
Complete Genome Sequence of vB_EcoP_SU7, a Podoviridae Coliphage with the Rare C3 Morphotype


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Phage Propagation and Purification
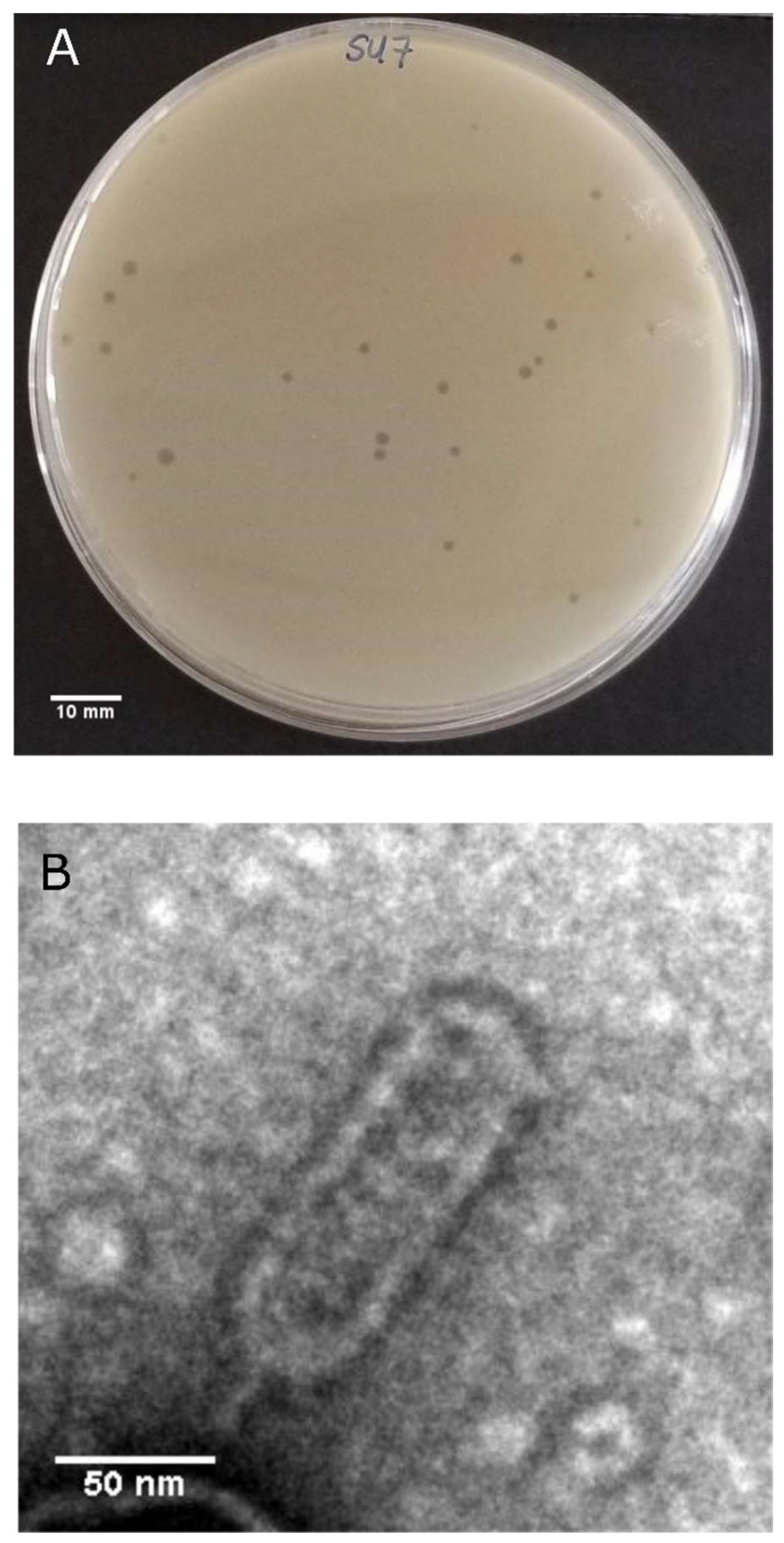
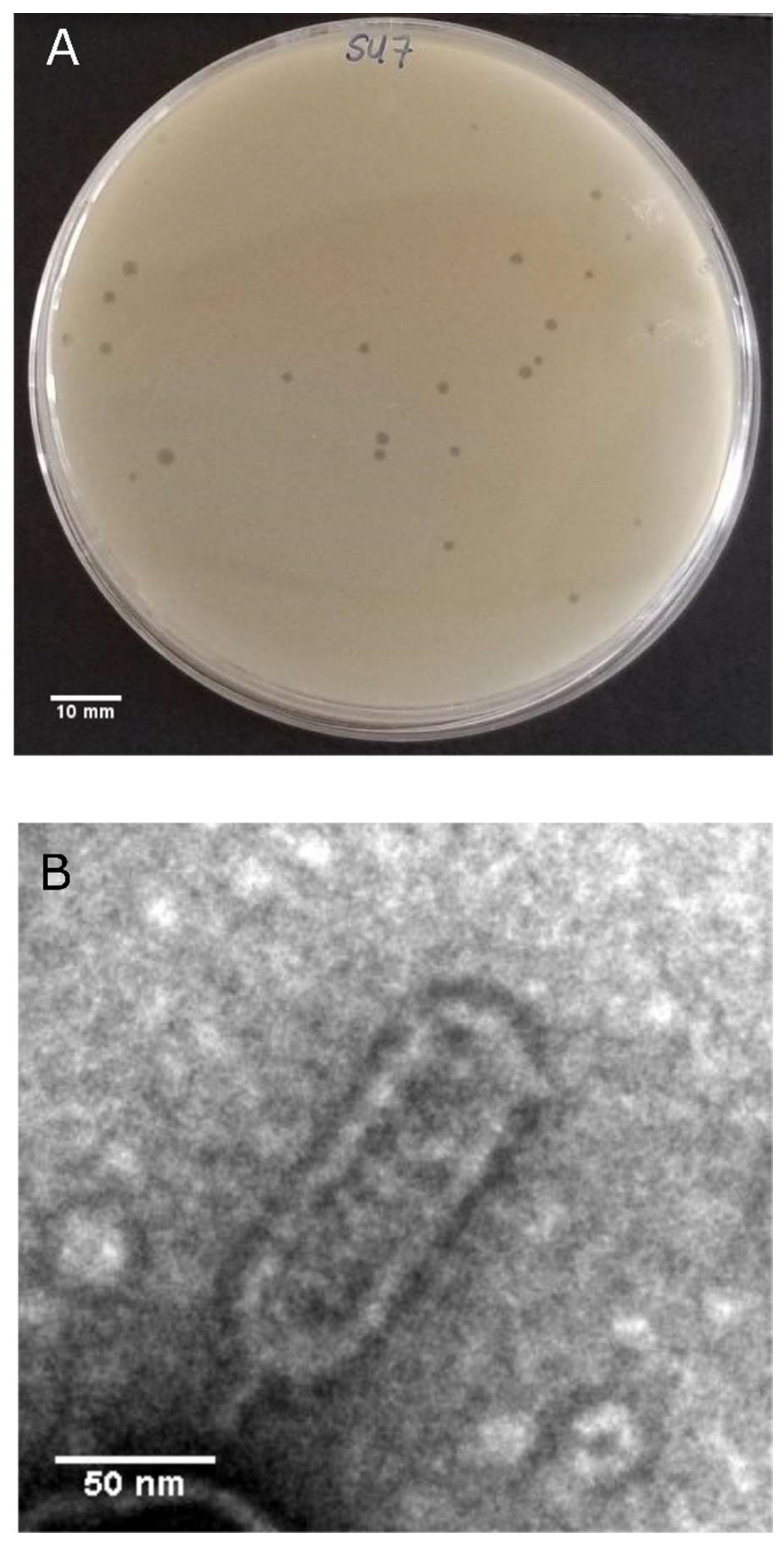
2.3. Plaque Morphology Determination
2.4. Host Range Analysis
2.5. Transmission Electron Microscopy (TEM)
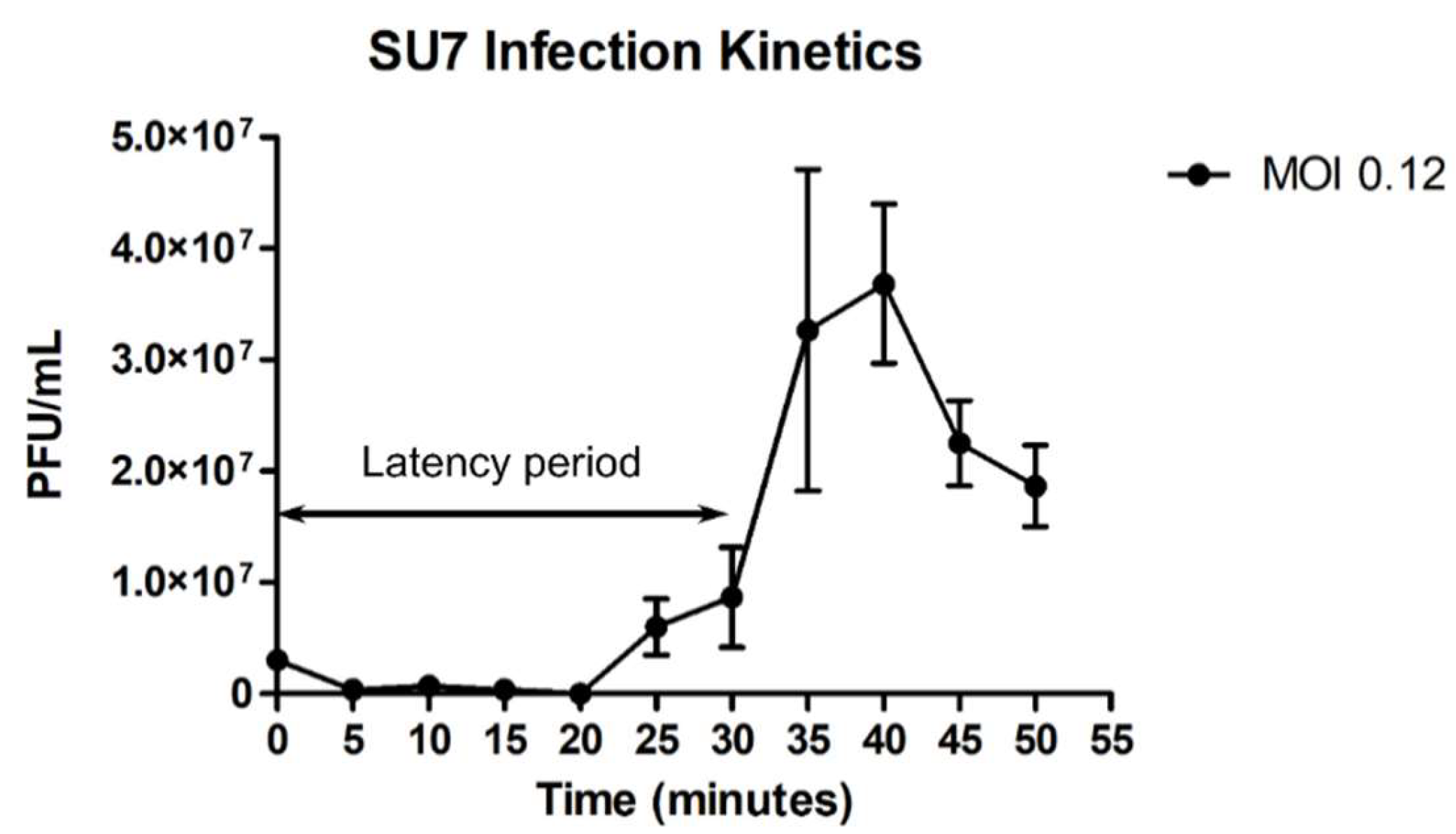
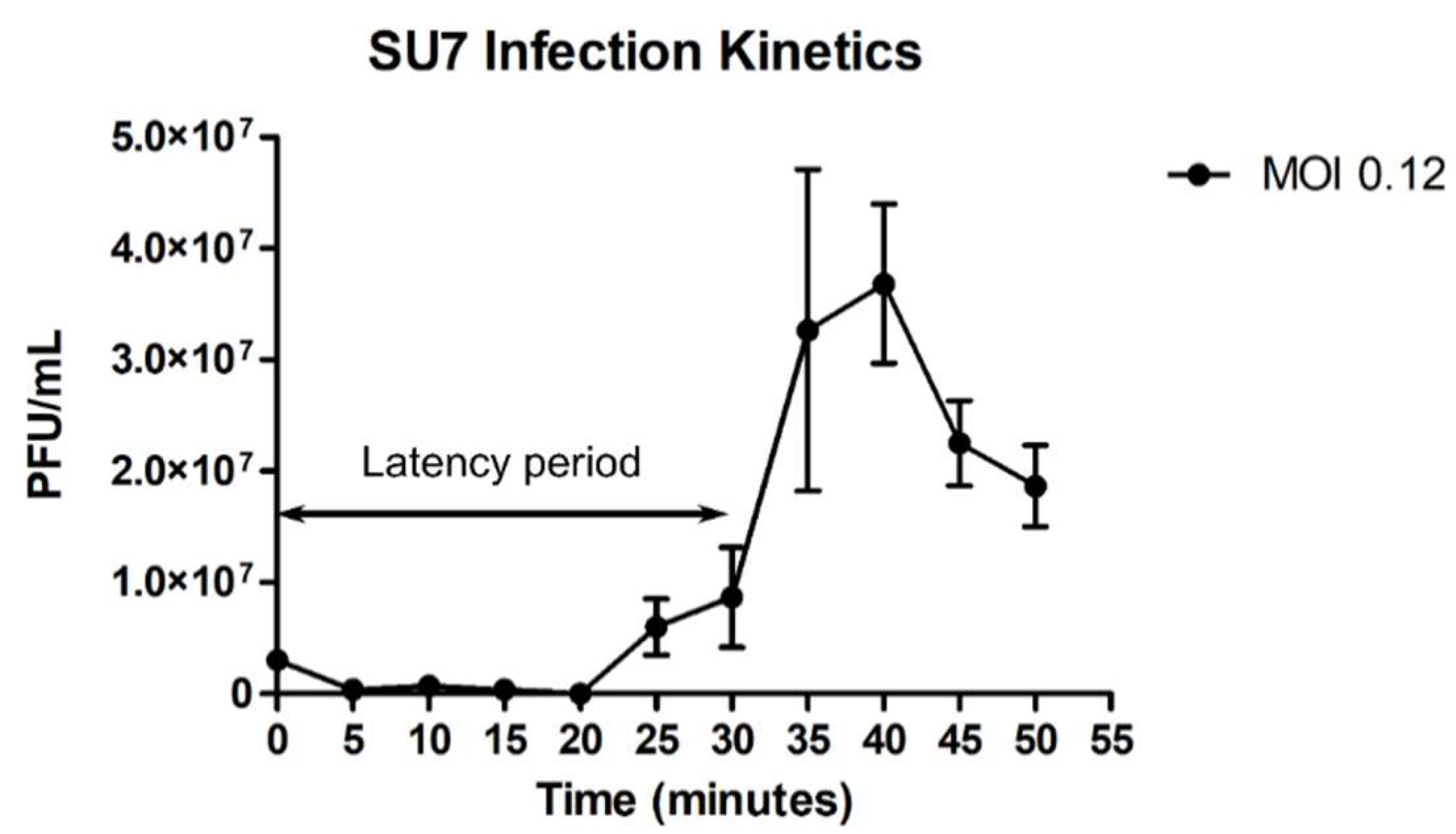
2.6. SU7 Infection Kinetics
2.7. Phage DNA Extraction
2.8. Genome Sequencing and Bioinformatics of the SU7 Genome
2.9. Phylogenetic Tree Construction
3. Results and Discussion
3.1. SU7 Plaque Morphology, Virion Morphology, Infection Kinetics, and Host Range
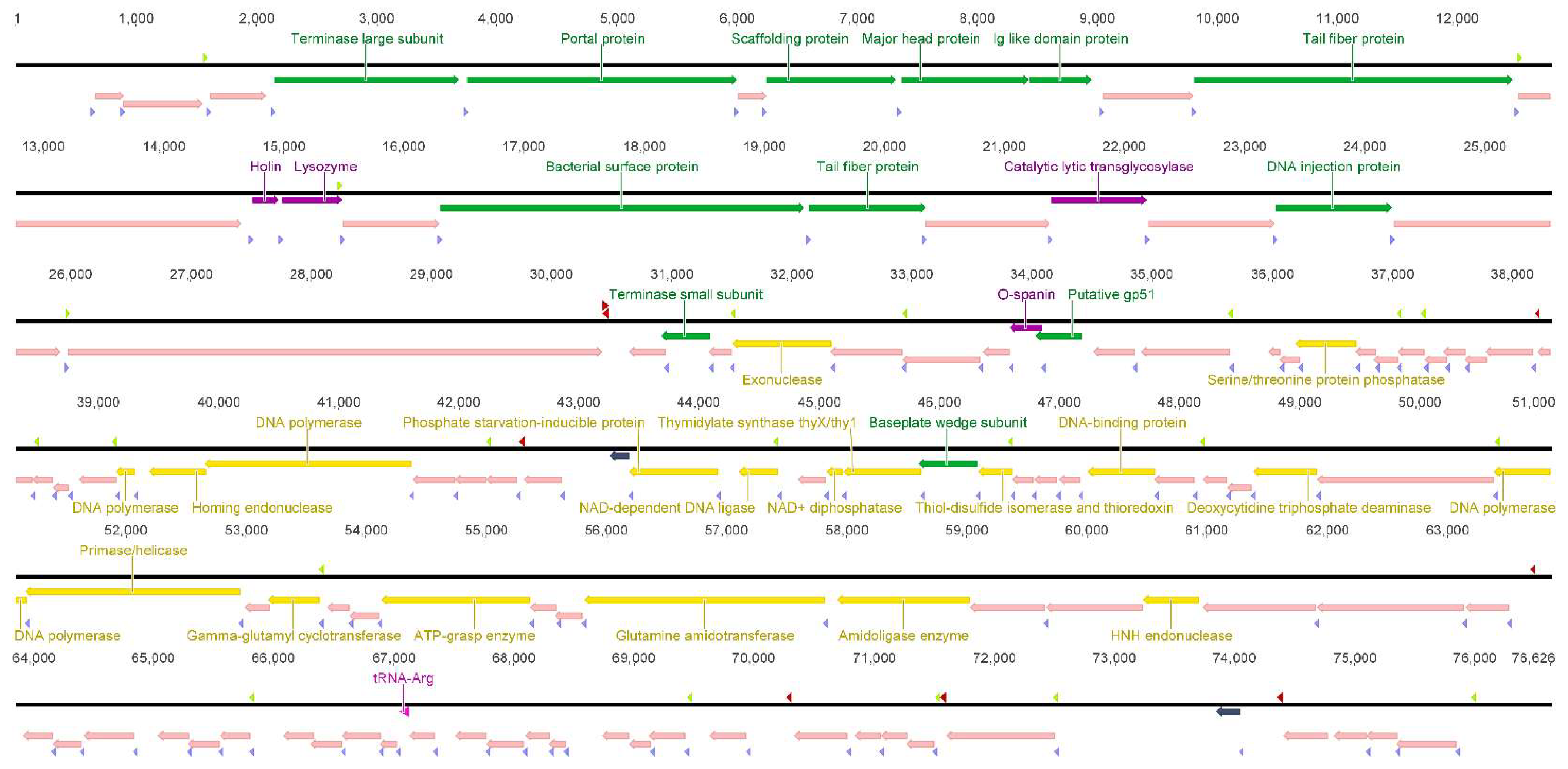
3.2. SU7 Genome Characterization
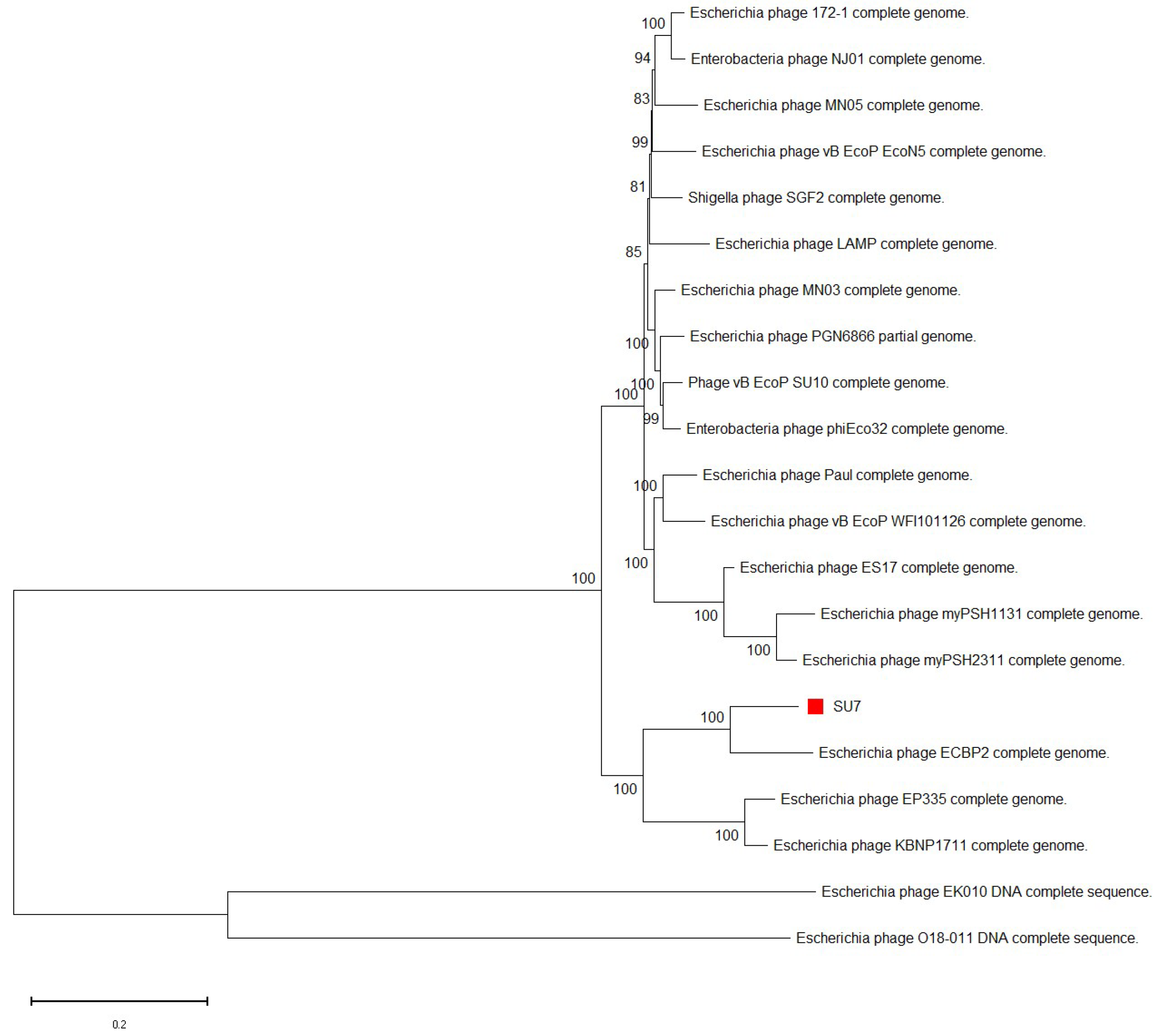
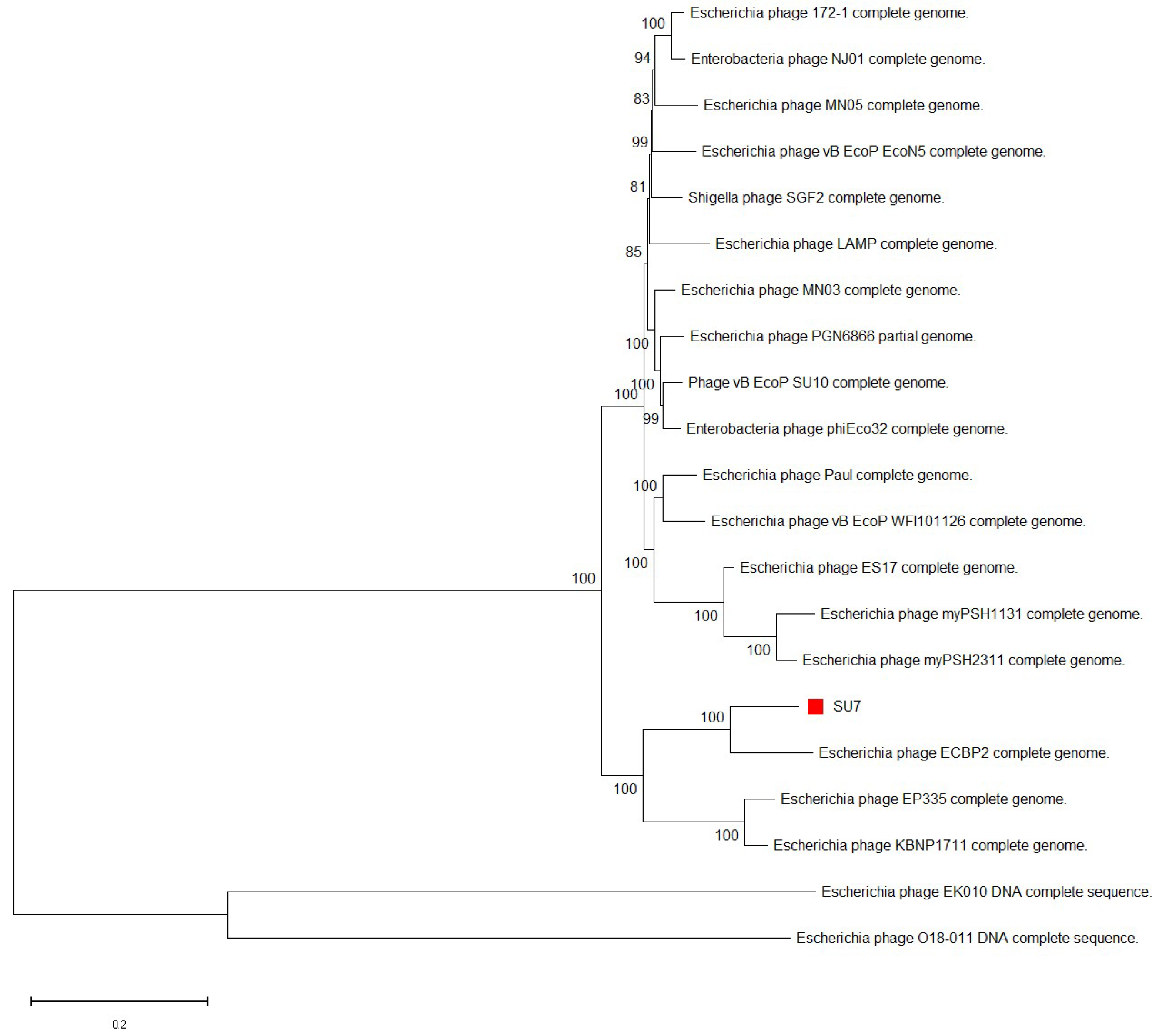
3.3. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conway, T.; Krogfelt, K.A.; Cohen, P.S. The Life of Commensal Escherichia coli in the Mammalian Intestine. EcoSal Plus 2004, 1. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.-D.; Zhao, Y.; Huang, J. Metabolic Modeling of CommonEscherichia coliStrains in Human Gut Microbiome. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.J., Jr.; Evans, D.G. Classification of Pathogenic Escherichia coli According to Serotype and the Pro-duction of Virulence Factors, with Special Reference to Colonization-Factor Antigens. Rev. Infect. Dis. 1983, 5, S692–S701. [Google Scholar]
- Bin Cha, S.; Na Yoo, A.; Lee, W.J.; Shin, M.K.; Jung, M.; Shin, S.W.; Cho, Y.W.; Yoo, H.S. Effect of Bacteriophage in Enterotoxigenic Escherichia coli (ETEC) Infected Pigs. J. Vet. Med. Sci. 2012, 74, 1037–1039. [Google Scholar] [CrossRef] [Green Version]
- Enterotoxigenic E. coli (ETEC)|E. coli|CDC. Available online: https://www.cdc.gov/ecoli/etec.html (accessed on 31 May 2021).
- Duan, Q.; Xia, P.; Nandre, R.; Zhang, W.; Zhu, G. Review of Newly Identified Functions Associated With the Heat-Labile Toxin of Enterotoxigenic Escherichia coli. Front. Cell. Infect. Microbiol. 2019, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Begum, Y.A.; Talukder, K.A.; Azmi, I.J.; Shahnaij, M.; Sheikh, A.; Sharmin, S.; Svennerholm, A.-M.; Qadri, F. Resistance Pattern and Molecular Characterization of Enterotoxigenic Escherichia coli (ETEC) Strains Isolated in Bangladesh. PLoS ONE 2016, 11, e0157415. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Futur. Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.S. Phage therapy—constraints and possibilities. Upsala J. Med. Sci. 2014, 119, 192–198. [Google Scholar] [CrossRef]
- Ochman, H.; Selander, R.K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 1984, 157, 690–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratia, A. The Numerical Relation between Lysogenic Bacteria and the Phage Particles which they carry. Ann. Inst. Pasteur 1936, 57, 652–676. [Google Scholar]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of Bacteriophages by Double Agar Overlay Plaque Assay. In Bacteriophages; Martha, R.J., Clokie, A.M.K., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 501, pp. 69–76. [Google Scholar]
- Sambrook, J.; Russell, D.W. Precipitation of Bacteriophage λ Particles from Large-scale Lysates. Cold Spring Harb. Protoc. 2006, 2006, 3966. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E. Phage Host Range and Efficiency of Plating. Adv. Struct. Saf. Stud. 2009, 501, 141–149. [Google Scholar] [CrossRef]
- Broers, A.N.; Panessa, B.J.; Gennaro, J.F. High-resolution scanning electron microscopy of bacteriophages 3C and T4. Science 1975, 189, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Koonjan, S.; Seijsing, F.; Cooper, C.J.; Nilsson, A.S. Infection Kinetics and Phylogenetic Analysis of vB_EcoD_SU57, a Virulent T1-Like Drexlerviridae Coliphage. Front. Microbiol. 2020, 11, 2439. [Google Scholar] [CrossRef]
- Babraham Bioinformatics. FastQC v. 0.11.2. 2014. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 January 2020).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Cuccuru, G.; Orsini, M.; Pinna, A.; Sbardellati, A.; Soranzo, N.; Travaglione, A.; Uva, P.; Zanetti, G.; Fotia, G. Orione, a web-based framework for NGS analysis in microbiology. Bioinformatics 2014, 30, 1928–1929. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galaxy|Pasteur. Available online: https://galaxy.pasteur.fr/?tool_id=toolshed.pasteur.fr%2Frepos%2Ffmareuil%2Fprokka%2Fprokka%2F1.14.5%2Bgalaxy0&version=1.14.5%2Bgalaxy0&__identifer=m5chf8bbb2 (accessed on 31 May 2021).
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Geneious Prime. Available online: http://www.geneious.com/ (accessed on 7 April 2020).
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solovyev, V.; Salamov, A. Automatic Annotation of Microbial Genomes and Metagenomic Sequences. In Meta-Genomics and Its Applications in Agriculture, Biomedicine, and Envrionmental Studies; Nova Science Publishers: Hauppauge, NY, USA, 2011; pp. 61–78. [Google Scholar]
- BPROM-Prediction of Bacterial Promoters. Available online: http://www.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb (accessed on 31 May 2021).
- Sampaio, M.; Rocha, M.; Oliveira, H.; Dias, O. Predicting promoters in phage genomes using PhagePromoter. Bioinformatics 2019, 35, 5301–5302. [Google Scholar] [CrossRef]
- Galaxy|Galaxy Docker Build. Available online: https://galaxy.bio.di.uminho.pt/?tool_id=toolshed.g2.bx.psu.edu%2Frepos%2Fmartasampaio%2Fphagepromoter%2Fget_proms%2F0.1.0&version=0.1.0&__identifer=axgcaazotdi (accessed on 31 May 2021).
- Naville, M.; Ghuillot-Gaudeffroy, A.; Marchais, A.; Gautheret, D. ARNold: A web tool for the prediction of Rho-independent transcription terminators. RNA Biol. 2011, 8, 11–13. [Google Scholar] [CrossRef] [Green Version]
- ARNold, Finding Terminators at I2BC-Web Server. Available online: http://rssf.i2bc.paris-saclay.fr/toolbox/arnold/ (accessed on 31 May 2021).
- Shine, J.; Dalgarno, L. The 3’-Terminal Sequence of Escherichia coli 16S Ribosomal RNA: Complementarity to Nonsense Triplets and Ribosome Binding Sites. Proc. Natl. Acad. Sci. USA 1974, 71, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Campbell, A.; Karlin, S. Correlations between Shine-Dalgarno Sequences and Gene Features Such as Predicted Expression Levels and Operon Structures. J. Bacteriol. 2002, 184, 5733–5745. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- EMBOSS: Geecee. Available online: https://www.bioinformatics.nl/cgi-bin/emboss/geecee (accessed on 31 May 2021).
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- ARAGORN Detects TRNA, MtRNA and TmRNA Genes. Available online: http://www.ansikte.se/ARAGORN/ (accessed on 31 May 2021).
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Galaxy Pasteur. Available online: https://galaxy.pasteur.fr/?tool_id=toolshed.pasteur.fr%2Frepos%2Ffmareuil%2Fphageterm%2FPhageTerm%2F1.0.12&version=1.0.12&__identifer=maucuexyo4 (accessed on 31 May 2021).
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Savalia, D.; Westblade, L.F.; Goel, M.; Florens, L.; Kemp, P.; Akulenko, N.; Pavlova, O.; Padovan, J.C.; Chait, B.T.; Washburn, M.; et al. Genomic and Proteomic Analysis of phiEco32, a Novel Escherichia coli Bacteriophage. J. Mol. Biol. 2008, 377, 774–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attoui, H.; Mertens, P.P.C.; Becnel, J.; Belaganahalli, S.; Bergoin, M.; Brussaard, C.P.; Chappell, J.D.; Ciarlet, M.; del Vas, M.; Dermody, T.S.; et al. Reoviridae. In Virus Taxonomy. Ninth Report of the International Committee on Taxonomy of Viruses, 1st ed.; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: London, UK, 2012; pp. 541–637. ISBN 978-012-384-684-6. [Google Scholar]
- Mirzaei, M.K.; Eriksson, H.; Kasuga, K.; Haggård-Ljungquist, E.; Nilsson, A.S. Genomic, Proteomic, Morphological, and Phylogenetic Analyses of vB_EcoP_SU10, a Podoviridae Phage with C3 Morphology. PLoS ONE 2014, 9, e116294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, A.; Saldana, R.; Moreland, R.; Gill, J.J.; Liu, M.; Ramsey, J. Complete Genome Sequence of Escherichia coli Phage Paul. Microbiol. Resour. Announc. 2019, 8, e01093-19. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, M.K.; Nilsson, A.S. Correction: Isolation of Phages for Phage Therapy: A Comparison of Spot Tests and Efficiency of Plating Analyses for Determination of Host Range and Efficacy. PLoS ONE 2015, 10, e0127606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurczak-Kurek, A.; Gąsior, T.; Nejman-Faleńczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef]
- Kleppen, H.P.; Holo, H.; Jeon, S.-R.; Nes, I.F.; Yoon, S.-S. Novel Podoviridae Family Bacteriophage Infecting Weissella cibaria Isolated from Kimchi. Appl. Environ. Microbiol. 2012, 78, 7299–7308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, K.; Uchiyama, J.; Kato, S.-I.; Ujihara, T.; Hoshiba, H.; Sugihara, S.; Muraoka, A.; Wakiguchi, H.; Matsuzaki, S. Morphological and genetic analysis of three bacteriophages ofSerratia marcescensisolated from environmental water. FEMS Microbiol. Lett. 2009, 291, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Iwashita, S.; Kanegasaki, S. Smooth specific phage adsorption: Endorhamnosidase activity of tail parts of P22. Biochem. Biophys. Res. Commun. 1973, 55, 403–409. [Google Scholar] [CrossRef]
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortright, K.E.; Chan, B.K.; Turner, P.E. High-throughput discovery of phage receptors using transposon insertion sequencing of bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 18670–18679. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Wang, X.; Xu, C.; Han, T.; Guo, X. Genetic Characterization of the O-Antigen and Development of a Molecular Serotyping Scheme for Enterobacter cloacae. Front. Microbiol. 2020, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wahab, L.; Gill, J.J. Development and Validation of a Microtiter Plate-Based Assay for Determination of Bacteriophage Host Range and Virulence. Viruses 2018, 10, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, P.; Abedon, S.T. Chapter 7—Bacteriophage Host Range and Bacterial Resistance. In Advances in Applied Microbiology; Academic Press: Salt Lake City, UT, USA, 2010; Volume 70, pp. 217–248. [Google Scholar]
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [Green Version]
- Albers, S.; Czech, A. Exploiting tRNAs to Boost Virulence. Life 2016, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.; Nilsson, A.; Ackermann, H.; Lavigne, R.; Kuhn, J.; Adriaenssens, E. To Include Four (4) New Species within the Genus Phieco32virus in the Family Podoviridae; International Committee for Taxonomy of Viruses (ICTV) Proposal (Taxoprop) No. 2015.014aB; International Committee for Taxonomy of Viruses (ICTV): London, UK, 2015. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Wang, Y.; Yang, H.; Shen, W.; Chen, X. Transcription regulation mechanisms of bacteriophages. Bioengineered 2014, 5, 300–304. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Genome Size (bp) | GC Content | No. of Genes | E-Value | Identity (%) | Query Cover (%) | Accession |
|---|---|---|---|---|---|---|---|
| Escherichia phage ECBP2 | 77,315 | 42.4 | 121 | 0 | 93.88 | 83 | JX415536.1 |
| Escherichia phage EK010 | 78,078 | 42.1 | 117 | 0 | 93.09 | 89 | LC553734.1 |
| Escherichia phage vB_EcoP_EcoN5 | 76,083 | 42.1 | 128 | 0 | 91.26 | 54 | MN715356.1 |
| Escherichia phage vB_EcoP_SU10 | 77,327 | 42.1 | 125 | 0 | 89.77 | 63 | KM044272.1 |
| Escherichia phage PGN6866 | 78,549 | 42.3 | 41 | 0 | 88.69 | 63 | MT127620.1 |
| Escherichia phage MN03 | 77,187 | 42.2 | 125 | 0 | 88.40 | 57 | MT129653.1 |
| Escherichia phage O18-011 | 75,646 | 42.1 | 121 | 0 | 87.88 | 56 | LC553735.1 |
| Escherichia virus phiEco32 | 77,554 | 42.3 | 129 | 0 | 87.37 | 61 | EU330206.1 |
| Escherichia phage MN05 | 76,899 | 42.2 | 127 | 0 | 87.00 | 52 | MT129655.1 |
| Escherichia phage ES17 | 75,007 | 42.1 | 123 | 0 | 86.25 | 56 | MN508615.2 |
| Escherichia phage 172-1 | 77,266 | 42.0 | 130 | 0 | 86.20 | 62 | KP308307.1 |
| Escherichia phage NJ01 | 77,448 | 42.0 | 109 | 0 | 86.18 | 61 | JX867715.1 |
| Escherichia phage myPSH2311 | 68,712 | 42.4 | 89 | 0 | 85.67 | 45 | MG976803.1 |
| Escherichia phage LAMP | 68,521 | 42.2 | 96 | 0 | 85.56 | 54 | MG673519.1 |
| Escherichia phage vB_EcoP_WFI101126 | 77,307 | 42.1 | 136 | 0 | 85.45 | 58 | MK373770.1 |
| Escherichia phage Paul | 79,429 | 42.0 | 134 | 0 | 85.18 | 58 | MN045231.1 |
| Escherichia phage EP335 | 76,622 | 42.5 | 126 | 0 | 84.69 | 57 | MG748548.1 |
| Escherichia phage KBNP1711 | 76,184 | 42.4 | 126 | 0 | 83.28 | 61 | KF981730.1 |
| Shigella phage SGF2 | 76,964 | 42.3 | 119 | 0 | 77.85 | 61 | MN148435.1 |
| Escherichia phage myPSH1131 | 76,163 | 42.3 | 97 | 0 | 75.90 | 44 | MG983840.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koonjan, S.; Cooper, C.J.; Nilsson, A.S. Complete Genome Sequence of vB_EcoP_SU7, a Podoviridae Coliphage with the Rare C3 Morphotype. Microorganisms 2021, 9, 1576. https://doi.org/10.3390/microorganisms9081576
Koonjan S, Cooper CJ, Nilsson AS. Complete Genome Sequence of vB_EcoP_SU7, a Podoviridae Coliphage with the Rare C3 Morphotype. Microorganisms. 2021; 9(8):1576. https://doi.org/10.3390/microorganisms9081576
Chicago/Turabian StyleKoonjan, Shazeeda, Callum J. Cooper, and Anders S. Nilsson. 2021. "Complete Genome Sequence of vB_EcoP_SU7, a Podoviridae Coliphage with the Rare C3 Morphotype" Microorganisms 9, no. 8: 1576. https://doi.org/10.3390/microorganisms9081576
APA StyleKoonjan, S., Cooper, C. J., & Nilsson, A. S. (2021). Complete Genome Sequence of vB_EcoP_SU7, a Podoviridae Coliphage with the Rare C3 Morphotype. Microorganisms, 9(8), 1576. https://doi.org/10.3390/microorganisms9081576