
Comparative Genomics Revealing Insights into Niche Separation of the Genus Methylophilus


Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Isolation and DNA Extraction
2.2. Genome Sequencing and Assembly
2.3. Gene Prediction and Annotation
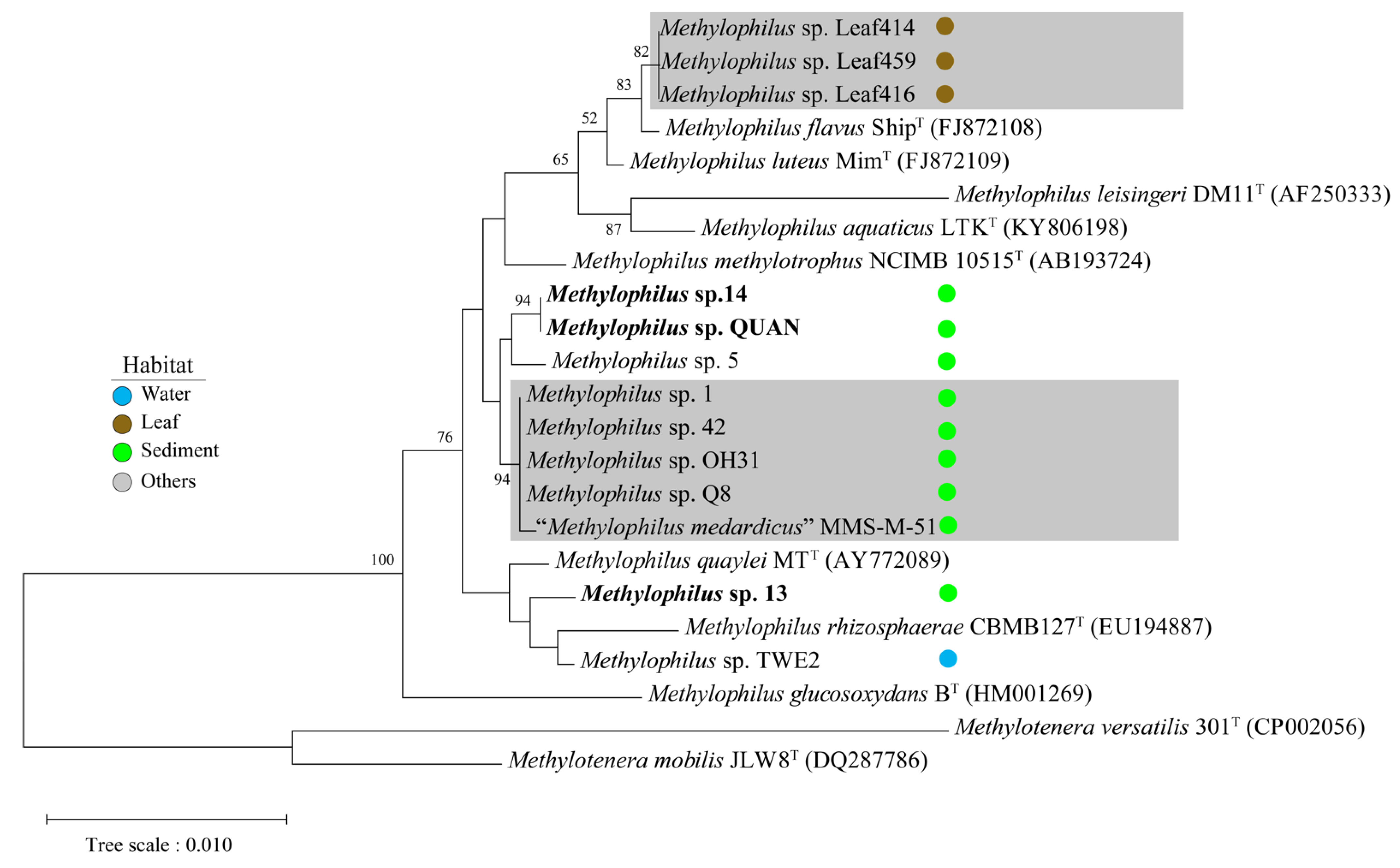
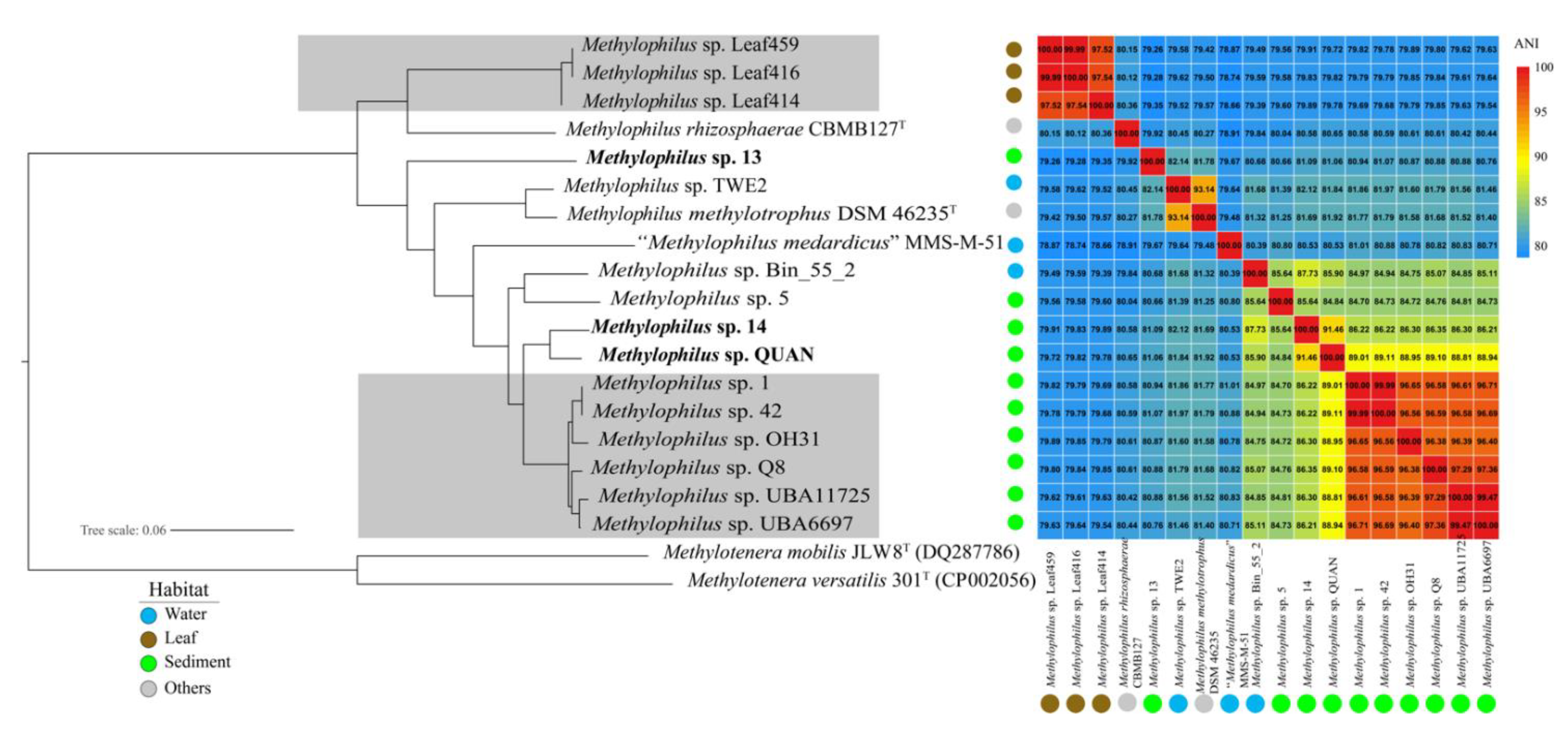
2.4. Genome Selection and Phylogenomic Analysis
2.5. Metabolic and Statistical Analysis
3. Results
3.1. General Genomic Features of the Three New Strains
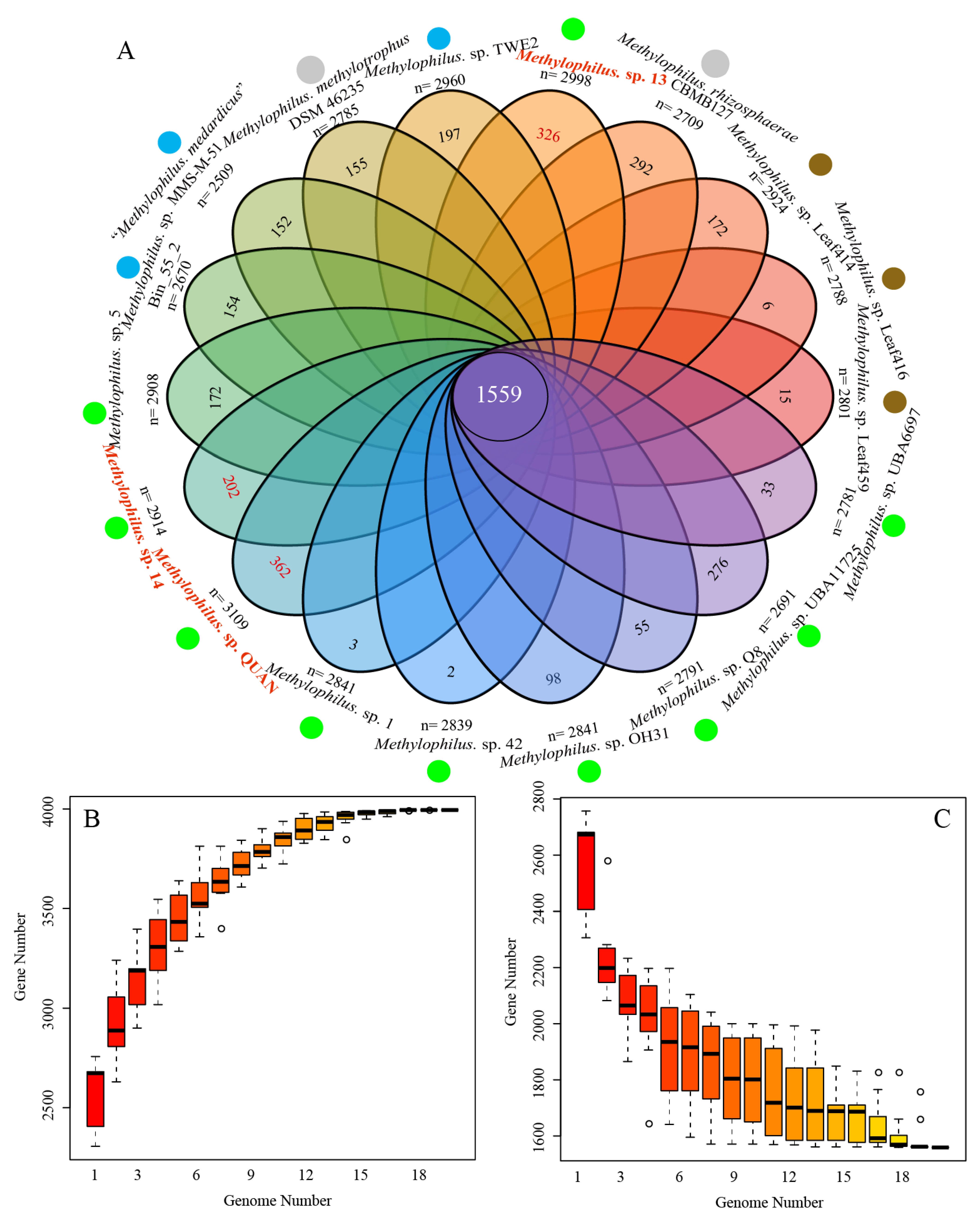
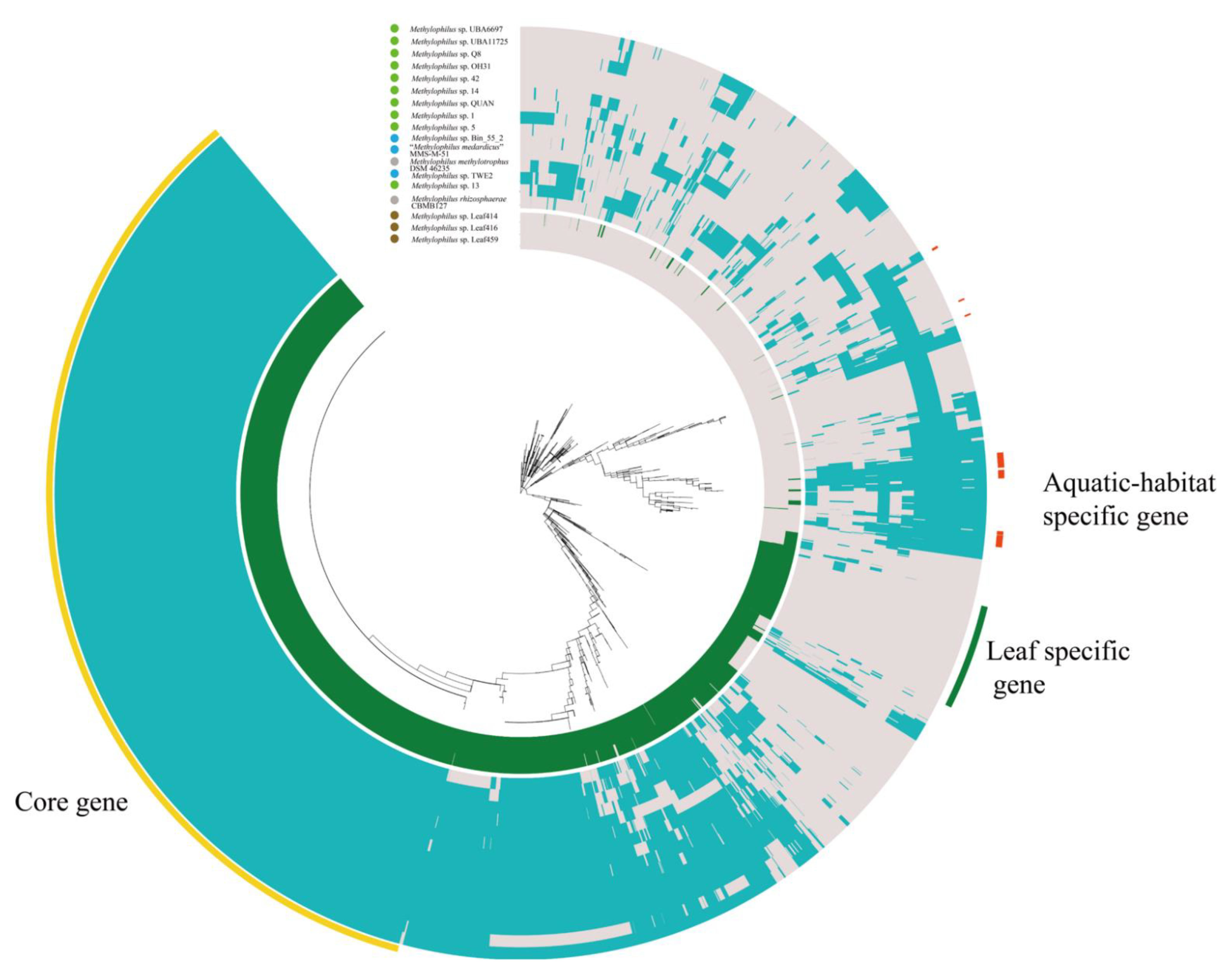
3.2. Pan-Genome and Orthology Analysis
3.3. Metabolic Analysis
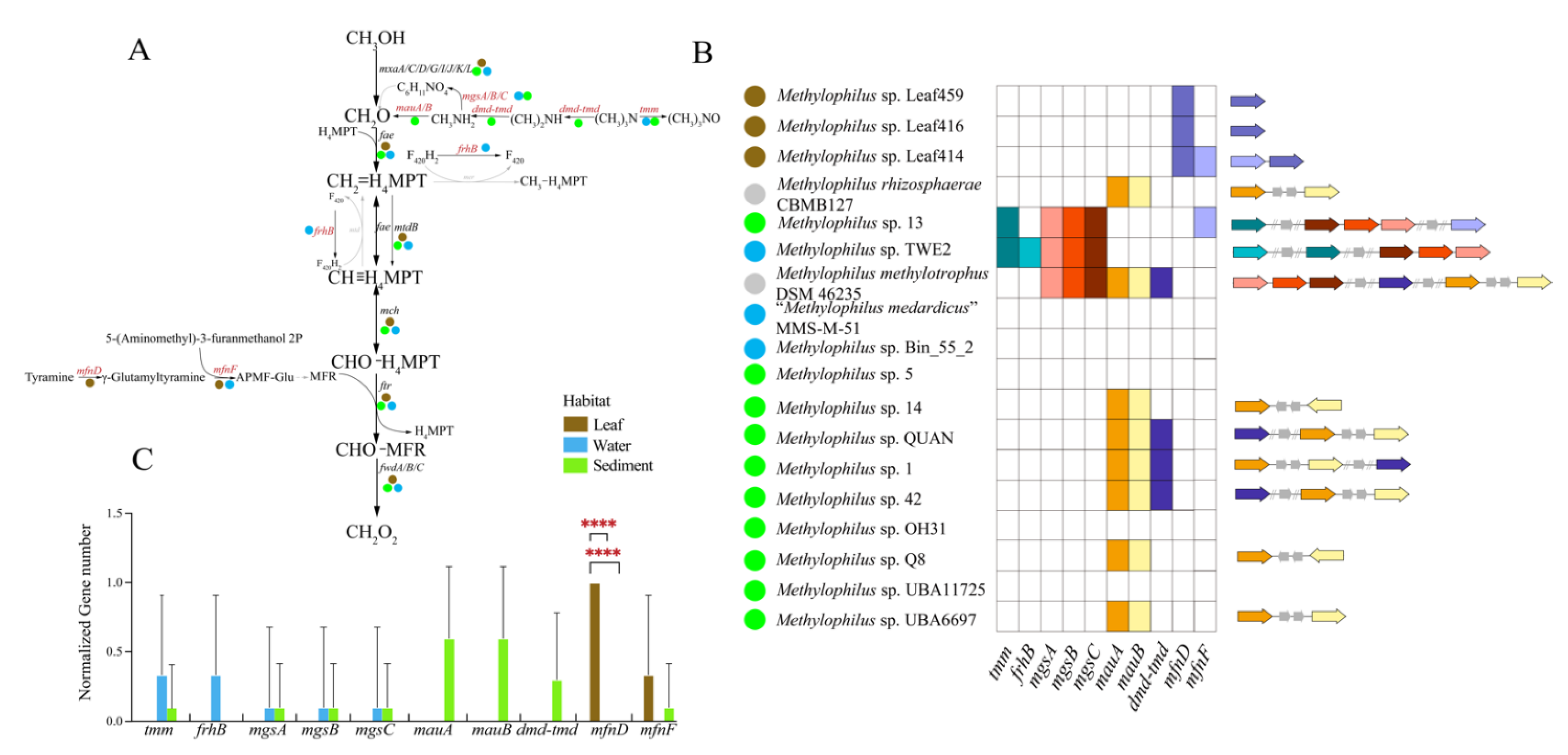
3.3.1. Methanol Metabolism
3.3.2. Carbohydrate-Active Enzymes Analysis
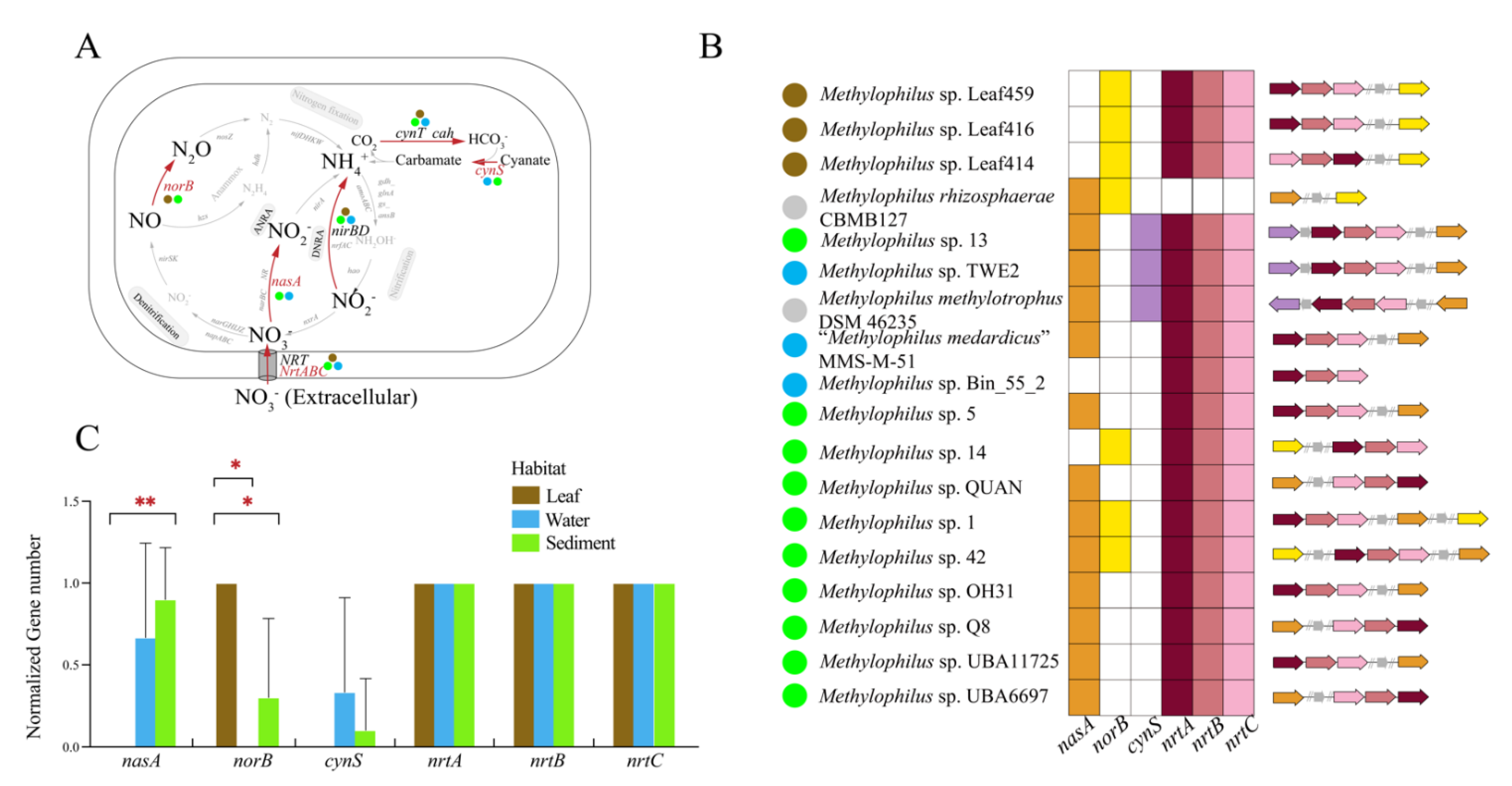
3.3.3. Nitrogen Metabolism
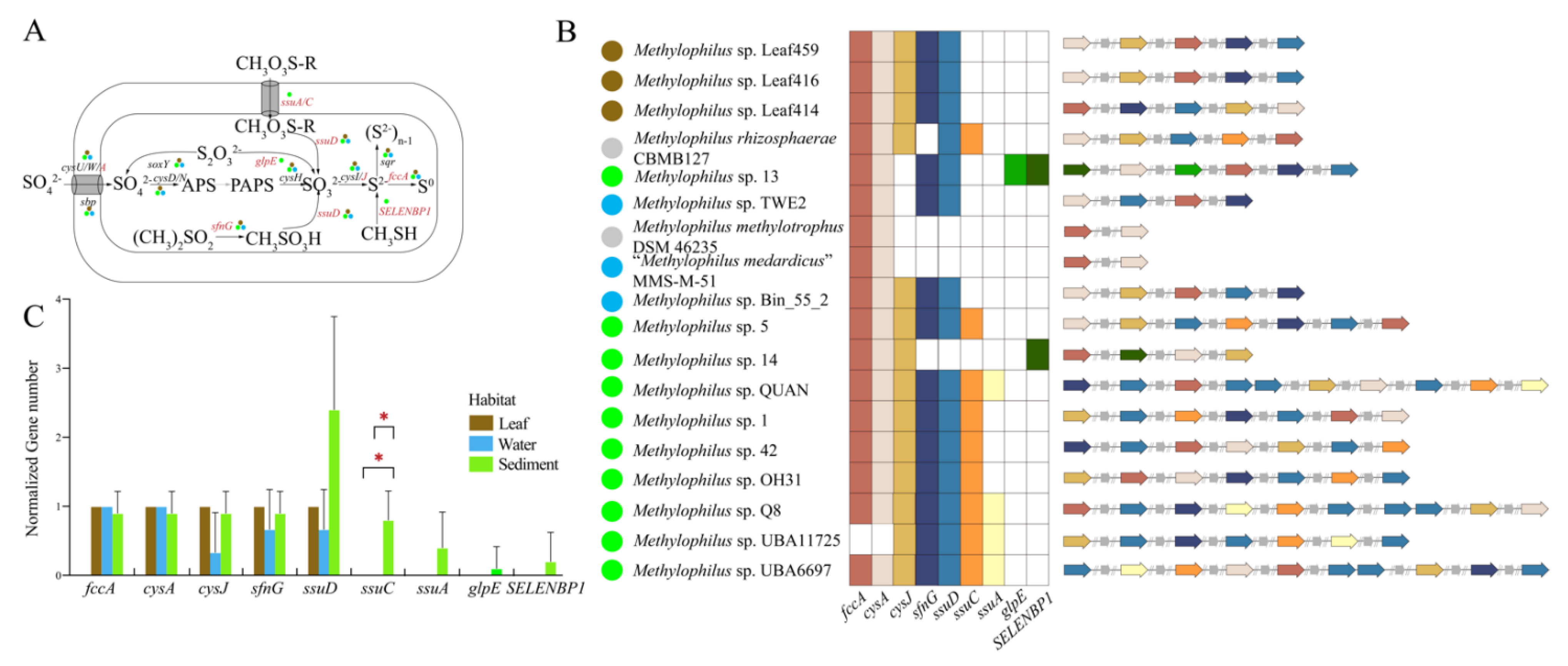
3.3.4. Sulfur Metabolism
3.3.5. Cellular Electron Transfer for Respiration
4. Discussion
4.1. Adaptation to Specific Habitats from Genomic View
4.2. The Elemental Metabolism Feature of Methylophilus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chistoserdova, L.; Kalyuzhnaya, M.G.; Lidstrom, M.E. The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 2009, 63, 477–499. [Google Scholar] [CrossRef]
- Dunfield, P.F.; Yuryev, A.; Senin, P.; Smirnova, A.V.; Stott, M.B.; Hou, S.; Ly, B.; Saw, J.H.; Zhou, Z.; Ren, Y.; et al. Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 2007, 450, 879–882. [Google Scholar] [CrossRef]
- Kim, I.T.; Lee, Y.E.; Jeong, Y.; Yoo, Y.S. A novel method to remove nitrogen from reject water in wastewater treatment plants using a methane- and methanol-dependent bacterial consortium. Water Res. 2020, 172, 115512. [Google Scholar] [CrossRef]
- Vuilleumier, S.; Ivos, N.; Dean, M.; Leisinger, T. Sequence variation in dichloromethane dehalogenases/glutathione S-transferases. Microbiology 2001, 147, 611–619. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsapekos, P.; Khoshnevisan, B.; Zhu, X.; Zha, X.; Angelidaki, I. Methane oxidising bacteria to upcycle effluent streams from anaerobic digestion of municipal biowaste. J. Environ. Manag. 2019, 251, 109590. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Lapidus, A.; Han, C.; Goodwin, L.; Saunders, L.; Brettin, T.; Tapia, R.; Gilna, P.; Lucas, S.; Richardson, P.M.; et al. Genome of Methylobacillus flagellatus, molecular basis for obligate methylotrophy, and polyphyletic origin of methylotrophy. J. Bacteriol. 2007, 189, 4020–4027. [Google Scholar] [CrossRef]
- Dai, Z.; Guo, F.; Ma, J.; Dong, W.; Zhou, J.; Jiang, M.; Zhang, W.; Xin, F. The Draft Genome Sequence of Methylophilus sp. D22, Capable of Growing Under High Concentration of Methanol. Curr. Microbiol. 2019, 76, 1520–1524. [Google Scholar] [CrossRef]
- Hirasawa, T.; Shimizu, H. Recent advances in amino acid production by microbial cells. Curr. Opin. Biotechnol. 2016, 42, 133–146. [Google Scholar] [CrossRef]
- Abalakina, E.G.; Tokmakova, I.L.; Gorshkova, N.V.; Gak, E.R.; Akhverdyan, V.Z.; Mashko, S.V.; Yomantas, Y.A. Phage Mu-driven two-plasmid system for integration of recombinant DNA in the Methylophilus methylotrophus genome. Appl. Microbiol. Biotechnol. 2008, 81, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Doronina, N.V.; Gogleva, A.A.; Trotsenko, Y.A. Methylophilusglucosoxydans sp. nov., a restricted facultative methylotroph from rice rhizosphere. Int. J. Syst. Evol. Microbiol. 2012, 62, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Anthony, C.; Zatman, L.J. The microbial oxidation of methanol. 2. The methanol-oxidizing enzyme of Pseudomonas sp. M 27. Biochem. J. 1964, 92, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Kalyuzhnaya, M.G. Current Trends in Methylotrophy. Trends Microbiol. 2018, 26, 703–714. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Madhaiyan, M.; Poonguzhali, S.; Kwon, S.W.; Sa, T.M. Methylophilus rhizosphaerae sp nov., a restricted facultative methylotroph isolated from rice rhizosphere soil. Int. J. Syst. Evol. Microbiol. 2009, 59, 2904–2908. [Google Scholar] [CrossRef]
- Jenkins, O.; Byrom, D.; Jones, D. Methylophilus—A New Genus of Methanol-Utilizing Bacteria. Int. J. Syst. Bacteriol. 1987, 37, 446–448. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.P.; Iobbi-Nivol, C.; Couillault, C.; Giordano, G.; Mejean, V. Molecular analysis of the trimethylamine N-oxide (TMAO) reductase respiratory system from a Shewanella species. J. Mol. Biol. 1998, 284, 421–433. [Google Scholar] [CrossRef]
- Lv, H.; Sahin, N.; Tani, A. Isolation and genomic characterization of Novimethylophilus kurashikiensis gen. nov. sp. nov., a new lanthanide-dependent methylotrophic species of Methylophilaceae. Environ. Microbiol. 2018, 20, 1204–1223. [Google Scholar] [CrossRef]
- He, Y.; Feng, X.; Fang, J.; Zhang, Y.; Xiao, X. Metagenome and Metatranscriptome Revealed a Highly Active and Intensive Sulfur Cycle in an Oil-Immersed Hydrothermal Chimney in Guaymas Basin. Front. Microbiol. 2015, 6, 1236. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Wang, H.; Zheng, Y.; Zhu, B.L.; Wu, X.; Zhao, F. Extracellular electron transfer of Methylophilus methylotrophs. Process Biochem. 2020, 94, 313–318. [Google Scholar] [CrossRef]
- Crundwell, F.K.; Holmes, P.R.; Fowler, T.A. How do bacteria interact with minerals? J. South. Afr. Inst. Min. Metall. 2000, 100, 399–401. [Google Scholar] [CrossRef]
- Wu, Y.D.; Liu, T.X.; Li, X.M.; Li, F.B. Exogenous Electron Shuttle-Mediated Extracellular Electron Transfer of Shewanella putrefaciens 200: Electrochemical Parameters and Thermodynamics. Environ. Sci. Technol. 2014, 48, 9306–9314. [Google Scholar] [CrossRef]
- van Staalduinen, L.M.; Jia, Z. Post-translational hydroxylation by 2OG/Fe(II)-dependent oxygenases as a novel regulatory mechanism in bacteria. Front. Microbiol. 2014, 5, 798. [Google Scholar] [CrossRef][Green Version]
- Loenarz, C.; Schofield, C.J. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 2011, 36, 7–18. [Google Scholar] [CrossRef]
- Rollefson, J.B.; Stephen, C.S.; Tien, M.; Bond, D.R. Identification of an Extracellular Polysaccharide Network Essential for Cytochrome Anchoring and Biofilm Formation in Geobacter sulfurreducens. J. Bacteriol. 2011, 193, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, B.; Chang, I.S. Tracking of Shewanella oneidensis MR-1 biofilm formation of a microbial electrochemical system via differential pulse voltammetry. Bioresour. Technol. 2018, 254, 357–361. [Google Scholar] [CrossRef]
- Jenal, U.; Reinders, A.; Lori, C. Cyclic di-GMP: Second messenger extraordinaire. Nat. Rev. Microbiol. 2017, 15, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Koga, R.; Kanaly, R.A.; Kouzuma, A.; Watanabe, K. Identification of a Diguanylate Cyclase That Facilitates Biofilm Formation on Electrodes by Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 2021, 87, e00201-21. [Google Scholar] [CrossRef] [PubMed]
- Kovacs-Simon, A.; Titball, R.W.; Michell, S.L. Lipoproteins of bacterial pathogens. Infect. Immun. 2011, 79, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Roussel-Jazede, V.; Grijpstra, J.; van Dam, V.; Tommassen, J.; van Ulsen, P. Lipidation of the autotransporter NalP of Neisseria meningitidis is required for its function in the release of cell-surface-exposed proteins. Microbiology 2013, 159, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Coutte, L.; Willery, E.; Antoine, R.; Drobecq, H.; Locht, C.; Jacob-Dubuisson, F. Surface anchoring of bacterial subtilisin important for maturation function. Mol. Microbiol. 2003, 49, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Salcher, M.M.; Schaefle, D.; Kaspar, M.; Neuenschwander, S.M.; Ghai, R. Evolution in action: Habitat transition from sediment to the pelagial leads to genome streamlining in Methylophilaceae. ISME J. 2019, 13, 2764–2777. [Google Scholar] [CrossRef]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Skovran, E.; Guo, X.; Sivam, D.; Lidstrom, M.E. Implementation of microarrays for Methylobacterium extorquens AM1. OMICS 2007, 11, 325–340. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stains | Taxonomy | Genome Assembly | Genome Size (Mb) | ScafNum | G+C Content (%) | Gene Number | 16S rRNA | tRNAs | Completeness | Contamination | Source Habitats |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Leaf459 | M. flavus | GCA_001425495.1 | 2.95 | 16 | 48.29 | 2801 | 5 | 30 | 99.57 | 0 | Arabidopsis leaf |
| Leaf416 | M. flavus | GCA_001424665.1 | 2.94 | 8 | 48.25 | 2788 | 1 | 40 | 100 | 0 | Arabidopsis leaf |
| Leaf414 | M. flavus | GCA_001425425.1 | 3.03 | 9 | 48.24 | 2924 | 1 | 44 | 100 | 0 | Arabidopsis leaf |
| CBMB127 * | M. rhizosphaerae | GCA_900100975.1 | 2.76 | 10 | 51.35 | 2709 | 1 | 41 | 100 | 0.03 | Rhizosphere soil of rice |
| 13 | M.quaylei | GCA_015354335.1 | 3.11 | 14 | 51.13 | 2998 | 1 | 41 | 100 | 0 | Lake Fuxian Sediment |
| TWE2 | M. quaylei | GCA_001183865.1 | 3.08 | 1 | 49.54 | 2960 | 2 | 40 | 100 | 0 | Tap water incubated with methane |
| DSM 46235 * | M. methylotrophus | GCA_000378225.1 | 2.86 | 14 | 49.61 | 2785 | 1 | 40 | 100 | 0 | Activated sludge |
| MMS-M-51 * | “M. medardicus” | GCA_006363915.1 | 2.60 | 1 | 49.82 | 2509 | 2 | 38 | 100 | 0 | Water column of Lake Medard, Czechia |
| Bin_55_2 | Inconclusive | GCA_008015755.1 | 2.75 | 40 | 50.49 | 2670 | 0 | 41 | 95.41 | 1.28 | Advanced Water Purification Facility |
| 5 | M. methylotrophus | GCA_000515275.1 | 3.03 | 1 | 50.25 | 2908 | 3 | 40 | 100 | 0.11 | Lake Washington Sediment |
| 14 | M. methylotrophus | GCA_015354345.1 | 3.02 | 15 | 50.48 | 2914 | 1 | 40 | 100 | 0.43 | Lake Fuxian Sediment |
| QUAN | M. methylotrophus | GCA_015354445.1 | 3.15 | 6 | 50.33 | 3109 | 1 | 42 | 100 | 0.85 | Lake Fuxian Sediment |
| 1 | M. methylotrophus | GCA_000374225.1 | 2.97 | 1 | 50.43 | 2841 | 2 | 40 | 100 | 0 | Lake Washington Sediment |
| 42 | M. methylotrophus | GCA_000384155.1 | 2.97 | 1 | 50.44 | 2839 | 1 | 44 | 100 | 0 | Lake Washington Sediment |
| OH31 | M. methylotrophus | GCA_000576615.1 | 2.93 | 16 | 50.59 | 2841 | 1 | 46 | 100 | 0 | Pond Sediment in Hokkaido, Japan |
| Q8 | M. methylotrophus | GCA_000800115.1 | 2.90 | 1 | 50.63 | 2791 | 3 | 42 | 100 | 0.03 | Lake Washington Sediment |
| UBA11725 | Inconclusive | GCA_003538435.1 | 2.22 | 303 | 50.75 | 2691 | 0 | 42 | 78.45 | 0 | Freshwater sediment |
| UBA6697 | Inconclusive | GCA_002454655.1 | 2.82 | 51 | 50.64 | 2781 | 0 | 25 | 97.93 | 0 | Costa Rican marine sediment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, N.; Tao, Y.; Gao, P.; Xu, Y.; Xing, P. Comparative Genomics Revealing Insights into Niche Separation of the Genus Methylophilus. Microorganisms 2021, 9, 1577. https://doi.org/10.3390/microorganisms9081577
Lin N, Tao Y, Gao P, Xu Y, Xing P. Comparative Genomics Revealing Insights into Niche Separation of the Genus Methylophilus. Microorganisms. 2021; 9(8):1577. https://doi.org/10.3390/microorganisms9081577
Chicago/Turabian StyleLin, Nana, Ye Tao, Peixin Gao, Yan Xu, and Peng Xing. 2021. "Comparative Genomics Revealing Insights into Niche Separation of the Genus Methylophilus" Microorganisms 9, no. 8: 1577. https://doi.org/10.3390/microorganisms9081577
APA StyleLin, N., Tao, Y., Gao, P., Xu, Y., & Xing, P. (2021). Comparative Genomics Revealing Insights into Niche Separation of the Genus Methylophilus. Microorganisms, 9(8), 1577. https://doi.org/10.3390/microorganisms9081577