem. 2020, 92, 14846–14852. [Google Scholar] [CrossRef]
- Li, F.; Ye, Q.; Chen, M.; Zhou, B.; Zhang, J.; Pang, R.; Xue, L.; Wang, J.; Zeng, H.; Wu, S.; et al. An ultrasensitive CRISPR/Cas12a based electrochemical biosensor for Listeria monocytogenes detection. Biosens. Bioelectron. 2021, 179, 113073. [Google Scholar] [CrossRef]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Yu, D.; Bao, M.; Korensky, G.; Chen, J.; Shin, M.; Kim, J.; Park, M.; Qin, P.; Du, K. High-throughput and all-solution phase African swine fever virus (ASFV) detection using CRISPR-Cas12a and fluorescence based point-of-care system. Biosens. Bioelectron. 2020, 154, 112068. [Google Scholar] [CrossRef]
- Xiong, D.; Dai, W.; Gong, J.; Li, G.; Liu, N.; Wu, W.; Pan, J.; Chen, C.; Jiao, Y.; Deng, H.; et al. Rapid detection of SARS-CoV-2 with CRISPR-Cas12a. PLoS. Biol. 2020, 18, e3000978. [Google Scholar] [CrossRef]
- Ai, J.-W.; Zhou, X.; Xu, T.; Yang, M.; Chen, Y.; He, G.; Pan, N.; Cai, Y.; Li, Y.; Wang, X.; et al. CRISPR-based rapid and ultra-sensitive diagnostic test for Mycobacterium tuberculosis. Emerg. Microbes Infect. 2019, 8, 1361–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ke, Y.; Liu, W.; Sun, Y.; Ding, X. A one-pot toolbox based on Cas12a/crRNA enables rapid foodborne pathogen detection at attomolar level. ACS Sens. 2020, 5, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yin, L.; Dong, Y.; Peng, L.; Liu, G.; Man, S.; Ma, L. CRISPR-Cas13a based bacterial detection platform: Sensing pathogen Staphylococcus aureus in food samples. Anal. Chim. Acta 2020, 1127, 225–233. [Google Scholar] [CrossRef]
- Lee, R.A.; Puig, H.; Nguyen, P.Q.; Angenent-Mari, N.M.; Donghia, N.M.; McGee, J.P.; Dvorin, J.D.; Klapperich, C.M.; Pollock, N.R.; Collins, J.J. Ultrasensitive CRISPR-based diagnostic for field-applicable detection of Plasmodium species in symptomatic and asymptomatic malaria. Proc. Natl. Acad. Sci. USA 2020, 117, 25722–25731. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, K.; Wang, Y.; Li, D.; Cui, Z.; Huang, J.; Zhang, S.; Li, X.; Zhang, L. CRISPR/Cas12a-based on-site diagnostics of Cryptosporidium parvum IId-subtype-family from human and cattle fecal samples. Parasit. Vectors 2021, 14, 208. [Google Scholar] [CrossRef] [PubMed]
- Lalitha, S.J.B.S. Primer Premier 5. Biotech Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, R.; Yang, M.; Peng, S.; Cheng, Y.; Chen, C. Conformational dynamics and cleavage sites of Cas12a are modulated by complementarity between crRNA and DNA. iScience 2019, 19, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Villena, I.; Aubert, D.; Gomis, P.; Ferté, H.; Inglard, J.-C.; Denis-Bisiaux, H.; Dondon, J.-M.; Pisano, E.; Ortis, N.; Pinon, J.-M. Evaluation of a strategy for Toxoplasma gondii oocyst detection in water. Appl. Environ. Microbiol. 2004, 70, 4035–4039. [Google Scholar] [CrossRef] [Green Version]
- Swierzy, I.J.; Muhammad, M.; Kroll, J.; Abelmann, A.; Tenter, A.M.; Lüder, C.G. Toxoplasma gondii within skeletal muscle cells: A critical interplay for food-borne parasite transmission. Int. J. Parasitol. 2014, 44, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Harito, J.B.; Campbell, A.T.; Tysnes, K.R.; Dubey, J.P.; Robertson, L.J. Lectin-magnetic separation (LMS) for isolation of Toxoplasma gondii oocysts from concentrated water samples prior to detection by microscopy or qPCR. Water Res. 2017, 114, 228–236. [Google Scholar] [CrossRef]
- Rousseau, A.; Escotte-Binet, S.; La Carbona, S.; Dumètre, A.; Chagneau, S.; Favennec, L.; Kubina, S.; Dubey, J.P.; Majou, D.; Bigot, A.; et al. Toxoplasma gondii oocyst infectivity assessed using a sporocyst-based cell culture assay combined with quantitative PCR for environmental applications. Appl. Environ. Microbiol. 2019, 85, e01189-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, D.; Wang, B.; Huang, H.; Meng, X.; Liu, C.; Xue, L.; Liu, F.; Wang, S. Sunlight based handheld smartphone spectrometer. Biosens. Bioelectron. 2019, 143, 111632. [Google Scholar] [CrossRef]
- Mohanraju, P.; Mougiakos, I.; Albers, J.; Mabuchi, M.; Fuchs, R.T.; Curcuru, J.L.; van Kranenburg, R.; Robb, G.B.; van der Oost, J. Development of a Cas12a-based genome editing tool for moderate thermophiles. CRISPR J. 2021, 4, 82–91. [Google Scholar] [CrossRef]
- Ding, X.; Yin, K.; Li, Z.; Lalla, R.V.; Ballesteros, E.; Sfeir, M.M.; Liu, C. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 2020, 11, 4711. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Xie, S.; Tian, R.; Wang, S.; Lu, Q.; Gao, D.; Lei, C.; Zhu, H.; Nie, Z. A CRISPR-Cas autocatalysis-driven feedback amplification network for supersensitive DNA diagnostics. Sci. Adv. 2021, 7, eabc7802. [Google Scholar] [CrossRef]
- Swarts, D.C. Making the cut(s): How Cas12a cleaves target and non-target DNA. Biochem. Soc. Trans. 2019, 47, 1499–1510. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Primer Name | Sequence (5′→3′) | Product Size (bp) |
|---|---|---|---|
| RAA | 529bp-RAA-F1 | GAAGGGACAGAAGTCGAAGGGGA | 171 |
| 529bp-RAA-R1 | GAAAAGCAGCCAAGCCGGAAACA | ||
| 529bp-RAA-F2 | TGGAGCCACAGAAGGGACAGAAGT | 184 | |
| 529bp-RAA-R2 | CAGGAAAAGCAGCCAAGCCGGAAA | ||
| 529bp-RAA-F3 | CAGAAGGGACAGAAGTCGAAGGGGA | 175 | |
| 529bp-RAA-R3 | AGGAAAAGCAGCCAAGCCGGAAACA | ||
| 529bp-RAA-F4 | GAGCCACAGAAGGGACAGAAGTCG | 186 | |
| 529bp-RAA-R4 | CCTCCAGGAAAAGCAGCCAAGCCG | ||
| Plasmid | 529bp-PF | GGAGGAAGACGAAAGTTG | 515 |
| 529bp-PR | ACAGTGCATCTGGATTCC | ||
| crRNA | crRNA1 | UAAUUUCUACUAAGUGUAGAUACTCGGGCCCAGCTGCGTCT | |
| crRNA2 | UAAUUUCUACUAAGUGUAGAUACAGGCAAGCTCGCCTGTGC | ||
| crRNA3 | UAAUUUCUACUAAGUGUAGAUCACCCUCCAGGAAAAGCAGCCA | ||
| crRNA4 | UAAUUUCUACUAAGUGUAGAUCTCGTGGTGATGGCGGAGAG | ||
| ssDNA-FQ | TgCas12a | 6FAM-CCGGAAAAAAAAAAAACCGG-BHQ1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Q.-N.; Wang, M.; Zheng, L.-B.; Lin, Z.-Q.; Ehsan, M.; Xiao, X.-X.; Zhu, X.-Q. RAA-Cas12a-Tg: A Nucleic Acid Detection System for Toxoplasma gondii Based on CRISPR-Cas12a Combined with Recombinase-Aided Amplification (RAA). Microorganisms 2021, 9, 1644. https://doi.org/10.3390/microorganisms9081644
Ma Q-N, Wang M, Zheng L-B, Lin Z-Q, Ehsan M, Xiao X-X, Zhu X-Q. RAA-Cas12a-Tg: A Nucleic Acid Detection System for Toxoplasma gondii Based on CRISPR-Cas12a Combined with Recombinase-Aided Amplification (RAA). Microorganisms. 2021; 9(8):1644. https://doi.org/10.3390/microorganisms9081644
Chicago/Turabian StyleMa, Qiao-Ni, Meng Wang, Lai-Bao Zheng, Zi-Qin Lin, Muhammad Ehsan, Xing-Xing Xiao, and Xing-Quan Zhu. 2021. "RAA-Cas12a-Tg: A Nucleic Acid Detection System for Toxoplasma gondii Based on CRISPR-Cas12a Combined with Recombinase-Aided Amplification (RAA)" Microorganisms 9, no. 8: 1644. https://doi.org/10.3390/microorganisms9081644
APA StyleMa, Q.-N., Wang, M., Zheng, L.-B., Lin, Z.-Q., Ehsan, M., Xiao, X.-X., & Zhu, X.-Q. (2021). RAA-Cas12a-Tg: A Nucleic Acid Detection System for Toxoplasma gondii Based on CRISPR-Cas12a Combined with Recombinase-Aided Amplification (RAA). Microorganisms, 9(8), 1644. https://doi.org/10.3390/microorganisms9081644