
Culture-Independent and Culture-Dependent Characterization of the Black Soldier Fly Gut Microbiome Reveals a Large Proportion of Culturable Bacteria with Potential for Industrial Applications

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Black Soldier Fly Breeding
2.2. Analysis of the BSFL Microbial Community by Amplicon Sequencing
2.3. Cultivation and Isolation of Gut Bacteria
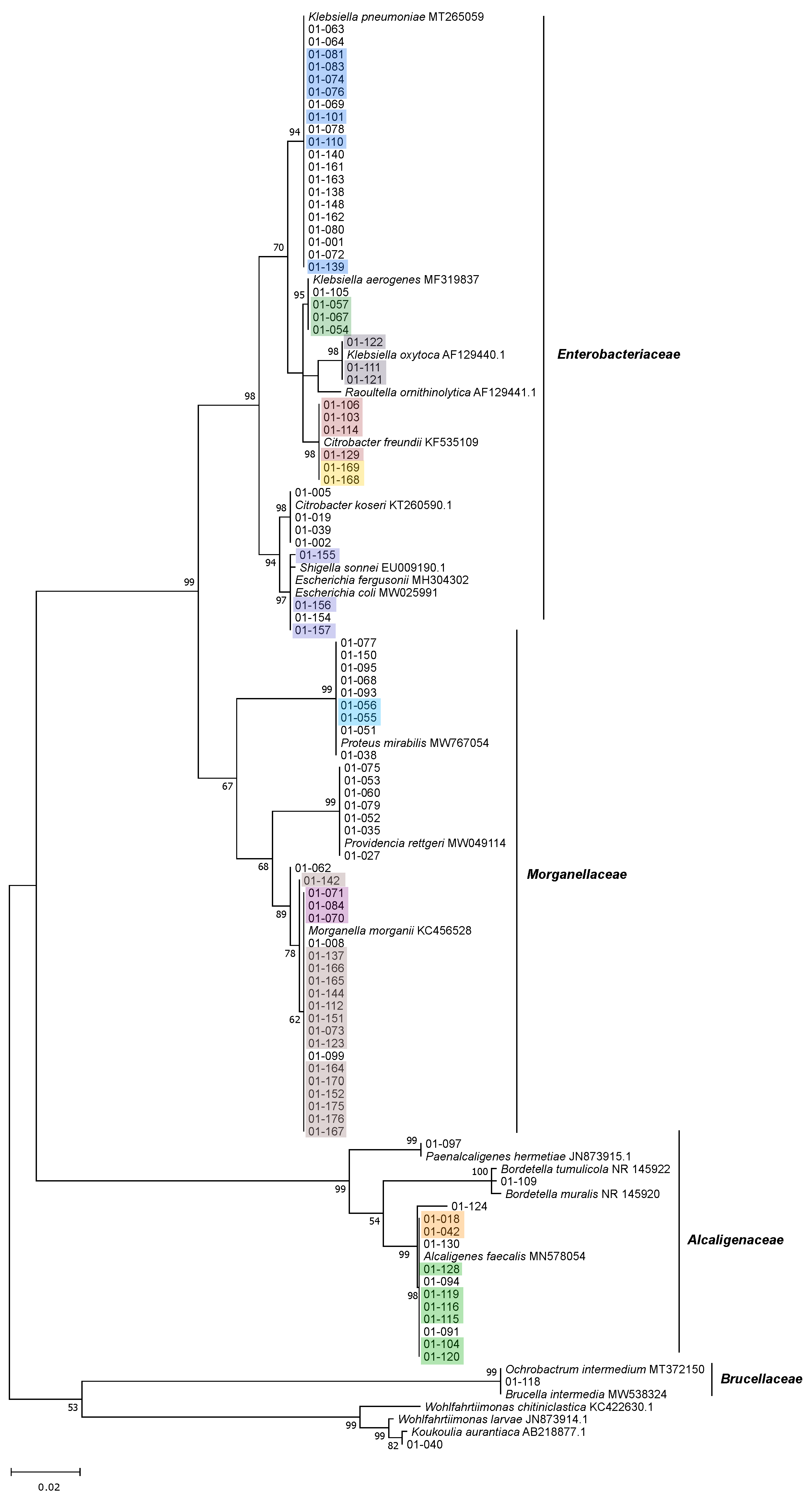
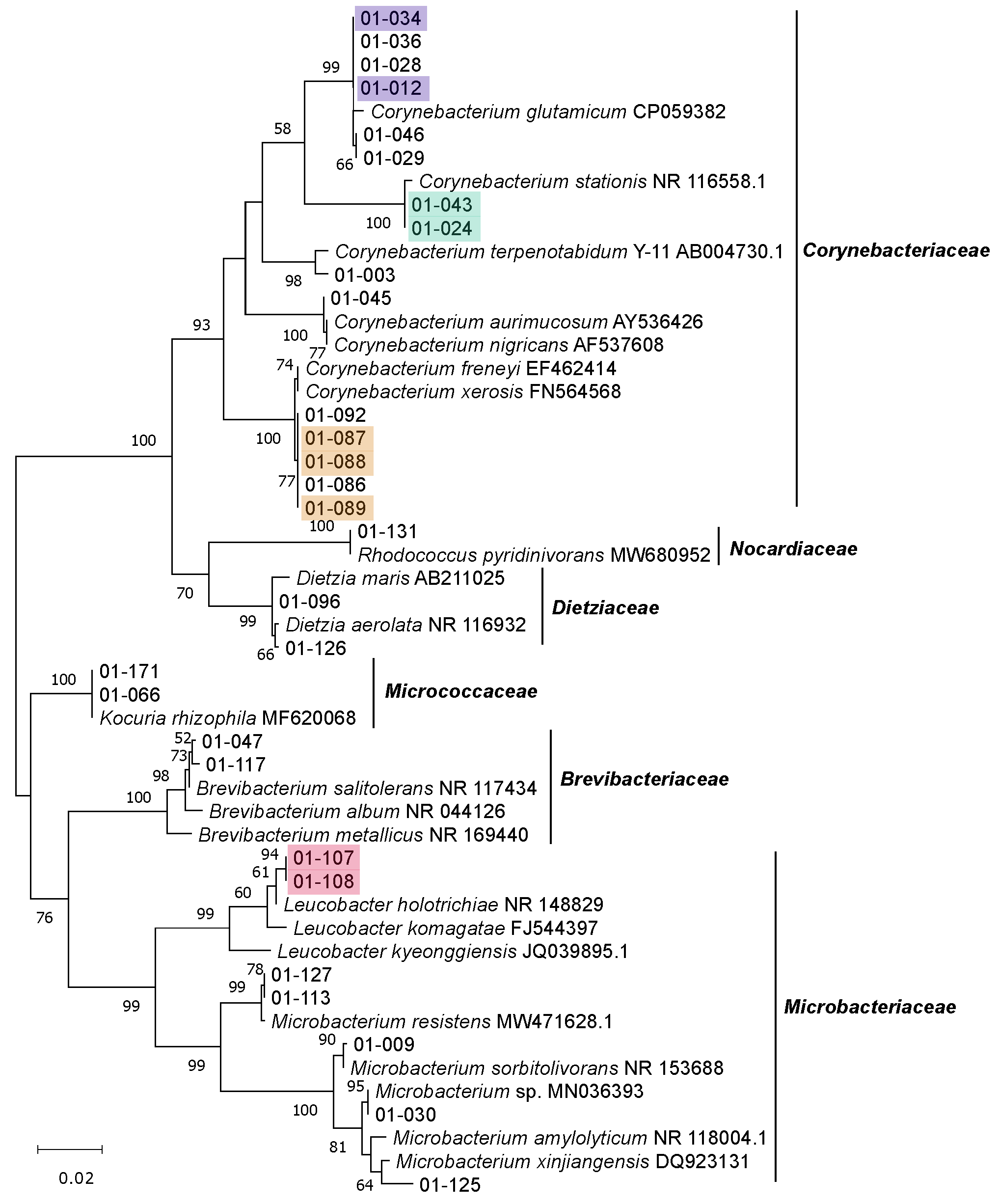
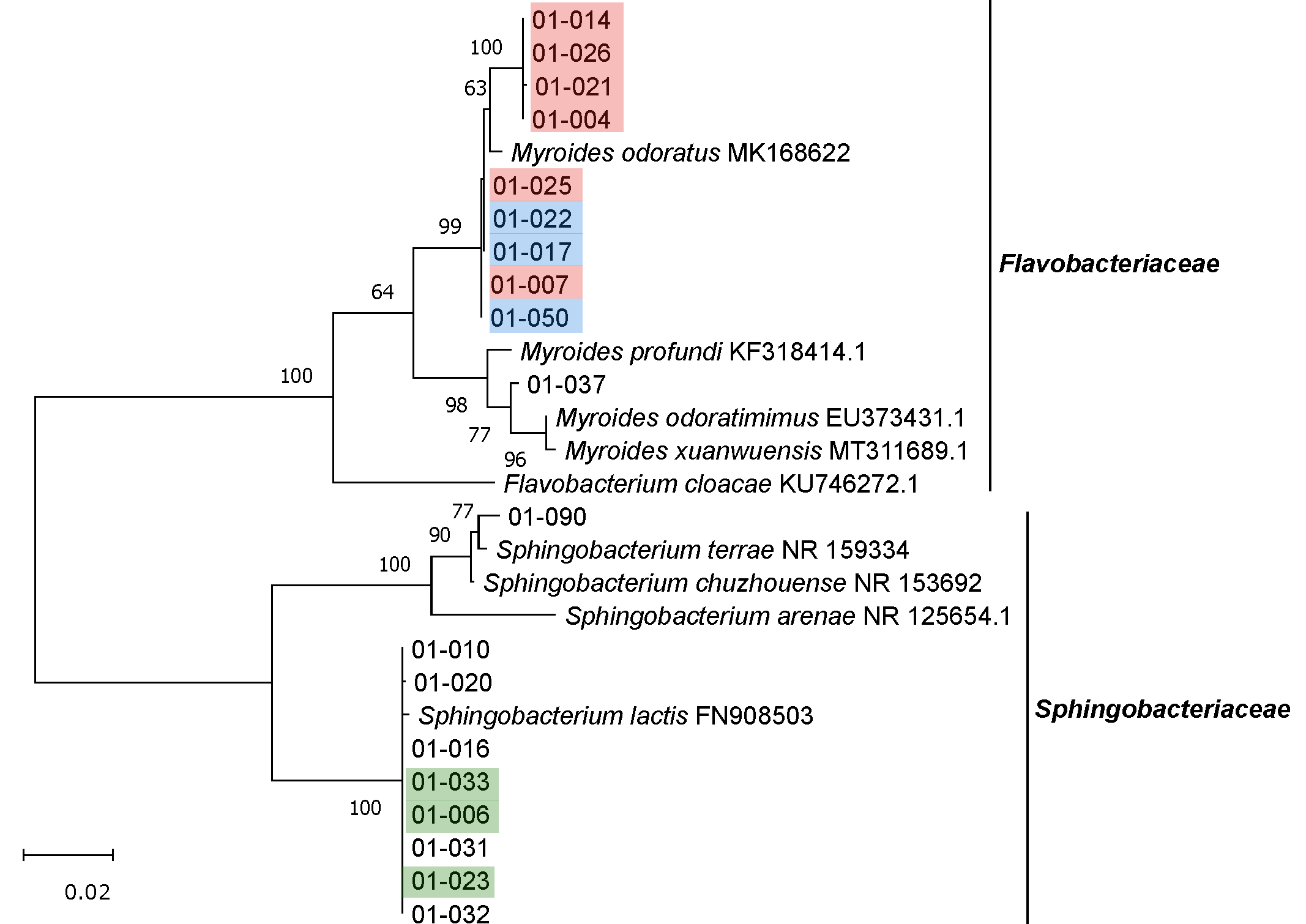
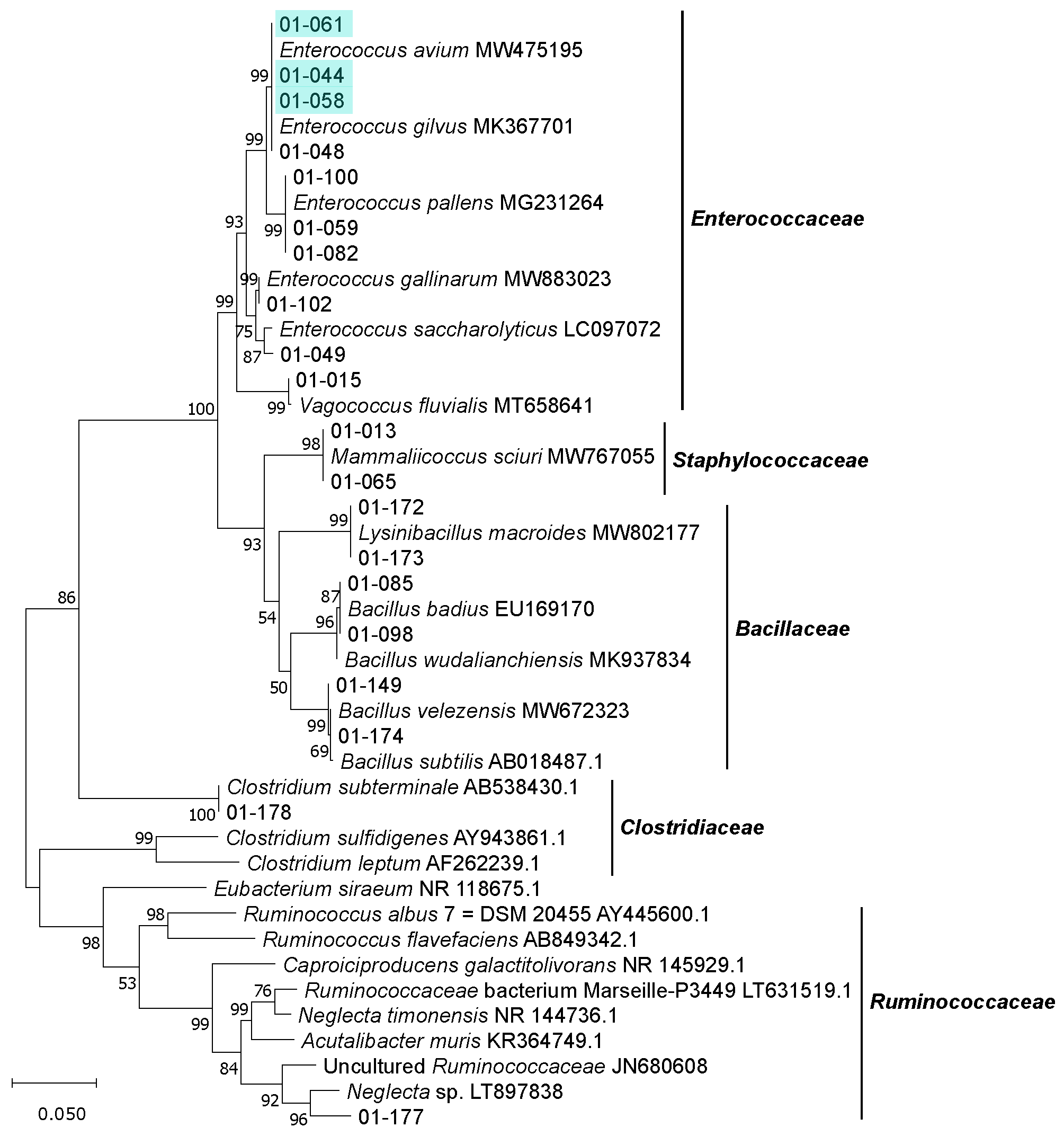
2.4. Phylogenetic Classification of Isolates and Comparison with Amplicon Sequences
2.5. Genomic Fingerprinting
2.6. Screening for Antimicrobial Activity
3. Results
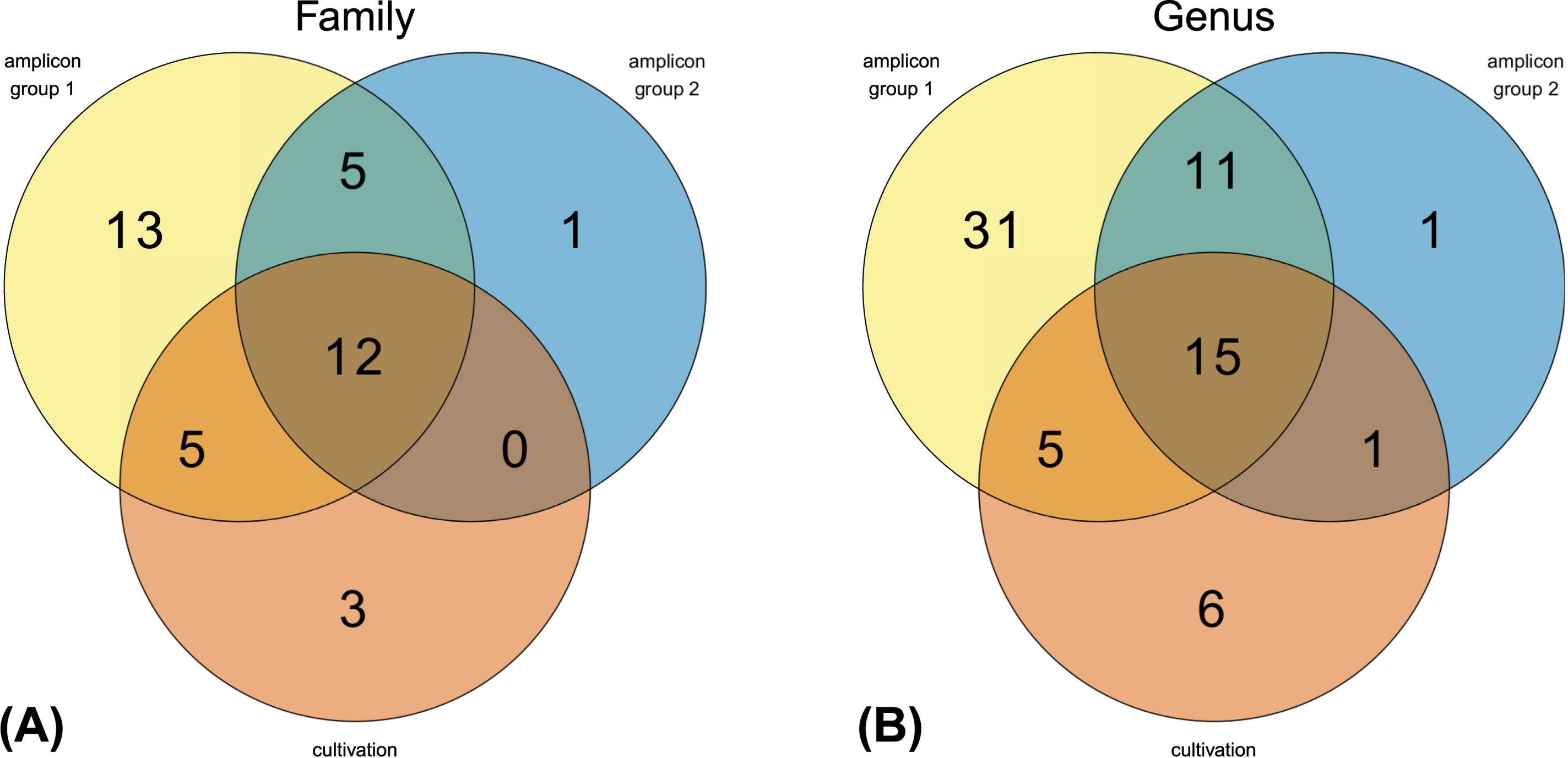
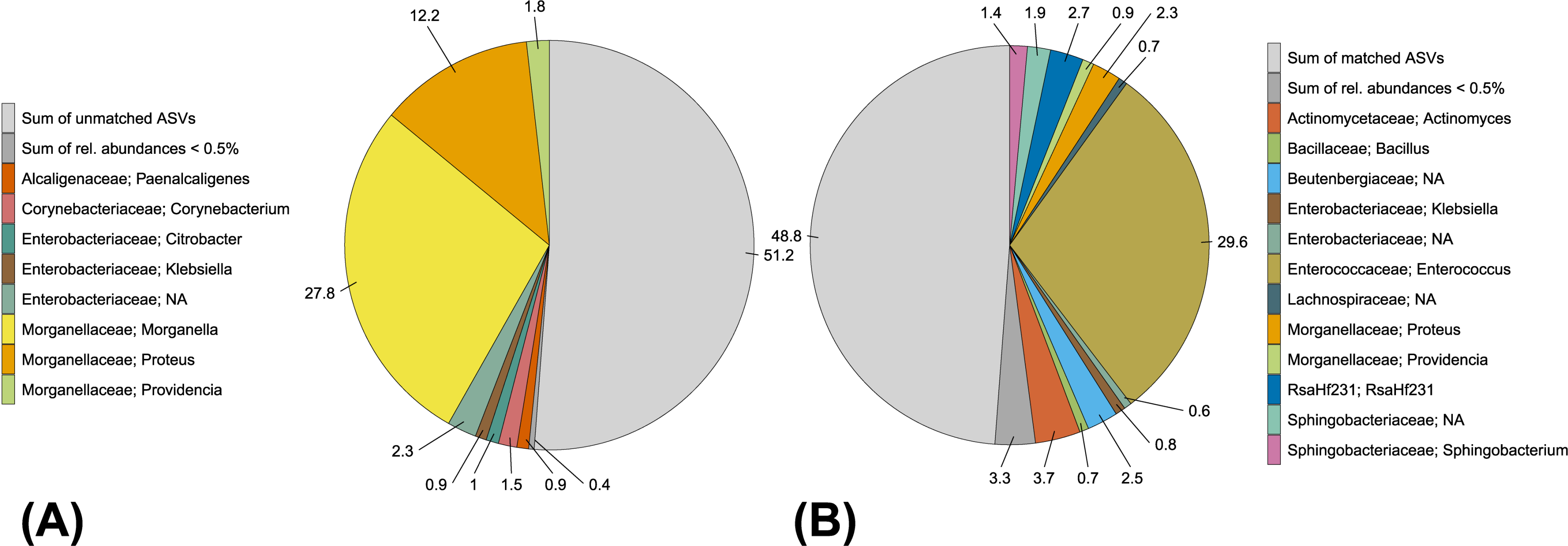
3.1. Analysis of the BSF Microbial Community by Amplicon Sequencing
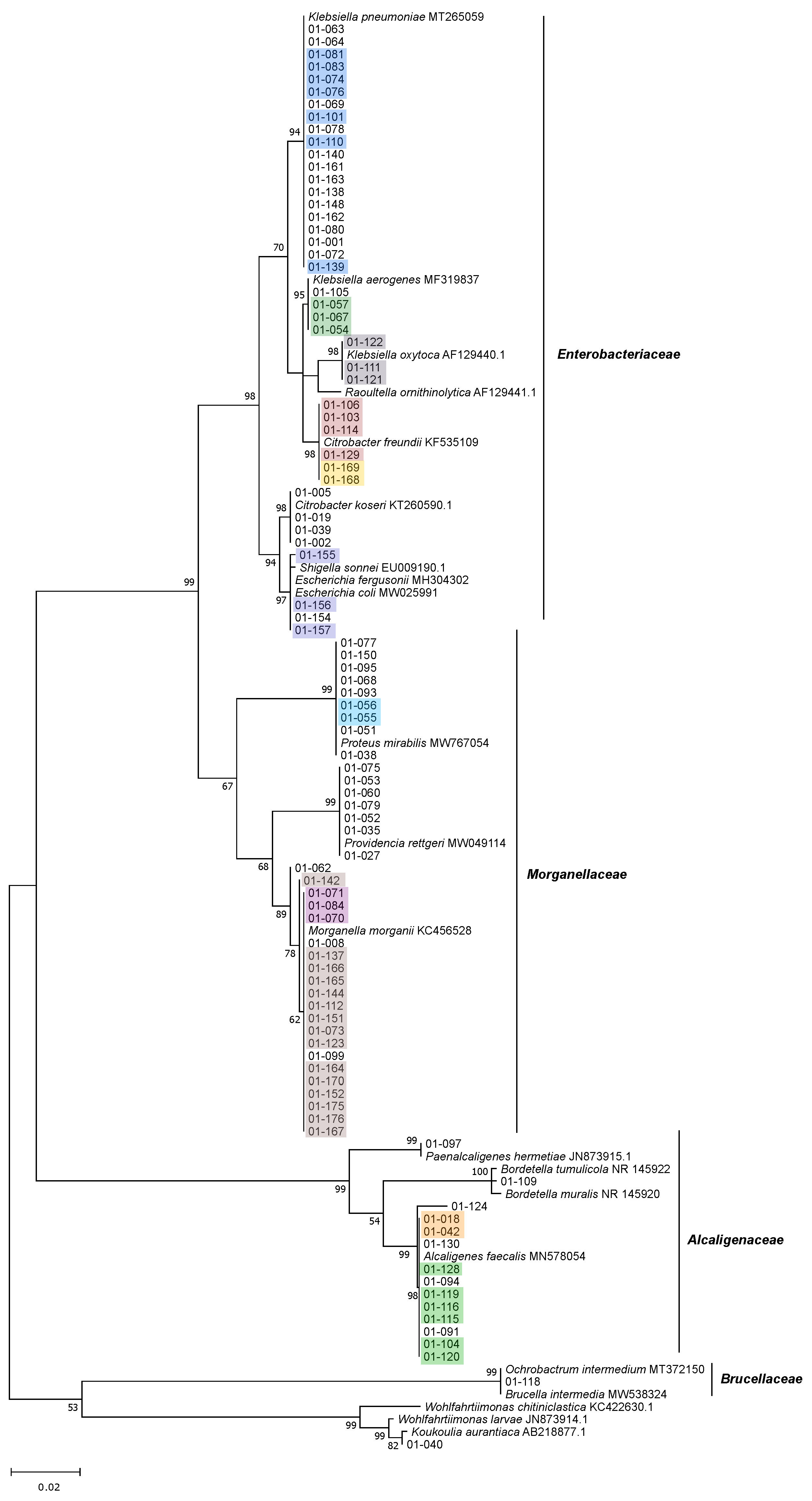
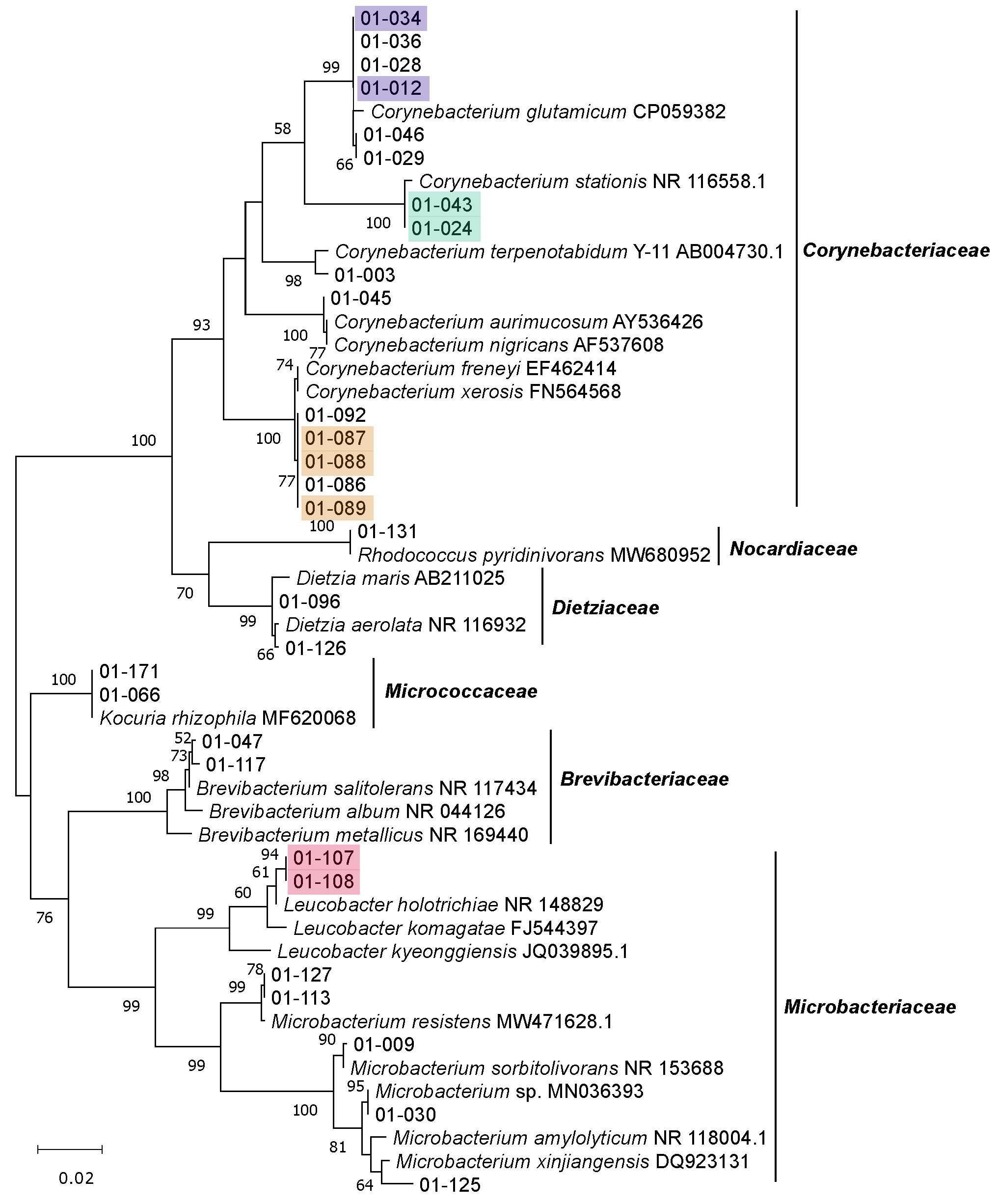
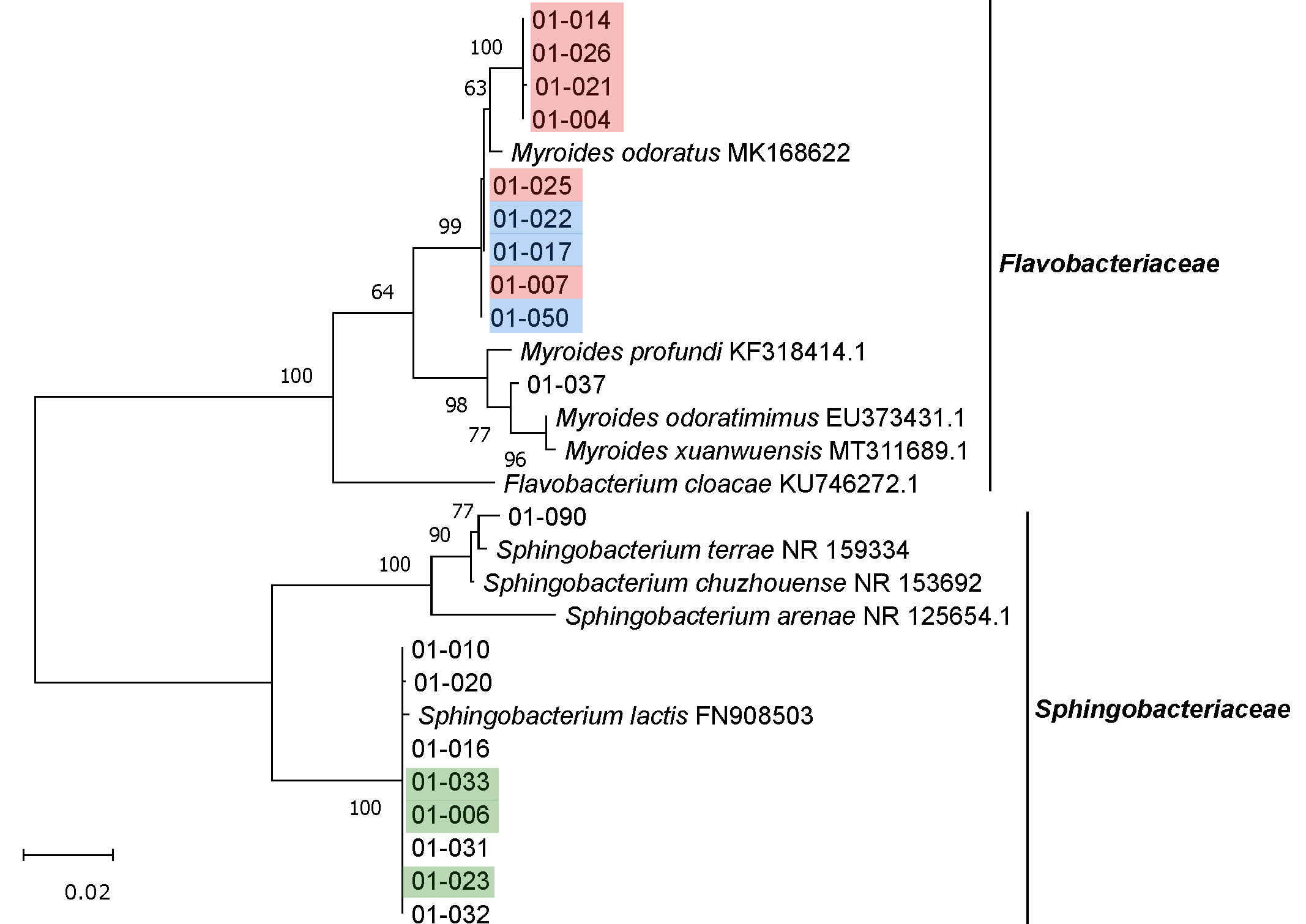
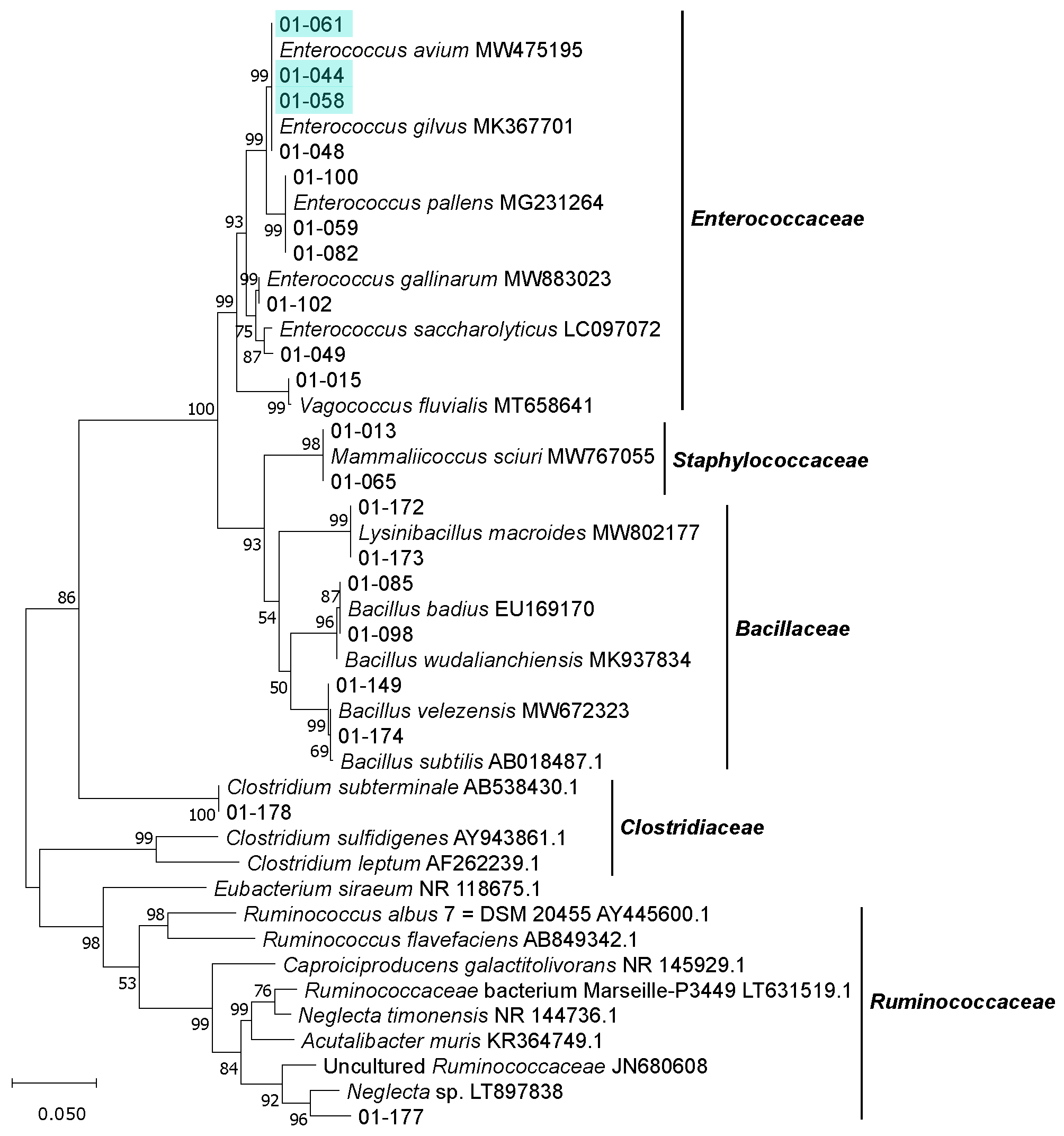
3.2. Isolation, Identification and Phylogenetic Analysis of Bacterial Isolates from BSFL Guts
3.3. The Culturable BSFL Gut Microbial Community
3.4. Genomic Fingerprinting of Bacterial Isolates from the Cultivation Approach
3.5. Screening for Antimicrobial Activity among the Isolates from the Cultivation Approach
4. Discussion
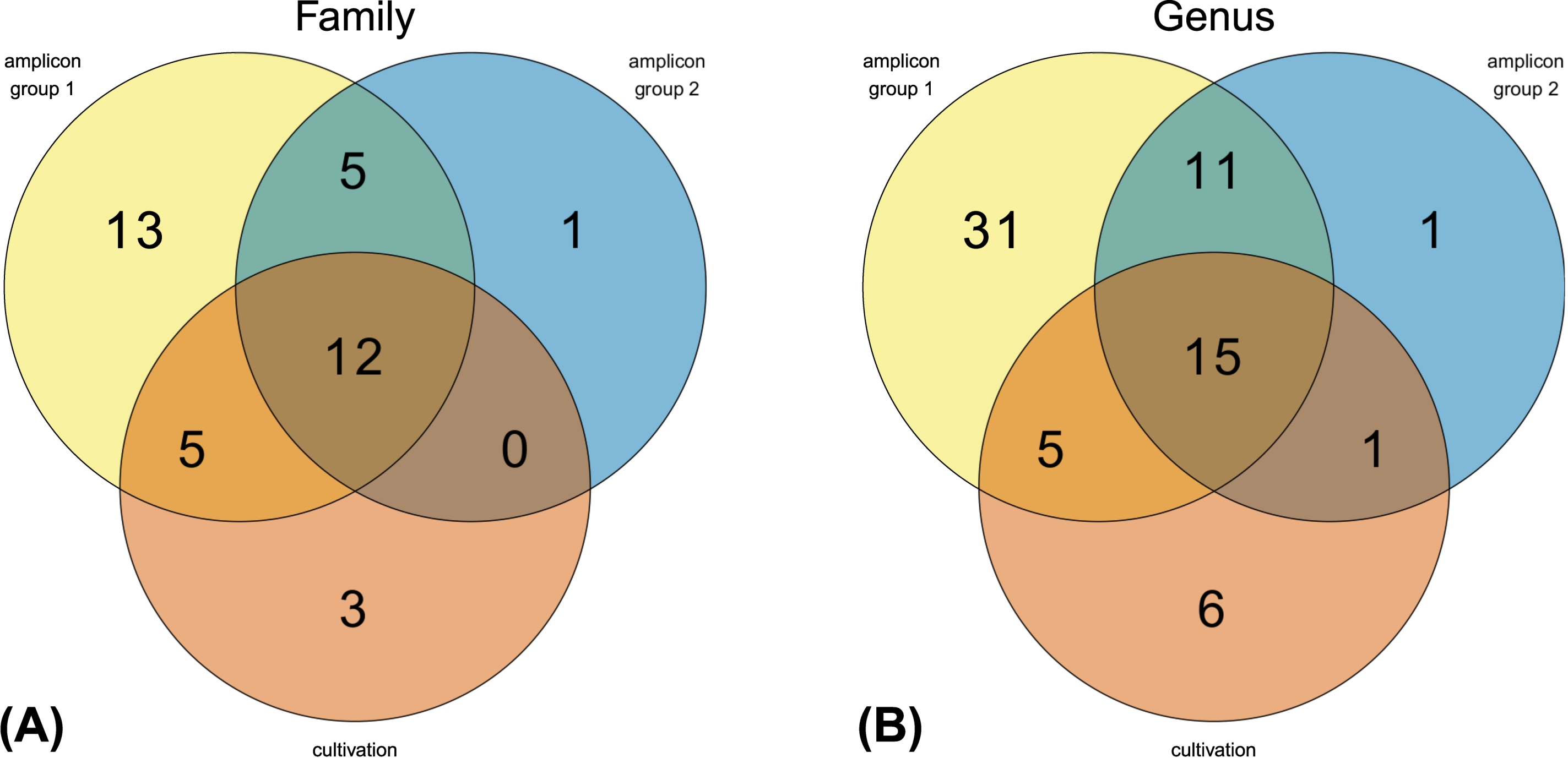
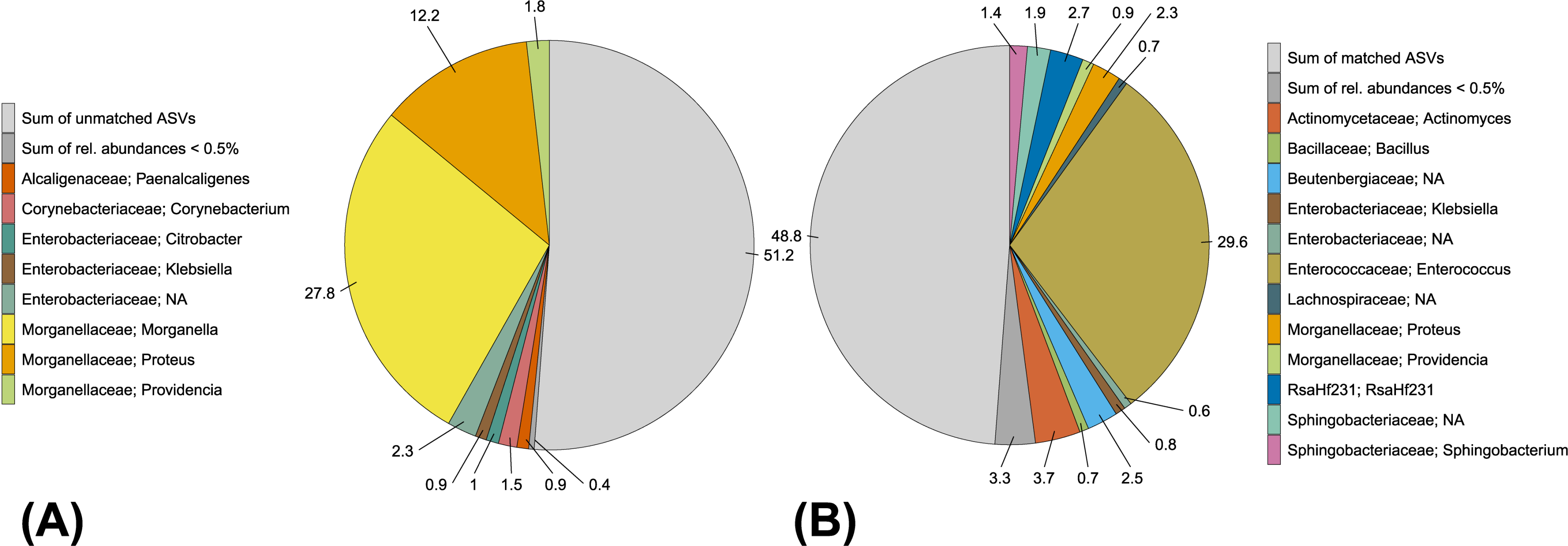
4.1. Amplicon Sequencing Versus Cultivation-Dependent Microbiome Analysis
4.2. Antimicrobial Activity of the Gut Microbiome
4.3. Possible Applications of BSFL Gut Bacterial Isolates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Huis, A. Potential of insects as food and feed in assuring food security. Annu. Rev. Entomol. 2013, 58, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Wolf, D.; Gutzeit, H.O. The black soldier fly, Hermetia illucens-a promising source for sustainable production of proteins, lipids and bioactive substances. Z. Naturforsch. C J. Biosci. 2017, 72, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Lalander, C.; Diener, S.; Zurbrügg, C.; Vinnerås, B. Effects of feedstock on larval development and process efficiency in waste treatment with black soldier fly (Hermetia illucens). J. Clean. Prod. 2019, 208, 211–219. [Google Scholar] [CrossRef]
- Bosch, G.; van der Fels-Klerx, H.J.; de Rijk, T.C.; Oonincx, D.G.A.B. Aflatoxin B1 Tolerance and Accumulation in Black Soldier Fly Larvae (Hermetia illucens) and Yellow Mealworms (Tenebrio molitor). Toxins 2017, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Wang, X.; Yan, Z.; Xu, X.; Xie, S.; Liang, J. Transformation of pig manure by passage through the gut of black soldier fly larvae (Hermetia illucens): Metal speciation, potential pathogens and metal-related functional profiling. Ecotoxicol. Environ. Saf. 2021, 211, 111925. [Google Scholar] [CrossRef]
- Tegtmeier, D.; Hurka, S.; Klüber, P.; Brinkrolf, K.; Heise, P.; Vilcinskas, A. Cottonseed Press Cake as a Potential Diet for Industrially Farmed Black Soldier Fly Larvae Triggers Adaptations of Their Bacterial and Fungal Gut Microbiota. Front. Microbiol. 2021, 12, 634503. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.C.; Islam, M.; Sheppard, C.; Liao, J.; Doyle, M.P. Reduction of Escherichia coli O157:H7 and Salmonella enterica serovar Enteritidis in chicken manure by larvae of the black soldier fly. J. Food Prot. 2004, 67, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Tomberlin, J.K.; Brady, J.A.; Sanford, M.R.; Yu, Z. Black soldier fly (Diptera: Stratiomyidae) larvae reduce Escherichia coli in dairy manure. Environ. Entomol. 2008, 37, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Lalander, C.H.; Fidjeland, J.; Diener, S.; Eriksson, S.; Vinnerås, B. High waste-to-biomass conversion and efficient Salmonella spp. reduction using black soldier fly for waste recycling. Agron. Sustain. Dev. 2015, 35, 261–271. [Google Scholar] [CrossRef]
- Jeon, H.; Park, S.; Choi, J.; Jeong, G.; Lee, S.-B.; Choi, Y.; Lee, S.-J. The intestinal bacterial community in the food waste-reducing larvae of Hermetia illucens. Curr. Microbiol. 2011, 62, 1390–1399. [Google Scholar] [CrossRef]
- Bruno, D.; Bonelli, M.; de Filippis, F.; Di Lelio, I.; Tettamanti, G.; Casartelli, M.; Ercolini, D.; Caccia, S. The Intestinal Microbiota of Hermetia illucens Larvae Is Affected by Diet and Shows a Diverse Composition in the Different Midgut Regions. Appl. Environ. Microbiol. 2019, 85, e01864-18. [Google Scholar] [CrossRef] [Green Version]
- Klammsteiner, T.; Walter, A.; Bogataj, T.; Heussler, C.D.; Stres, B.; Steiner, F.M.; Schlick-Steiner, B.C.; Arthofer, W.; Insam, H. The Core Gut Microbiome of Black Soldier Fly (Hermetia illucens) Larvae Raised on Low-Bioburden Diets. Front. Microbiol. 2020, 11, 993. [Google Scholar] [CrossRef] [PubMed]
- Wynants, E.; Frooninckx, L.; Crauwels, S.; Verreth, C.; de Smet, J.; Sandrock, C.; Wohlfahrt, J.; van Schelt, J.; Depraetere, S.; Lievens, B.; et al. Assessing the Microbiota of Black Soldier Fly Larvae (Hermetia illucens) Reared on Organic Waste Streams on Four Different Locations at Laboratory and Large Scale. Microb. Ecol. 2019, 77, 913–930. [Google Scholar] [CrossRef]
- Shelomi, M.; Wu, M.-K.; Chen, S.-M.; Huang, J.-J.; Burke, C.G. Microbes Associated With Black Soldier Fly (Diptera: Stratiomiidae) Degradation of Food Waste. Environ. Entomol. 2020, 49, 405–411. [Google Scholar] [CrossRef]
- Zheng, L.; Crippen, T.L.; Singh, B.; Tarone, A.M.; Dowd, S.; Yu, Z.; Wood, T.K.; Tomberlin, J.K. A survey of bacterial diversity from successive life stages of black soldier fly (Diptera: Stratiomyidae) by using 16S rDNA pyrosequencing. J. Med. Entomol. 2013, 50, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, Y.; Glaeser, S.P.; Mvie, J.; Bartz, J.-O.; Müller, A.; Gutzeit, H.O.; Vilcinskas, A.; Kämpfer, P. The gut and feed residue microbiota changing during the rearing of Hermetia illucens larvae. Antonie Van Leeuwenhoek 2020, 113, 1323–1344. [Google Scholar] [CrossRef] [PubMed]
- Callegari, M.; Jucker, C.; Fusi, M.; Leonardi, M.G.; Daffonchio, D.; Borin, S.; Savoldelli, S.; Crotti, E. Hydrolytic Profile of the Culturable Gut Bacterial Community Associated With Hermetia illucens. Front. Microbiol. 2020, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Gorrens, E.; van Moll, L.; Frooninckx, L.; de Smet, J.; van Campenhout, L. Isolation and Identification of Dominant Bacteria From Black Soldier Fly Larvae (Hermetia illucens) Envisaging Practical Applications. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Tegtmeier, D.; Thompson, C.L.; Schauer, C.; Brune, A. Oxygen Affects Gut Bacterial Colonization and Metabolic Activities in a Gnotobiotic Cockroach Model. Appl. Environ. Microbiol. 2016, 82, 1080–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, R.J.; Dillon, V.M. The gut bacteria of insects: Nonpathogenic interactions. Annu. Rev. Entomol. 2004, 49, 71–92. [Google Scholar] [CrossRef]
- Kundu, P.; Manna, B.; Majumder, S.; Ghosh, A. Species-wide Metabolic Interaction Network for Understanding Natural Lignocellulose Digestion in Termite Gut Microbiota. Sci. Rep. 2019, 9, 16329. [Google Scholar] [CrossRef] [Green Version]
- Egert, M.; Wagner, B.; Lemke, T.; Brune, A.; Friedrich, M.W. Microbial community structure in midgut and hindgut of the humus-feeding larva of Pachnoda ephippiata (Coleoptera: Scarabaeidae). Appl. Environ. Microbiol. 2003, 69, 6659–6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, T.; Dietrich, C.; Scheffrahn, R.H.; Brune, A. High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl. Environ. Microbiol. 2012, 78, 4691–4701. [Google Scholar] [CrossRef] [Green Version]
- Schauer, C.; Thompson, C.L.; Brune, A. The bacterial community in the gut of the Cockroach Shelfordella lateralis reflects the close evolutionary relatedness of cockroaches and termites. Appl. Environ. Microbiol. 2012, 78, 2758–2767. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.-H.; Roh, S.W.; Whon, T.W.; Jung, M.-J.; Kim, M.-S.; Park, D.-S.; Yoon, C.; Nam, Y.-D.; Kim, Y.-J.; Choi, J.-H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Bauer, E.; Lampert, N.; Mikaelyan, A.; Köhler, T.; Maekawa, K.; Brune, A. Physicochemical conditions, metabolites and community structure of the bacterial microbiota in the gut of wood-feeding cockroaches (Blaberidae: Panesthiinae). FEMS Microbiol. Ecol. 2015, 91, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brune, A.; Dietrich, C. The Gut Microbiota of Termites: Digesting the Diversity in the Light of Ecology and Evolution. Annu. Rev. Microbiol. 2015, 69, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.; Charnley, K. Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res. Microbiol. 2002, 153, 503–509. [Google Scholar] [CrossRef]
- Dillon, R.J.; Vennard, C.T.; Buckling, A.; Charnley, A.K. Diversity of locust gut bacteria protects against pathogen invasion. Ecol. Lett. 2005, 8, 1291–1298. [Google Scholar] [CrossRef]
- Vogel, H.; Shukla, S.P.; Engl, T.; Weiss, B.; Fischer, R.; Steiger, S.; Heckel, D.G.; Kaltenpoth, M.; Vilcinskas, A. The digestive and defensive basis of carcass utilization by the burying beetle and its microbiota. Nat. Commun. 2017, 8, 15186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rozen, D.E. Gut microbiota in the burying beetle, Nicrophorus vespilloides, provide colonization resistance against larval bacterial pathogens. Ecol. Evol. 2018, 8, 1646–1654. [Google Scholar] [CrossRef] [Green Version]
- Heise, P.; Liu, Y.; Degenkolb, T.; Vogel, H.; Schäberle, T.F.; Vilcinskas, A. Antibiotic-Producing Beneficial Bacteria in the Gut of the Burying Beetle Nicrophorus vespilloides. Front. Microbiol. 2019, 10, 1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Chen, B.; Sun, C.; Ishida, K.; Hertweck, C.; Boland, W. Symbiont-Derived Antimicrobials Contribute to the Control of the Lepidopteran Gut Microbiota. Cell Chem. Biol. 2017, 24, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Schmid-Hempel, P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. USA 2011, 108, 19288–19292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, M.K.; Liu, T.; Awasthi, S.K.; Duan, Y.; Pandey, A.; Zhang, Z. Manure pretreatments with black soldier fly Hermetia illucens L. (Diptera: Stratiomyidae): A study to reduce pathogen content. Sci. Total Environ. 2020, 737, 139842. [Google Scholar] [CrossRef] [PubMed]
- Vogel, H.; Müller, A.; Heckel, D.G.; Gutzeit, H.; Vilcinskas, A. Nutritional immunology: Diversification and diet-dependent expression of antimicrobial peptides in the black soldier fly Hermetia illucens. Dev. Comp. Immunol. 2018, 78, 141–148. [Google Scholar] [CrossRef]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Buffie, C.G.; Pamer, E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Barros, L.M.; Gutjahr, A.L.N.; Ferreira-Keppler, R.L.; Martins, R.T. Morphological description of the immature stages of Hermetia illucens (Linnaeus, 1758) (Diptera: Stratiomyidae). Microsc. Res. Tech. 2019, 82, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, C.; Al-Soud, W.A.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Werner, J.J.; Koren, O.; Hugenholtz, P.; DeSantis, T.Z.; Walters, W.A.; Caporaso, J.G.; Angenent, L.T.; Knight, R.; Ley, R.E. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012, 6, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- García-López, M.; Meier-Kolthoff, J.P.; Tindall, B.J.; Gronow, S.; Woyke, T.; Kyrpides, N.C.; Hahnke, R.L.; Göker, M. Analysis of 1000 Type-Strain Genomes Improves Taxonomic Classification of Bacteroidetes. Front. Microbiol. 2019, 10, 2083. [Google Scholar] [CrossRef] [Green Version]
- Oren, A.; Garrity, G.M. List of new names and new combinations previously effectively, but not validly, published. Int. J. Syst. Evol. Microbiol. 2020, 70, 2960–2966. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Richter, M.; Peplies, J.; Euzeby, J.; Amann, R.; Schleifer, K.-H.; Ludwig, W.; Glöckner, F.O.; Rosselló-Móra, R. The All-Species Living Tree project: A 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol. 2008, 31, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Chaudhary, D.K.; Khulan, A.; Kim, J. Development of a novel cultivation technique for uncultured soil bacteria. Sci. Rep. 2019, 9, 6666. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Dubin, K.; Littmann, E.R.; Moody, T.U.; Fontana, E.; Seok, R.; Leiner, I.M.; Taur, Y.; Peled, J.U.; van den Brink, M.R.M.; et al. Inhibiting antibiotic-resistant Enterobacteriaceae by microbiota-mediated intracellular acidification. J. Exp. Med. 2019, 216, 84–98. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Q.; Chen, X.; Jiang, C. Isolation and Cultivation Methods of Actinobacteria. In Actinobacteria-Basics and Biotechnological Applications; Dhanasekaran, D., Jiang, Y., Eds.; InTechOpen: London, UK, 2016; ISBN 978-953-51-2248-7. [Google Scholar]
- Cruden, D.L.; Markovetz, A.J. Carboxymethyl Cellulose Decomposition by Intestinal Bacteria of Cockroaches. Appl. Environ. Microbiol. 1979, 38, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Strassert, J.F.H.; Desai, M.S.; Radek, R.; Brune, A. Identification and localization of the multiple bacterial symbionts of the termite gut flagellate Joenia annectens. Microbiology 2010, 156, 2068–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riyanti; Balansa, W.; Liu, Y.; Sharma, A.; Mihajlovic, S.; Hartwig, C.; Leis, B.; Rieuwpassa, F.J.; Ijong, F.G.; Wägele, H.; et al. Selection of sponge-associated bacteria with high potential for the production of antibacterial compounds. Sci. Rep. 2020, 10, 19614. [Google Scholar] [CrossRef]
- Lee, J.K.; Lee, Y.Y.; Park, K.H.; Sim, J.; Choi, Y.; Lee, S.-J. Wohlfahrtiimonas larvae sp. nov., isolated from the larval gut of Hermetia illucens (Diptera: Stratiomyidae). Antonie Van Leeuwenhoek 2014, 105, 15–21. [Google Scholar] [CrossRef]
- Bessis, S.; Ndongo, S.; Lagier, J.-C.; Raoult, D.; Fournier, P.-E. ‘Neglecta timonensis’ gen. nov., sp. nov., a new human-associated species. New Microbes New Infect. 2016, 13, 13–14. [Google Scholar] [CrossRef] [Green Version]
- Lagkouvardos, I.; Pukall, R.; Abt, B.; Foesel, B.U.; Meier-Kolthoff, J.P.; Kumar, N.; Bresciani, A.; Martínez, I.; Just, S.; Ziegler, C.; et al. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016, 1, 16131. [Google Scholar] [CrossRef] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Marcy, Y.; Ouverney, C.; Bik, E.M.; Lösekann, T.; Ivanova, N.; Martin, H.G.; Szeto, E.; Platt, D.; Hugenholtz, P.; Relman, D.A.; et al. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. USA 2007, 104, 11889–11894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, S.; Kitade, O.; Inoue, T.; Kawai, M.; Kanuka, M.; Hiroshima, K.; Hongoh, Y.; Constantino, R.; Uys, V.; Zhong, J.; et al. Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mol. Ecol. 2007, 16, 1257–1266. [Google Scholar] [CrossRef]
- de Smet, J.; Wynants, E.; Cos, P.; van Campenhout, L. Microbial Community Dynamics during Rearing of Black Soldier Fly Larvae (Hermetia illucens) and Impact on Exploitation Potential. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanga, C.M.; Waweru, J.W.; Tola, Y.H.; Onyoni, A.A.; Khamis, F.M.; Ekesi, S.; Paredes, J.C. Organic Waste Substrates Induce Important Shifts in Gut Microbiota of Black Soldier Fly (Hermetia illucens L.): Coexistence of Conserved, Variable, and Potential Pathogenic Microbes. Front. Microbiol. 2021, 12, 635881. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Lee, J.K.; Park, K.H.; Kim, S.-Y.; Roh, S.W.; Lee, S.-B.; Choi, Y.; Lee, S.-J. Paenalcaligenes hermetiae sp. nov., isolated from the larval gut of Hermetia illucens (Diptera: Stratiomyidae), and emended description of the genus Paenalcaligenes. Int. J. Syst. Evol. Microbiol. 2013, 63, 4224–4229. [Google Scholar] [CrossRef]
- Stackebrandt, E. The Family Lachnospiraceae. In The Prokaryotes: Firmicutes and Tenericutes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 197–201. ISBN 978-3-642-30120-9. [Google Scholar]
- Yassin, A.-A.F. The Family Actinomycetaceae. In The Prokaryotes: Actinobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 21–103. ISBN 978-3-642-30138-4. [Google Scholar]
- Dehority, B.A. Carbon Dioxide Requirement of Various Species of Rumen Bacteria. J. Bacteriol. 1971, 105, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Schmitt-Wagner, D.; Stingl, U.; Brune, A. Niche heterogeneity determines bacterial community structure in the termite gut (Reticulitermes santonensis). Environ. Microbiol. 2005, 7, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Lee, S.-C.; Cho, Y.-Y.; Oh, H.-J.; Ko, Y.H. Isolation of Cellulolytic Bacillus subtilis Strains from Agricultural Environments. ISRN Microbiol. 2012, 2012, 650563. [Google Scholar] [CrossRef] [Green Version]
- Logan, N.A.; de Vos, P.; Genus, I. Bacillus Cohn 1872. In Bergey’s Manual of Systematic Bacteriology, 2nd ed.; de Vos, P., Whitman, W.B., Parte, A.C., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 21–128. ISBN 9780387950419. [Google Scholar]
- Ma, L.; Lu, Y.; Yan, H.; Wang, X.; Yi, Y.; Shan, Y.; Liu, B.; Zhou, Y.; Lü, X. Screening of cellulolytic bacteria from rotten wood of Qinling (China) for biomass degradation and cloning of cellulases from Bacillus methylotrophicus. BMC Biotechnol. 2020, 20, 2. [Google Scholar] [CrossRef] [Green Version]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Raimondi, S.; Spampinato, G.; Macavei, L.I.; Lugli, L.; Candeliere, F.; Rossi, M.; Maistrello, L.; Amaretti, A. Effect of Rearing Temperature on Growth and Microbiota Composition of Hermetia illucens. Microorganisms 2020, 8, 902. [Google Scholar] [CrossRef]
- Ladha, J.K.; Barraquio, W.L.; Watanabe, I. Isolation and identification of nitrogen-fixing Enterobacter cloacae and Klebsiella planticola associated with rice plants. Can. J. Microbiol. 1983, 29, 1301–1308. [Google Scholar] [CrossRef]
- Grimont, P.A.D.; Grimont, F. Klebsiella. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Rainey, F., Kämpfer, P., Trujillo, M., Chun, J., DeVos, P., Hedlund, B., Dedysh, S., Eds.; Wiley: New York, NY, USA, 2015; pp. 1–26. ISBN 9781118960608. [Google Scholar]
- Boyaval, P.; Boyaval, E.; Desmazeaud, M.J. Survival of Brevibacterium linens during nutrient starvation and intracellular changes. Arch. Microbiol. 1985, 141, 128–132. [Google Scholar] [CrossRef]
- Siva-Jothy, M.T.; Moret, Y.; Rolff, J. Insect Immunity: An Evolutionary Ecology Perspective. In Advances in Insect Physiology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 32, pp. 1–48. ISBN 9780120242320. [Google Scholar]
- Rabbee, M.F.; Ali, M.S.; Choi, J.; Hwang, B.S.; Jeong, S.C.; Baek, K.-H. Bacillus velezensis: A Valuable Member of Bioactive Molecules within Plant Microbiomes. Molecules 2019, 24, 1046. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef]
- Wang, Y.; Rozen, D.E. Gut Microbiota Colonization and Transmission in the Burying Beetle Nicrophorus vespilloides throughout Development. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [Green Version]
- Branca, G.; Galasso, C.; Cornaglia, G.; Fontana, R.; Satta, G. A simple method to detect bacteriolytic enzymes produced by enterobacteriaceae. Microbios 1996, 86, 205–212. [Google Scholar]
- Janda, J.M.; Abbott, S.L. Morganella. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Rainey, F., Kämpfer, P., Trujillo, M., Chun, J., DeVos, P., Hedlund, B., Dedysh, S., Eds.; Wiley: New York, NY, USA, 2015; pp. 1–7. ISBN 9781118960608. [Google Scholar]
- Park, K.H.; Kwak, K.W.; Nam, S.H.; Choi, J.; Lee, S.H.; Kim, H.G.; Kim, S.H. Antibacterial activity of larval extract from the black soldier fly Hermetia illucens (Diptera: Stratiomyidae) against plant pathogens. J. Entomol. Zool. Stud. 2015, 3, 176–179. [Google Scholar]
- Himathongkham, S.; Riemann, H. Destruction of Salmonella typhimurium, Escherichia coli O157:H7 and Listeria monocytogenes in chicken manure by drying and/or gassing with ammonia. FEMS Microbiol. Lett. 1999, 171, 179–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonelli, M.; Bruno, D.; Caccia, S.; Sgambetterra, G.; Cappellozza, S.; Jucker, C.; Tettamanti, G.; Casartelli, M. Structural and Functional Characterization of Hermetia illucens Larval Midgut. Front. Physiol. 2019, 10, 204. [Google Scholar] [CrossRef]
- van Moll, L.; de Smet, J.; Cos, P.; van Campenhout, L. Microbial symbionts of insects as a source of new antimicrobials: A review. Crit. Rev. Microbiol. 2021, 1–18. [Google Scholar] [CrossRef]
- Guzman, J.; Vilcinskas, A. Bacteria associated with cockroaches: Health risk or biotechnological opportunity? Appl. Microbiol. Biotechnol. 2020, 104, 10369–10387. [Google Scholar] [CrossRef] [PubMed]
- Rawski, M.; Mazurkiewicz, J.; Kierończyk, B.; Józefiak, D. Black Soldier Fly Full-Fat Larvae Meal is More Profitable than Fish Meal and Fish Oil in Siberian Sturgeon Farming: The Effects on Aquaculture Sustainability, Economy and Fish GIT Development. Animals 2021, 11, 604. [Google Scholar] [CrossRef] [PubMed]
- Smetana, S.; Schmitt, E.; Mathys, A. Sustainable use of Hermetia illucens insect biomass for feed and food: Attributional and consequential life cycle assessment. Resour. Conserv. Recycl. 2019, 144, 285–296. [Google Scholar] [CrossRef]
- Weththasinghe, P.; Lagos, L.; Cortés, M.; Hansen, J.Ø.; Øverland, M. Dietary Inclusion of Black Soldier Fly (Hermetia Illucens) Larvae Meal and Paste Improved Gut Health but Had Minor Effects on Skin Mucus Proteome and Immune Response in Atlantic Salmon (Salmo Salar). Front. Immunol. 2021, 12, 599530. [Google Scholar] [CrossRef]
- Henry, M.A.; Gasco, L.; Chatzifotis, S.; Piccolo, G. Does dietary insect meal affect the fish immune system? The case of mealworm, Tenebrio molitor on European sea bass, Dicentrarchus labrax. Dev. Comp. Immunol. 2018, 81, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Gasco, L.; Finke, M.; van Huis, A. Can diets containing insects promote animal health? J. Insects Food Feed 2018, 4, 1–4. [Google Scholar] [CrossRef]
- Cardinaletti, G.; Randazzo, B.; Messina, M.; Zarantoniello, M.; Giorgini, E.; Zimbelli, A.; Bruni, L.; Parisi, G.; Olivotto, I.; Tulli, F. Effects of Graded Dietary Inclusion Level of Full-Fat Hermetia illucens Prepupae Meal in Practical Diets for Rainbow Trout (Oncorhynchus mykiss). Animals 2019, 9, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarantoniello, M.; Zimbelli, A.; Randazzo, B.; Compagni, M.D.; Truzzi, C.; Antonucci, M.; Riolo, P.; Loreto, N.; Osimani, A.; Milanović, V.; et al. Black Soldier Fly (Hermetia illucens) reared on roasted coffee by-product and Schizochytrium sp. as a sustainable terrestrial ingredient for aquafeeds production. Aquaculture 2020, 518, 734659. [Google Scholar] [CrossRef]
- Vargas-Abúndez, A.J.; Randazzo, B.; Foddai, M.; Sanchini, L.; Truzzi, C.; Giorgini, E.; Gasco, L.; Olivotto, I. Insect meal based diets for clownfish: Biometric, histological, spectroscopic, biochemical and molecular implications. Aquaculture 2019, 498, 1–11. [Google Scholar] [CrossRef]
- Li, Y.; Bruni, L.; Jaramillo-Torres, A.; Gajardo, K.; Kortner, T.M.; Krogdahl, Å. Differential response of digesta- and mucosa-associated intestinal microbiota to dietary insect meal during the seawater phase of Atlantic salmon. Anim. Microbiome 2021, 3, 8. [Google Scholar] [CrossRef]
- Bruni, L.; Pastorelli, R.; Viti, C.; Gasco, L.; Parisi, G. Characterisation of the intestinal microbial communities of rainbow trout (Oncorhynchus mykiss) fed with Hermetia illucens (black soldier fly) partially defatted larva meal as partial dietary protein source. Aquaculture 2018, 487, 56–63. [Google Scholar] [CrossRef]
- Yang, F.; Tomberlin, J.K.; Jordan, H.R. Starvation Alters Gut Microbiome in Black Soldier Fly (Diptera: Stratiomyidae) Larvae. Front. Microbiol. 2021, 12, 601253. [Google Scholar] [CrossRef] [PubMed]
- Thimm, T.; Hoffmann, A.; Borkott, H.; Munch, J.C.; Tebbe, C.C. The gut of the soil microarthropod Folsomia candida (Collembola) is a frequently changeable but selective habitat and a vector for microorganisms. Appl. Environ. Microbiol. 1998, 64, 2660–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husseneder, C.; Grace, J.K.; Oishi, D.E. Use of genetically engineered Escherichia coli to monitor ingestion, loss, and transfer of bacteria in termites. Curr. Microbiol. 2005, 50, 119–123. [Google Scholar] [CrossRef]
- Husseneder, C.; Grace, J.K. Genetically engineered termite gut bacteria (Enterobacter cloacae) deliver and spread foreign genes in termite colonies. Appl. Microbiol. Biotechnol. 2005, 68, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Cheng, P.; Chen, Y.; Li, Y.; Yang, Z.; Chen, Y.; Tomberlin, J.K. Inoculating poultry manure with companion bacteria influences growth and development of black soldier fly (Diptera: Stratiomyidae) larvae. Environ. Entomol. 2011, 40, 30–35. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. aureus | E. coli | P. aeruginosa | |||||
|---|---|---|---|---|---|---|---|
| Strain No. | Closest Relative (Based on 16S rRNA Sequencing) | Inhibition | Zone of Inhibition (mm) | Inhibition | Zone of Inhibition (mm) | Inhibition | Zone of Inhibition (mm) |
| 01-003 | Corynebacterium terpenotabidum | − | − | − | − | (+) | 1.50 ± 0.71 |
| 01-008 | Morganella morganii | − | − | − | − | + | 3.25 ± 0.35 |
| 01-018 | Alcaligenes faecalis | + | 2.75 ± 0.35 | + | 3.50 ± 0.00 | − | − |
| 01-027 | Providencia rettgeri | − | − | − | − | (+) | 3.00 ± 0.00 |
| 01-035 | Providencia rettgeri | − | − | − | − | (+) | 2.50 ± 0.00 |
| 01-040 | Wohlfahrtiimonas larvae | (+) | 1.00 ± 0.00 | − | − | + | 2.00 ± 0.00 |
| 01-052 | Providencia rettgeri | − | − | − | − | (+) | 3.00 ± 0.00 |
| 01-053 | Providencia rettgeri | − | − | − | − | (+) | 2.00 ± 0.00 |
| 01-054 | Klebsiella aerogenes | − | − | + | 3.75 ± 0.35 | − | − |
| 01-060 | Providencia rettgeri | − | − | − | − | (+) | 3.00 ± 0.00 |
| 01-079 | Providencia rettgeri | − | − | − | − | (+) | 2.00 ± 0.00 |
| 01-105 | Klebsiella aerogenes | − | − | + | 3.25 ± 0.35 | − | − |
| 01-115 | Alcaligenes faecalis | + | 2.00 ± 0.00 | − | − | − | − |
| 01-124 | Alcaligenes faecalis | − | − | + | 2.75 ± 0.35 | + | 3.50 ± 0.71 |
| 01-149 | Bacillus velezensis | + | 2.75 ± 0.35 | + | 1.50 ± 0.00 | + | 5.25 ± 1.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tegtmeier, D.; Hurka, S.; Mihajlovic, S.; Bodenschatz, M.; Schlimbach, S.; Vilcinskas, A. Culture-Independent and Culture-Dependent Characterization of the Black Soldier Fly Gut Microbiome Reveals a Large Proportion of Culturable Bacteria with Potential for Industrial Applications. Microorganisms 2021, 9, 1642. https://doi.org/10.3390/microorganisms9081642
Tegtmeier D, Hurka S, Mihajlovic S, Bodenschatz M, Schlimbach S, Vilcinskas A. Culture-Independent and Culture-Dependent Characterization of the Black Soldier Fly Gut Microbiome Reveals a Large Proportion of Culturable Bacteria with Potential for Industrial Applications. Microorganisms. 2021; 9(8):1642. https://doi.org/10.3390/microorganisms9081642
Chicago/Turabian StyleTegtmeier, Dorothee, Sabine Hurka, Sanja Mihajlovic, Maren Bodenschatz, Stephanie Schlimbach, and Andreas Vilcinskas. 2021. "Culture-Independent and Culture-Dependent Characterization of the Black Soldier Fly Gut Microbiome Reveals a Large Proportion of Culturable Bacteria with Potential for Industrial Applications" Microorganisms 9, no. 8: 1642. https://doi.org/10.3390/microorganisms9081642
APA StyleTegtmeier, D., Hurka, S., Mihajlovic, S., Bodenschatz, M., Schlimbach, S., & Vilcinskas, A. (2021). Culture-Independent and Culture-Dependent Characterization of the Black Soldier Fly Gut Microbiome Reveals a Large Proportion of Culturable Bacteria with Potential for Industrial Applications. Microorganisms, 9(8), 1642. https://doi.org/10.3390/microorganisms9081642