
Application and Perspectives of MALDI–TOF Mass Spectrometry in Clinical Microbiology Laboratories


Abstract
1. Background
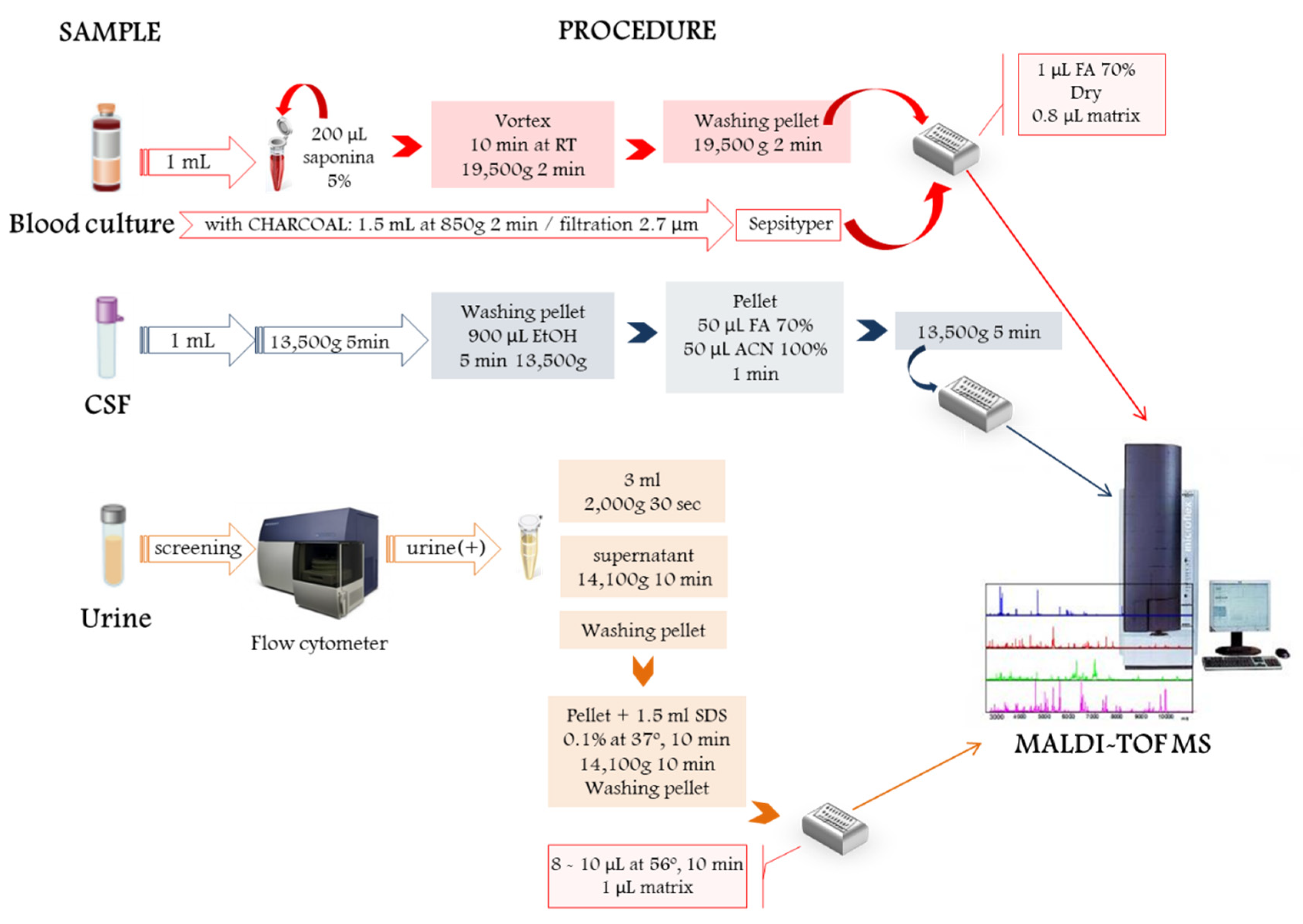
2. Direct Microbial Identification from Human Samples
3. Microbial Identification Using Reference Databases and Open Free Libraries
4. MS Big Data and Machine Learning Applied to Clinical Diagnosis
5. The State-of-the-Art Combining Approaches
6. Current Challenges: Viral Identification and Antimicrobial Resistance
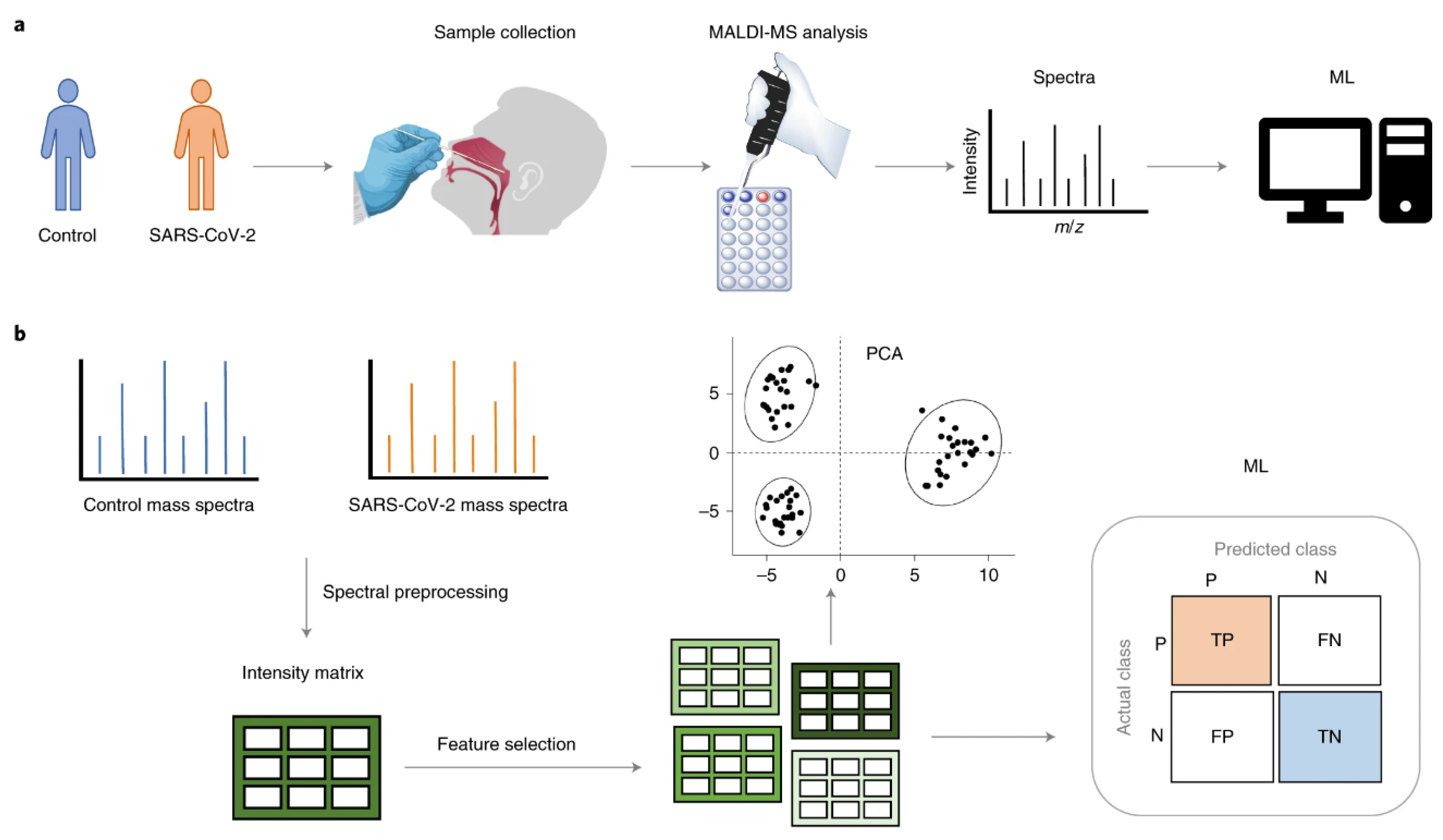
6.1. Building Virus Spectral Libraries, Getting a Fast Diagnosis of SARS-2
6.2. Identifying Mechanisms of Resistance
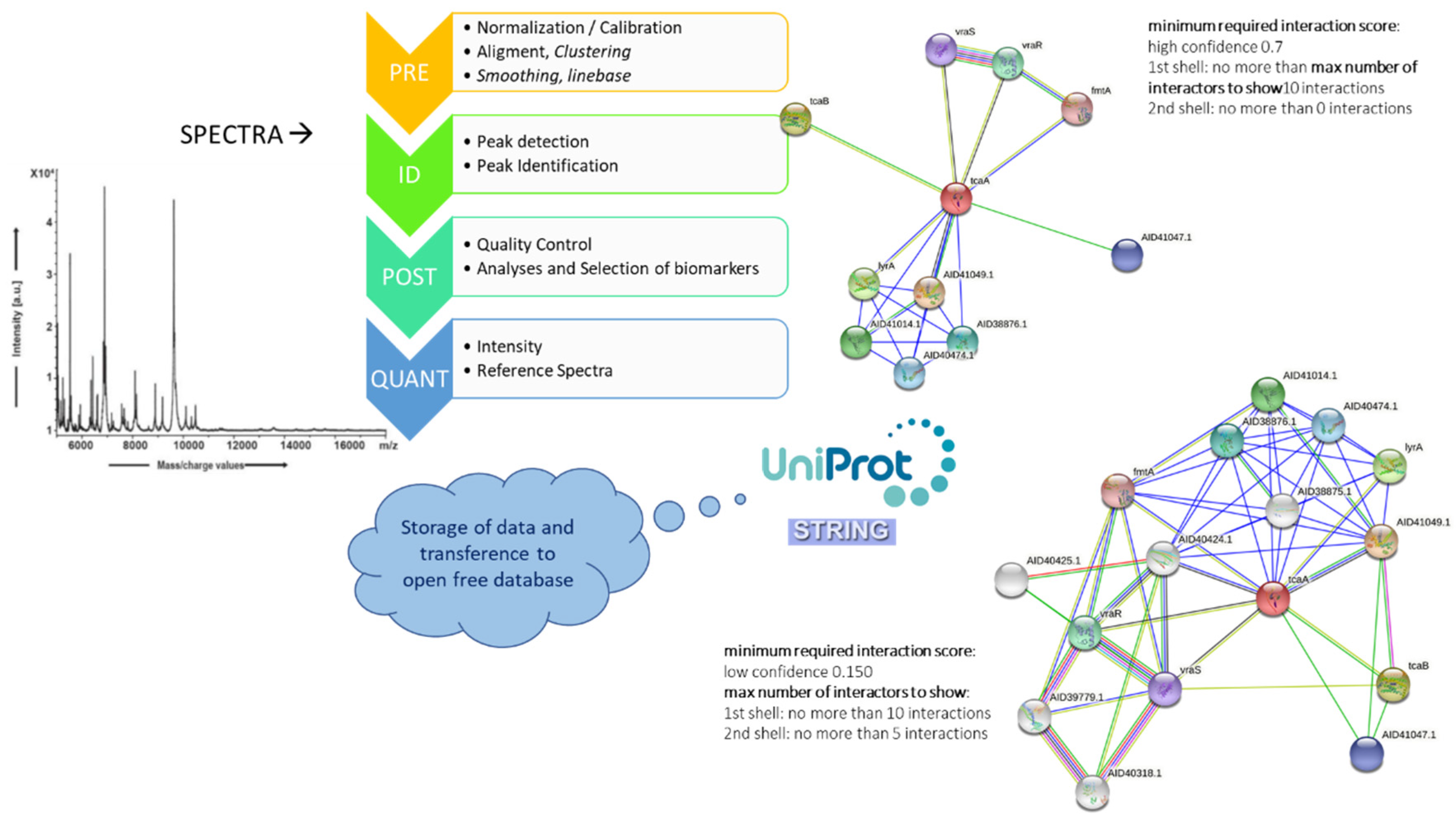
7. An MS-Systems Biology Approach: The Future of Identification
8. Conclusions
9. Websites
- BioNumerics: http://www.applied-maths.com/, accessed on 19 July 2021
- BRUKER BioTyper: https://www.bruker.com/es/service/support-upgrades/software-downloads/mass-spectrometry.html, accessed on 19 July 2021
- Institute for System Biology: https://www.systemsbiology.org/, accessed on 19 July 2021
- MATLAB: http://es.mathworks.com/products/matlab/, accessed on 19 July 2021
- Matrix Science: http://www.matrixscience.com/, accessed on 19 July 2021
- Seattle Proteome Center: http://www.proteomecenter.org/, accessed on 19 July 2021
- Swiss Institute of BioInformatics: http://www.isb-sib.ch/, accessed on 19 July 2021
- The European Bioinformatics Institute (EMBL-EBI): http://www.ebi.ac.uk/, accessed on 19 July 2021
- UniProt: http://www.uniprot.org/, accessed on 19 July 2021
- VITEK-MS: http://www.vitekms.com/, accessed on 19 July 2021
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffiths, J. A brief history of mass spectrometry. Anal. Chem. 2008, 80, 5678–5683. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Holland, R.D.; Wilkes, J.G.; Rafii, F.; Sutherland, J.B.; Persons, C.C.; Voorhees, K.J.; Lay, J.O., Jr. Rapid identification of intact whole bacteria based on spectral patterns using matrix-assisted laser desorption/ionization with time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. RCM 1996, 10, 1227–1232. [Google Scholar] [CrossRef]
- Dieckmann, R.; Helmuth, R.; Erhard, M.; Malorny, B. Rapid classification and identification of salmonellae at the species and subspecies levels by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 2008, 74, 7767–7778. [Google Scholar] [CrossRef] [PubMed]
- Valentine, N.B.; Wahl, J.H.; Kingsley, M.T.; Wahl, K.L. Direct surface analysis of fungal species by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2002, 16, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, D.R.; Schwab, K.J.; Cole, R.N.; Halden, R.U. Detection of norovirus capsid protein in authentic standards and in stool extracts by matrix-assisted laser desorption ionization and nanospray mass spectrometry. Appl. Environ. Microbiol. 2006, 72, 2749–2755. [Google Scholar] [CrossRef]
- Perera, M.R.; Vanstone, V.A.; Jones, M.G. A novel approach to identify plant parasitic nematodes using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2005, 19, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Gregorich, Z.R.; Ge, Y. Top-down proteomics in health and disease: Challenges and opportunities. Proteomics 2014, 14, 1195–1210. [Google Scholar] [CrossRef]
- Altun, O.; Botero-Kleiven, S.; Carlsson, S.; Ullberg, M.; Ozenci, V. Rapid identification of bacteria from positive blood culture bottles by MALDI-TOF MS following short-term incubation on solid media. J. Med. Microbiol. 2015, 64, 1346–1352. [Google Scholar] [CrossRef]
- Clark, A.E.; Kaleta, E.J.; Arora, A.; Wolk, D.M. Matrix-assisted laser desorption ionization-time of flight mass spectrometry: A fundamental shift in the routine practice of clinical microbiology. Clin. Microbiol. Rev. 2013, 26, 547–603. [Google Scholar] [CrossRef] [PubMed]
- Greco, V.; Piras, C.; Pieroni, L.; Ronci, M.; Putignani, L.; Roncada, P.; Urbani, A. Applications of MALDI-TOF mass spectrometry in clinical proteomics. Expert. Rev. Proteom. 2018, 15, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Abián, J.; Carrascal, M.; Gay, M. . Introducción a la Espectrometría de Masas Para la Caracterización de Péptidos y Proteínas en Proteómica; ProteoRed: Barcelona, Spain, 2008; Volume 2, p. 30. [Google Scholar]
- Fagerquist, C.K.; Garbus, B.R.; Miller, W.G.; Williams, K.E.; Yee, E.; Bates, A.H.; Boyle, S.; Harden, L.A.; Cooley, M.B.; Mandrell, R.E. Rapid identification of protein biomarkers of Escherichia coli O157:H7 by matrix-assisted laser desorption ionization-time-of-flight-time-of-flight mass spectrometry and top-down proteomics. Anal. Chem. 2010, 82, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, T.R.; Goldstein, J.E.; Schumaker, S. MALDI TOF MS profiling of bacteria at the strain level: A review. Mass Spectrom. Rev. 2013, 32, 188–217. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef]
- Cuenod, A.; Foucault, F.; Pfluger, V.; Egli, A. Factors Associated With MALDI-TOF Mass Spectral Quality of Species Identification in Clinical Routine Diagnostics. Front. Cell Infect. Microbiol. 2021, 11, 646648. [Google Scholar] [CrossRef]
- De Carolis, E.; Vella, A.; Vaccaro, L.; Torelli, R.; Spanu, T.; Fiori, B.; Posteraro, B.; Sanguinetti, M. Application of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. J. Infect. Dev. Ctries 2014, 8, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Jamal, W.Y.; Ahmad, S.; Khan, Z.U.; Rotimi, V.O. Comparative evaluation of two matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) systems for the identification of clinically significant yeasts. Int. J. Infect. Dis. 2014, 26, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Deak, E.; Charlton, C.L.; Bobenchik, A.M.; Miller, S.A.; Pollett, S.; McHardy, I.H.; Wu, M.T.; Garner, O.B. Comparison of the Vitek MS and Bruker Microflex LT MALDI-TOF MS platforms for routine identification of commonly isolated bacteria and yeast in the clinical microbiology laboratory. Diagn. Microbiol. Infect. Dis. 2015, 81, 27–33. [Google Scholar] [CrossRef]
- Lavergne, R.A.; Chauvin, P.; Valentin, A.; Fillaux, J.; Roques-Malecaze, C.; Arnaud, S.; Menard, S.; Magnaval, J.F.; Berry, A.; Cassaing, S.; et al. An extraction method of positive blood cultures for direct identification of Candida species by Vitek MS matrix-assisted laser desorption ionization time of flight mass spectrometry. Med. Mycol. 2013, 51, 652–656. [Google Scholar] [CrossRef]
- Teke, L.; Barış, A.; Bayraktar, B. Comparative evaluation of the Bruker Biotyper and Vitek MS matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) systems for non-albicans Candida and uncommon yeast isolates. J. Microbiol. Methods 2021, 185, 106232. [Google Scholar] [CrossRef]
- Ge, M.-C.; Kuo, A.-J.; Liu, K.-L.; Wen, Y.-H.; Chia, J.-H.; Chang, P.-Y.; Lee, M.-H.; Wu, T.-L.; Chang, S.-C.; Lu, J.-J. Routine identification of microorganisms by matrix-assisted laser desorption ionization time-of-flight mass spectrometry: Success rate, economic analysis, and clinical outcome. J. Microbiol. Immunol. Infect. 2017, 50, 662–668. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, S.; Umemura, H.; Nakayama, T. Current Status of Matrix-Assisted Laser Desorption/Ionization-Time-of-Flight Mass Spectrometry (MALDI-TOF MS) in Clinical Diagnostic Microbiology. Molecules 2020, 25, 4775. [Google Scholar] [CrossRef] [PubMed]
- Nomura, F.; Tsuchida, S.; Murata, S.; Satoh, M.; Matsushita, K. Mass spectrometry-based microbiological testing for blood stream infection. Clin. Proteom. 2020, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Weinert, K.; Wagner, C.; Gunzl, B.; Wieser, A.; Maier, T.; Kostrzewa, M. Novel, improved sample preparation for rapid, direct identification from positive blood cultures using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. J. Mol. Diagn. 2011, 13, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, A.; Kasinathan, V.; Hyman, J.; Walsh, J.; Drake, T.; Wang, Y.F. Rapid identification of bacteria and yeasts from positive-blood-culture bottles by using a lysis-filtration method and matrix-assisted laser desorption ionization-time of flight mass spectrum analysis with the SARAMIS database. J. Clin. Microbiol. 2013, 51, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Broyer, P.; Perrot, N.; Rostaing, H.; Blaze, J.; Pinston, F.; Gervasi, G.; Charles, M.H.; Dachaud, F.; Dachaud, J.; Moulin, F.; et al. An Automated Sample Preparation Instrument to Accelerate Positive Blood Cultures Microbial Identification by MALDI-TOF Mass Spectrometry (Vitek((R))MS). Front. Microbiol. 2018, 9, 911. [Google Scholar] [CrossRef]
- Ashizawa, K.; Murata, S.; Terada, T.; Ito, D.; Bunya, M.; Watanabe, K.; Teruuchi, Y.; Tsuchida, S.; Satoh, M.; Nishimura, M.; et al. Applications of copolymer for rapid identification of bacteria in blood culture broths using matrix-assisted laser desorption ionization time-of-flight mass spectrometry. J. Microbiol. Methods 2017, 139, 54–60. [Google Scholar] [CrossRef]
- Tsuchida, S.; Murata, S.; Miyabe, A.; Satoh, M.; Takiwaki, M.; Ashizawa, K.; Terada, T.; Ito, D.; Matsushita, K.; Nomura, F. Application of the biocopolymer preparation system, rapid BACpro® II kit, for mass-spectrometry-based bacterial identification from positive blood culture bottles by the MALDI Biotyper system. J. Microbiol. Methods 2018, 152, 86–91. [Google Scholar] [CrossRef]
- Jakovljev, A.; Bergh, K. Development of a rapid and simplified protocol for direct bacterial identification from positive blood cultures by using matrix assisted laser desorption ionization time-of- flight mass spectrometry. BMC Microbiol. 2015, 15, 258. [Google Scholar] [CrossRef]
- Chen, J.H.; Ho, P.L.; Kwan, G.S.; She, K.K.; Siu, G.K.; Cheng, V.C.; Yuen, K.Y.; Yam, W.C. Direct bacterial identification in positive blood cultures by use of two commercial matrix-assisted laser desorption ionization-time of flight mass spectrometry systems. J. Clin. Microbiol. 2013, 51, 1733–1739. [Google Scholar] [CrossRef]
- Riederer, K.; Cruz, K.; Shemes, S.; Szpunar, S.; Fishbain, J.T. MALDI-TOF identification of Gram-negative bacteria directly from blood culture bottles containing charcoal: Sepsityper(R) kits versus centrifugation-filtration method. Diagn. Microbiol. Infect. Dis. 2015, 82, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Gray, T.J.; Thomas, L.; Olma, T.; Iredell, J.R.; Chen, S.C. Rapid identification of Gram-negative organisms from blood culture bottles using a modified extraction method and MALDI-TOF mass spectrometry. Diagn. Microbiol. Infect. Dis. 2013, 77, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Idelevich, E.A.; Storck, L.M.; Sparbier, K.; Drews, O.; Kostrzewa, M.; Becker, K. Rapid Direct Susceptibility Testing from Positive Blood Cultures by the Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry-Based Direct-on-Target Microdroplet Growth Assay. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [PubMed]
- Bishop, B.; Geffen, Y.; Plaut, A.; Kassis, O.; Bitterman, R.; Paul, M.; Neuberger, A. The use of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for rapid bacterial identification in patients with smear-positive bacterial meningitis. Clin. Microbiol. Infect. 2018, 24, 171–174. [Google Scholar] [CrossRef]
- Zboromyrska, Y.; Bosch, J.; Aramburu, J.; Cuadros, J.; Garcia-Riestra, C.; Guzman-Puche, J.; Liebana Martos, C.; Loza, E.; Munoz-Algarra, M.; Ruiz de Alegria, C.; et al. A multicentre study investigating parameters which influence direct bacterial identification from urine. PLoS ONE 2018, 13, e0207822. [Google Scholar] [CrossRef]
- Ferreira, L.; Sanchez-Juanes, F.; Munoz-Bellido, J.L.; Gonzalez-Buitrago, J.M. Rapid method for direct identification of bacteria in urine and blood culture samples by matrix-assisted laser desorption ionization time-of-flight mass spectrometry: Intact cell vs. extraction method. Clin. Microbiol. Infect. 2011, 17, 1007–1012. [Google Scholar] [CrossRef]
- Demarco, M.L.; Burnham, C.A. Diafiltration MALDI-TOF mass spectrometry method for culture-independent detection and identification of pathogens directly from urine specimens. Am. J. Clin. Pathol. 2014, 141, 204–212. [Google Scholar] [CrossRef]
- Sanchez-Juanes, F.; Siller Ruiz, M.; Moreno Obregon, F.; Criado Gonzalez, M.; Hernandez Egido, S.; de Frutos Serna, M.; Gonzalez-Buitrago, J.M.; Munoz-Bellido, J.L. Pretreatment of urine samples with SDS improves direct identification of urinary tract pathogens with matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 2014, 52, 335–338. [Google Scholar] [CrossRef]
- Kim, Y.; Park, K.G.; Lee, K.; Park, Y.J. Direct Identification of Urinary Tract Pathogens From Urine Samples Using the Vitek MS System Based on Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry. Ann. Lab. Med. 2015, 35, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Veron, L.; Mailler, S.; Girard, V.; Muller, B.H.; L’Hostis, G.; Ducruix, C.; Lesenne, A.; Richez, A.; Rostaing, H.; Lanet, V.; et al. Rapid urine preparation prior to identification of uropathogens by MALDI-TOF MS. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Idelevich, E.A.; Schule, I.; Grunastel, B.; Wullenweber, J.; Peters, G.; Becker, K. Rapid identification of microorganisms from positive blood cultures by MALDI-TOF mass spectrometry subsequent to very short-term incubation on solid medium. Clin. Microbiol. Infect. 2014, 20, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Hong, S.K.; Han, G.H.; Kim, M.; Yong, D.; Lee, K. Same-Day Identification and Antimicrobial Susceptibility Testing of Bacteria in Positive Blood Culture Broths Using Short-Term Incubation on Solid Medium with the MicroFlex LT, Vitek-MS, and Vitek2 Systems. Ann. Lab. Med. 2018, 38, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Florio, W.; Cappellini, S.; Giordano, C.; Vecchione, A.; Ghelardi, E.; Lupetti, A. A new culture-based method for rapid identification of microorganisms in polymicrobial blood cultures by MALDI-TOF MS. BMC Microbiol. 2019, 19, 267. [Google Scholar] [CrossRef] [PubMed]
- Royo-Cebrecos, C.; Gudiol, C.; Ardanuy, C.; Pomares, H.; Calvo, M.; Carratalà, J. A fresh look at polymicrobial bloodstream infection in cancer patients. PLoS ONE 2017, 12, e0185768. [Google Scholar] [CrossRef]
- Scohy, A.; Noël, A.; Boeras, A.; Brassinne, L.; Laurent, T.; Rodriguez-Villalobos, H.; Verroken, A. Evaluation of the Bruker® MBT Sepsityper IVD module for the identification of polymicrobial blood cultures with MALDI-TOF MS. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2145–2152. [Google Scholar] [CrossRef]
- Faron, M.L.; Buchan, B.W.; Ledeboer, N.A. Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry for Use with Positive Blood Cultures: Methodology, Performance, and Optimization. J. Clin. Microbiol. 2017, 55, 3328–3338. [Google Scholar] [CrossRef] [PubMed]
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Croxatto, A.; Prod’hom, G.; Greub, G. Applications of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. FEMS Microbiol. Rev. 2012, 36, 380–407. [Google Scholar] [CrossRef]
- Levesque, S.; Dufresne, P.J.; Soualhine, H.; Domingo, M.C.; Bekal, S.; Lefebvre, B.; Tremblay, C. A Side by Side Comparison of Bruker Biotyper and VITEK MS: Utility of MALDI-TOF MS Technology for Microorganism Identification in a Public Health Reference Laboratory. PLoS ONE 2015, 10, e0144878. [Google Scholar] [CrossRef]
- Wilen, C.B.; McMullen, A.R.; Burnham, C.A. Comparison of Sample Preparation Methods, Instrumentation Platforms, and Contemporary Commercial Databases for Identification of Clinically Relevant Mycobacteria by Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry. J. Clin. Microbiol. 2015, 53, 2308–2315. [Google Scholar] [CrossRef]
- Brown-Elliott, B.A.; Fritsche, T.R.; Olson, B.J.; Vasireddy, S.; Vasireddy, R.; Iakhiaeva, E.; Alame, D.; Wallace, R.J.; Branda, J.A. Comparison of Two Commercial Matrix-Assisted Laser Desorption/Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS) Systems for Identification of Nontuberculous Mycobacteria. Am. J. Clin. Pathol. 2019, 152, 527–536. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, J.; Chen, R.; Hu, L.; Xia, Q.; Wu, W.; Wang, J.; Hu, F. Multicenter evaluation of three different MALDI-TOF MS systems for identification of clinically relevant filamentous fungi. Med. Mycol. 2020, 59, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Camoez, M.; Sierra, J.M.; Dominguez, M.A.; Ferrer-Navarro, M.; Vila, J.; Roca, I. Automated categorization of methicillin-resistant Staphylococcus aureus clinical isolates into different clonal complexes by MALDI-TOF mass spectrometry. Clin. Microbiol. Infect. 2015, 22, 161.e1–161.e7. [Google Scholar] [CrossRef] [PubMed]
- Deol, P.; Girard, V.; Hyman, J.; Miller, E.; Dussoulier, R.; Mailler, S.; Schrenzel, J.; Beni, A.M.; Ninet Bescher, B.; Walsh, J.; et al. Identification of Mycobacteria by VITEK® MS MatrixAssisted Laser Desorption Ionization—Time of Flight Mass Spectrometry; BioMerieux, Ed.; BioMerieux: Marcy-l’Étoile, France, 2013; Volume 1, p. 32. Available online: http://www.biomerieux-diagnostics.com/vitek-ms (accessed on 19 July 2021).
- Mirande, C.; Canard, I.; Perrot, N.; Welker, M.; Van Belkum, A.; Chatellier, S. Matrix-Assisted Laser Desorption Ionization—Time of Flight Mass Spectrometry for Rapid Antibiotic Resistance Detection; BioMerieux, Ed.; BioMerieux: Marcy-l’Étoile, France, 2013; Volume 1, p. 32. Available online: http://www.biomerieux-diagnostics.com/vitek-ms (accessed on 19 July 2021).
- Angeletti, S.; Dicuonzo, G.; Lo Presti, A.; Cella, E.; Crea, F.; Avola, A.; Vitali, M.A.; Fagioni, M.; De Florio, L. MALDI-TOF mass spectrometry and blakpc gene phylogenetic analysis of an outbreak of carbapenem-resistant K. pneumoniae strains. New Microbiol. 2015, 38, 541–550. [Google Scholar] [PubMed]
- Morgenthaler, N.G.; Kostrzewa, M. Rapid identification of pathogens in positive blood culture of patients with sepsis: Review and meta-analysis of the performance of the sepsityper kit. Int. J. Microbiol. 2015, 2015, 827416. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; She, K.K.; Wong, O.Y.; Teng, J.L.; Yam, W.C.; Lau, S.K.; Woo, P.C.; Cheng, V.C.; Yuen, K.Y. Use of MALDI Biotyper plus ClinProTools mass spectra analysis for correct identification of Streptococcus pneumoniae and Streptococcus mitis/oralis. J. Clin. Pathol. 2015, 68, 652–656. [Google Scholar] [CrossRef]
- Tadros, M.; Cabrera, A.; Matukas, L.M.; Muller, M. Evaluation of Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry and ClinPro Tools as a Rapid Tool for Typing Streptococcus pyogenes. Open Forum. Infect. Dis. 2019, 6, ofz441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ding, J.; Rao, X.; Yu, J.; Chu, M.; Ren, W.; Wang, L.; Xue, W. Analysis of methicillin-resistant Staphylococcus aureus major clonal lineages by Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS). J. Microbiol. Methods 2015, 117, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Lien, F.; Liu, T.-P.; Chen, C.-H.; Chen, C.-J.; Lu, J.-J. Application of a MALDI-TOF analysis platform (ClinProTools) for rapid and preliminary report of MRSA sequence types in Taiwan. PeerJ 2018, 6, e5784. [Google Scholar] [CrossRef]
- Shan, W.; Li, J.; Fang, Y.; Wang, X.; Gu, D.; Zhang, R. Rapid Identification of Methicillin-Resistant Staphylococcus aureus (MRSA) by the Vitek MS Saramis system. Curr. Microbiol. 2016, 72, 29–32. [Google Scholar] [CrossRef]
- Kehrmann, J.; Schoerding, A.K.; Murali, R.; Wessel, S.; Koehling, H.L.; Mosel, F.; Buer, J. Performance of Vitek MS in identifying nontuberculous mycobacteria from MGIT liquid medium and Lowenstein-Jensen solid medium. Diagn. Microbiol. Infect. Dis. 2016, 84, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.R.; Harris, R.M. Mass Spectrometry and Microbial Diagnostics in the Clinical Laboratory. Clin. Lab. Med. 2021, 41, 285–295. [Google Scholar] [CrossRef]
- Dortet, L.; Bonnin, R.A.; Pennisi, I.; Gauthier, L.; Jousset, A.B.; Dabos, L.; Furniss, R.C.D.; Mavridou, D.A.I.; Bogaerts, P.; Glupczynski, Y.; et al. Rapid detection and discrimination of chromosome- and MCR-plasmid-mediated resistance to polymyxins by MALDI-TOF MS in Escherichia coli: The MALDIxin test. J. Antimicrob. Chemother. 2018, 73, 3359–3367. [Google Scholar] [CrossRef]
- Dortet, L.; Potron, A.; Bonnin, R.A.; Plesiat, P.; Naas, T.; Filloux, A.; Larrouy-Maumus, G. Rapid detection of colistin resistance in Acinetobacter baumannii using MALDI-TOF-based lipidomics on intact bacteria. Sci. Rep. 2018, 8, 16910. [Google Scholar] [CrossRef]
- Ryzhov, V.; Hathout, Y.; Fenselau, C. Rapid characterization of spores of Bacillus cereus group bacteria by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry. Appl. Environ. Microbiol. 2000, 66, 3828–3834. [Google Scholar] [CrossRef]
- Giebel, R.A.; Fredenberg, W.; Sandrin, T.R. Characterization of environmental isolates of Enterococcus spp. by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Water Res. 2008, 42, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Valcárcel Cases, M.; Gómez Hens, A. Hibridación Instrumental en Técnicas Analíticas de Separación; Editorial Reverté, S.A.: Barcelona, Spain, 2003; Volume 1, p. 725. [Google Scholar]
- Perez-Riverol, Y.; Xu, Q.W.; Wang, R.; Uszkoreit, J.; Griss, J.; Sanchez, A.; Reisinger, F.; Csordas, A.; Ternent, T.; Del-Toro, N.; et al. PRIDE Inspector Toolsuite: Moving Toward a Universal Visualization Tool for Proteomics Data Standard Formats and Quality Assessment of ProteomeXchange Datasets. Mol. Cell. Proteom. MCP 2016, 15, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Uszkoreit, J.; Plohnke, N.; Rexroth, S.; Marcus, K.; Eisenacher, M. The bacterial proteogenomic pipeline. BMC Genom. 2014, 15 (Suppl. 9), S19. [Google Scholar] [CrossRef]
- Chong, Y.K.; Ho, C.C.; Leung, S.Y.; Lau, S.K.P.; Woo, P.C.Y. Clinical Mass Spectrometry in the Bioinformatics Era: A Hitchhiker’s Guide. Comput. Struct. Biotechnol. J. 2018, 16, 316–334. [Google Scholar] [CrossRef]
- Mantini, D.; Petrucci, F.; Pieragostino, D.; Del Boccio, P.; Sacchetta, P.; Candiano, G.; Ghiggeri, G.M.; Lugaresi, A.; Federici, G.; Di Ilio, C.; et al. A computational platform for MALDI-TOF mass spectrometry data: Application to serum and plasma samples. J. Proteom. 2010, 73, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Nahnsen, S.; Bielow, C.; Reinert, K.; Kohlbacher, O. Tools for label-free peptide quantification. Mol. Cell. Proteom. MCP 2013, 12, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Niyompanich, S.; Srisanga, K.; Jaresitthikunchai, J.; Roytrakul, S.; Tungpradabkul, S. Utilization of Whole-Cell MALDI-TOF Mass Spectrometry to Differentiate Burkholderia pseudomallei Wild-Type and Constructed Mutants. PLoS ONE 2015, 10, e0144128. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr. Metab. 2013, 1, 92–107. [Google Scholar] [CrossRef]
- Santos, T.; Capelo, J.L.; Santos, H.M.; Oliveira, I.; Marinho, C.; Goncalves, A.; Araujo, J.E.; Poeta, P.; Igrejas, G. Use of MALDI-TOF mass spectrometry fingerprinting to characterize Enterococcus spp. and Escherichia coli isolates. J. Proteom. 2015, 127, 321–331. [Google Scholar] [CrossRef]
- Landgrebe, J.; Wurst, W.; Welzl, G. Permutation-validated principal components analysis of microarray data. Genome Biol. 2002, 3, research0019.1. [Google Scholar] [CrossRef] [PubMed]
- Werth, M.T.; Halouska, S.; Shortridge, M.D.; Zhang, B.; Powers, R. Analysis of metabolomic PCA data using tree diagrams. Anal. Biochem. 2010, 399, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Gibb, S.; Strimmer, K. MALDIquant: A versatile R package for the analysis of mass spectrometry data. Bioinformatics 2012, 28, 2270–2271. [Google Scholar] [CrossRef]
- Lopez-Fernandez, H.; Santos, H.M.; Capelo, J.L.; Fdez-Riverola, F.; Glez-Pena, D.; Reboiro-Jato, M. Mass-Up: An all-in-one open software application for MALDI-TOF mass spectrometry knowledge discovery. BMC Bioinform. 2015, 16, 318. [Google Scholar] [CrossRef]
- Montoro Bustos, A.R.; Ruiz Encinar, J.; Sanz-Medel, A. Mass spectrometry for the characterisation of nanoparticles. Anal. Bioanal. Chem. 2013, 405, 5637–5643. [Google Scholar] [CrossRef] [PubMed]
- Torres-Sangiao, E.; Holban, A.M.; Gestal, M.C. Advanced Nanobiomaterials: Vaccines, Diagnosis and Treatment of Infectious Diseases. Molecules 2016, 21, 867. [Google Scholar] [CrossRef]
- Torres Sangiao, E.; Holban, A.M.; Gestal, M.C. Applications of Nanodiamonds in the Detection and Therapy of Infectious Diseases. Materials 2019, 12, 1639. [Google Scholar] [CrossRef]
- Yang, Y.; Long, C.L.; Li, H.P.; Wang, Q.; Yang, Z.G. Analysis of silver and gold nanoparticles in environmental water using single particle-inductively coupled plasma-mass spectrometry. Sci. Total Environ. 2016, 563, 996–1007. [Google Scholar] [CrossRef]
- Lopez-Cortes, R.; Formigo, J.; Reboiro-Jato, M.; Fdez-Riverola, F.; Blanco, F.J.; Lodeiro, C.; Oliveira, E.; Capelo, J.L.; Santos, H.M. A methodological approach based on gold-nanoparticles followed by matrix assisted laser desorption ionization time of flight mass spectrometry for the analysis of urine profiling of knee osteoarthritis. Talanta 2016, 150, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Shrivas, K.; Wu, H.F. Applications of silver nanoparticles capped with different functional groups as the matrix and affinity probes in surface-assisted laser desorption/ionization time-of-flight and atmospheric pressure matrix-assisted laser desorption/ionization ion trap mass spectrometry for rapid analysis of sulfur drugs and biothiols in human urine. Rapid Commun. Mass Spectrom. RCM 2008, 22, 2863–2872. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Hsu, W.; Wang, C.H.; Chen, Y.J.; Fang, J.M. Rapid and specific influenza virus detection by functionalized magnetic nanoparticles and mass spectrometry. J. Nanobiotechnology 2011, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Magro, M.; Terzo, M.; Zaccarin, M.; Da Dalt, L.; Bonaiuto, E.; Baratella, D.; Gabai, G.; Vianello, F. Protein corona as a proteome fingerprint: The example of hidden biomarkers for cow mastitis. Colloids Surf. B Biointerfaces 2016, 140, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Trevisan, J.; Bassan, P.; Bhargava, R.; Butler, H.J.; Dorling, K.M.; Fielden, P.R.; Fogarty, S.W.; Fullwood, N.J.; Heys, K.A.; et al. Using Fourier transform IR spectroscopy to analyze biological materials. Nat. Protoc. 2014, 9, 1771–1791. [Google Scholar] [CrossRef]
- Vatanshenassan, M.; Boekhout, T.; Mauder, N.; Robert, V.; Maier, T.; Meis, J.F.; Berman, J.; Then, E.; Kostrzewa, M.; Hagen, F. Evaluation of Microsatellite Typing, ITS Sequencing, AFLP Fingerprinting, MALDI-TOF MS, and Fourier-Transform Infrared Spectroscopy Analysis of Candida auris. J. Fungi 2020, 6, 146. [Google Scholar] [CrossRef]
- Naumann, D.; Helm, D.; Labischinski, H. Microbiological characterizations by FT-IR spectroscopy. Nature 1991, 351, 81–82. [Google Scholar] [CrossRef]
- Martak, D.; Valot, B.; Sauget, M.; Cholley, P.; Thouverez, M.; Bertrand, X.; Hocquet, D. Fourier-Transform InfraRed Spectroscopy Can Quickly Type Gram-Negative Bacilli Responsible for Hospital Outbreaks. Front. Microbiol. 2019, 10, 1440. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, I.; Sebastian, K.; Mauder, N.; Kostrzewa, M.; Burckhardt, F.; Zimmermann, S. Analysis of Streptococcus pneumoniae using Fourier-transformed infrared spectroscopy allows prediction of capsular serotype. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1883–1890. [Google Scholar] [CrossRef]
- Cordovana, M.; Mauder, N.; Kostrzewa, M.; Wille, A.; Rojak, S.; Hagen, R.M.; Ambretti, S.; Pongolini, S.; Soliani, L.; Justesen, U.S.; et al. Classification of Salmonella enterica of the (Para-)Typhoid Fever Group by Fourier-Transform Infrared (FTIR) Spectroscopy. Microorganisms 2021, 9, 853. [Google Scholar] [CrossRef]
- Alexandrov, T. MALDI imaging mass spectrometry: Statistical data analysis and current computational challenges. BMC Bioinform. 2012, 13 (Suppl. 16), S11. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, R.M.; Farmer, T.B.; Gile, J. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal. Chem. 1997, 69, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Attia, A.S.; Schroeder, K.A.; Seeley, E.H.; Wilson, K.J.; Hammer, N.D.; Colvin, D.C.; Manier, M.L.; Nicklay, J.J.; Rose, K.L.; Gore, J.C.; et al. Monitoring the inflammatory response to infection through the integration of MALDI IMS and MRI. Cell Host Microbe 2012, 11, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Watrous, J.D.; Dorrestein, P.C. Imaging mass spectrometry in microbiology. Nat. Rev. Microbiol. 2011, 9, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Chaurand, P.; Cornett, D.S.; Angel, P.M.; Caprioli, R.M. From whole-body sections down to cellular level, multiscale imaging of phospholipids by MALDI mass spectrometry. Mol. Cell. Proteom. MCP 2011, 10, S1–S11. [Google Scholar] [CrossRef]
- Schwamborn, K.; Krieg, R.C.; Uhlig, S.; Ikenberg, H.; Wellmann, A. MALDI imaging as a specific diagnostic tool for routine cervical cytology specimens. Int. J. Mol. Med. 2011, 27, 417–421. [Google Scholar] [CrossRef][Green Version]
- Yi, X.; Li, J.; Yu, S.; Zhang, A.; Xu, J.; Yi, J.; Zou, J.; Nie, X.; Huang, J.; Wang, J. A new PCR-based mass spectrometry system for high-risk HPV, part I: Methods. Am. J. Clin. Pathol. 2011, 136, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Cricca, M.; Marasco, E.; Alessandrini, F.; Fazio, C.; Prossomariti, A.; Savini, C.; Venturoli, S.; Chieco, P.; De Carolis, S.; Bonafe, M.; et al. High-throughput genotyping of high-risk Human Papillomavirus by MALDI-TOF Mass Spectrometry-based method. New Microbiol. 2015, 38, 211–223. [Google Scholar]
- Calderaro, A.; Arcangeletti, M.C.; Rodighiero, I.; Buttrini, M.; Gorrini, C.; Motta, F.; Germini, D.; Medici, M.C.; Chezzi, C.; De Conto, F. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry applied to virus identification. Sci. Rep. 2014, 4, 6803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xiao, Y.; Du, J.; Ren, L.; Wang, J.; Peng, J.; Jin, Q. Application of Multiplex PCR Coupled with Matrix-Assisted Laser Desorption Ionization-Time of Flight Analysis for Simultaneous Detection of 21 Common Respiratory Viruses. J. Clin. Microbiol. 2015, 53, 2549–2554. [Google Scholar] [CrossRef]
- Downard, K.M. Proteotyping for the rapid identification of influenza virus and other biopathogens. Chem. Soc. Rev. 2013, 42, 8584–8595. [Google Scholar] [CrossRef] [PubMed]
- Zurcher, S.; Mooser, C.; Luthi, A.U.; Muhlemann, K.; Barbani, M.T.; Mohacsi, P.; Garzoni, C.; Gorgievski-Hrisoho, M.; Schaller, A.; Flatz, L. Sensitive and rapid detection of ganciclovir resistance by PCR based MALDI-TOF analysis. J. Clin. Virol. 2012, 54, 359–363. [Google Scholar] [CrossRef]
- Nachtigall, F.M.; Pereira, A.; Trofymchuk, O.S.; Santos, L.S. Detection of SARS-CoV-2 in nasal swabs using MALDI-MS. Nat. Biotechnol. 2020, 38, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, A.; De Conto, F.; Buttrini, M.; Piccolo, G.; Montecchini, S.; Maccari, C.; Martinelli, M.; Di Maio, A.; Ferraglia, F.; Pinardi, F.; et al. Human respiratory viruses, including SARS-CoV-2, circulating in the winter season 2019–2020 in Parma, Northern Italy. Int. J. Infect. Dis. 2021, 102, 79–84. [Google Scholar] [CrossRef]
- Milewska, A.; Ner-Kluza, J.; Dabrowska, A.; Bodzon-Kulakowska, A.; Pyrc, K.; Suder, P. MASS SPECTROMETRY IN VIROLOGICAL SCIENCES. Mass Spectrom. Rev. 2020, 39, 499–522. [Google Scholar] [CrossRef]
- Calderaro, A.; Arcangeletti, M.C.; Rodighiero, I.; Buttrini, M.; Montecchini, S.; Vasile Simone, R.; Medici, M.C.; Chezzi, C.; De Conto, F. Identification of different respiratory viruses, after a cell culture step, by matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS). Sci. Rep. 2016, 6, 36082. [Google Scholar] [CrossRef]
- Rybicka, M.; Miłosz, E.; Bielawski, K.P. Superiority of MALDI-TOF Mass Spectrometry over Real-Time PCR for SARS-CoV-2 RNA Detection. Viruses 2021, 13, 730. [Google Scholar] [CrossRef]
- Hernandez, M.M.; Banu, R.; Shrestha, P.; Patel, A.; Chen, F.; Cao, L.; Fabre, S.; Tan, J.; Lopez, H.; Chiu, N.; et al. RT-PCR/MALDI-TOF mass spectrometry-based detection of SARS-CoV-2 in saliva specimens. J. Med. Virol. 2021, 93, 5481–5486. [Google Scholar] [CrossRef]
- Iles, R.K.; Zmuidinaite, R.; Iles, J.K.; Carnell, G.; Sampson, A.; Heeney, J.L. Development of a Clinical MALDI-ToF Mass Spectrometry Assay for SARS-CoV-2: Rational Design and Multi-Disciplinary Team Work. Diagnostics 2020, 10, 746. [Google Scholar] [CrossRef] [PubMed]
- Cartelle Gestal, M.; Dedloff, M.R.; Torres-Sangiao, E. Computational Health Engineering Applied to Model Infectious Diseases and Antimicrobial Resistance Spread. Appl. Sci. 2019, 9, 2486. [Google Scholar] [CrossRef]
- Tran, N.K.; Howard, T.; Walsh, R.; Pepper, J.; Loegering, J.; Phinney, B.; Salemi, M.R.; Rashidi, H.H. Novel application of automated machine learning with MALDI-TOF-MS for rapid high-throughput screening of COVID-19: A proof of concept. Sci. Rep. 2021, 11, 8219. [Google Scholar] [CrossRef] [PubMed]
- Hrabak, J.; Chudackova, E.; Walkova, R. Matrix-assisted laser desorption ionization-time of flight (maldi-tof) mass spectrometry for detection of antibiotic resistance mechanisms: From research to routine diagnosis. Clin. Microbiol. Rev. 2013, 26, 103–114. [Google Scholar] [CrossRef]
- Machen, A.; Drake, T.; Wang, Y.F. Same day identification and full panel antimicrobial susceptibility testing of bacteria from positive blood culture bottles made possible by a combined lysis-filtration method with MALDI-TOF VITEK mass spectrometry and the VITEK2 system. PLoS ONE 2014, 9, e87870. [Google Scholar] [CrossRef] [PubMed]
- Nix, I.D.; Idelevich, E.A.; Storck, L.M.; Sparbier, K.; Drews, O.; Kostrzewa, M.; Becker, K. Detection of Methicillin Resistance in Staphylococcus aureus From Agar Cultures and Directly From Positive Blood Cultures Using MALDI-TOF Mass Spectrometry-Based Direct-on-Target Microdroplet Growth Assay. Front. Microbiol. 2020, 11, 232. [Google Scholar] [CrossRef]
- Correa-Martinez, C.L.; Idelevich, E.A.; Sparbier, K.; Kuczius, T.; Kostrzewa, M.; Becker, K. Development of a MALDI-TOF MS-based screening panel for accelerated differential detection of carbapenemases in Enterobacterales using the direct-on-target microdroplet growth assay. Sci. Rep. 2020, 10, 4988. [Google Scholar] [CrossRef]
- Correa-Martinez, C.L.; Idelevich, E.A.; Sparbier, K.; Kostrzewa, M.; Becker, K. Rapid Detection of Extended-Spectrum beta-Lactamases (ESBL) and AmpC beta-Lactamases in Enterobacterales: Development of a Screening Panel Using the MALDI-TOF MS-Based Direct-on-Target Microdroplet Growth Assay. Front. Microbiol. 2019, 10, 13. [Google Scholar] [CrossRef]
- Idelevich, E.A.; Becker, K. MALDI-TOF mass spectrometry for antimicrobial susceptibility testing. J. Clin. Microbiol. 2021, JCM0181419. [Google Scholar] [CrossRef]
- Boyle, J.; Rovira, H.; Cavnor, C.; Burdick, D.; Killcoyne, S.; Shmulevich, I. Adaptable data management for systems biology investigations. BMC Bioinform. 2009, 10, 79. [Google Scholar] [CrossRef]
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554. [Google Scholar] [CrossRef]
- Kshirsagar, M.; Carbonell, J.; Klein-Seetharaman, J. Multitask learning for host-pathogen protein interactions. Bioinformatics 2013, 29, i217–i226. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Fuchs, S.; Zuhlke, D.; Pane-Farre, J.; Kusch, H.; Wolf, C.; Reiss, S.; Binh, L.T.N.; Albrecht, D.; Riedel, K.; Hecker, M.; et al. Aureolib—A proteome signature library: Towards an understanding of staphylococcus aureus pathophysiology. PLoS ONE 2013, 8, e70669. [Google Scholar] [CrossRef]
- Oughtred, R.; Chatr-Aryamontri, A.; Breitkreutz, B.J.; Chang, C.S.; Rust, J.M.; Theesfeld, C.L.; Heinicke, S.; Breitkreutz, A.; Chen, D.; Hirschman, J.; et al. BioGRID: A Resource for Studying Biological Interactions in Yeast. Cold Spring Harb. Protoc. 2016, 2016, pdb-top080754. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Desiere, F.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.I.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.N.; Aebersold, R. The PeptideAtlas project. Nucleic Acids Res. 2006, 34, D655–D658. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [PubMed]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.I.; Kim, J.W.; Kang, G.S.; Kim, H.S.; Yoo, J.S.; Lee, Y.S. Prevalence of amino acid changes in the yvqF, vraSR, graSR, and tcaRAB genes from vancomycin intermediate resistant Staphylococcus aureus. J. Microbiol. 2013, 51, 160–165. [Google Scholar] [CrossRef]
- McCallum, N.; Meier, P.S.; Heusser, R.; Berger-Bachi, B. Mutational analyses of open reading frames within the vraSR operon and their roles in the cell wall stress response of Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Gardete, S.; Tomasz, A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J. Clin. Investig. 2014, 124, 2836–2840. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Statistical Analysis | Spectra Analysis | Accepted Formats | URL | Reference |
|---|---|---|---|---|---|
| BioNumerics | ANOVA, MANOVA, PCA, MDS, SOM and other statistical, parametric and non-parametric tests. Dendograms, cluster analysis, bioclustering, generation of phylogenetic trees. QA/QC. | Creation, identification and classification (spectrum libraries). Pre-processing: optimization, normalization, alignment, subtraction, smoothing. Peak detection, identification and quantification. | mzML, *.btmsp, *.txt RAW | https://www.applied-maths.com/applications/maldi-tof-bacterial-identification (accessed on 19 July 2021) | Bionumerics™ software (Applied Maths BVBA, Sint-Martens-Lantem, Belgium). |
| MaldiQUANT | Computational framework in R language: statistical analysis, dendograms, clustering, probability distributions, quality control, etc. | Pre-processing: optimization, normalization, alignment, subtraction, smoothing. Peak detection, identification, and quantification. | mzML, mzXLM, imzML *.csv, *.fid, *.tab | http://strimmerlab.org/software/maldiquant/ (accessed on 19 July 2021) | Gibb S and Strimmer [82] |
| Mass-Up | PCA, classification analysis, biomarker discovery, clustering and bioclustering. QA/QC. | Preprocessing: intensity transformation, optimization, alignment, subtraction, smoothing and peak analysis. Peak detection and identification. | mzML, mzXLM, *.csv, *.muc | http://www.sing-group.org/mass-up/ (accessed on 19 July 2021) | López-Fernandez et al. [83] |
| MATLAB | Regression, ANOVA, PCA, multivariate analysis, probability distributions, cluster analysis. | Pre-processing: optimization, smoothing, alignment, signal statistics, peak analysis, envelope extraction. Spectral analysis. | *.txt, *.xls, *.xlsx | http://es.mathworks.com/products/matlab-online/ (accessed on 19 July 2021) | MATLAB® software (MathWorks Inc., Natick, MA, USA) |
| PEAKS | Algorithms and support for analysis. | Pre-processing: optimization, normalization, alignment, subtraction, smoothing, peak analysis. Peak detection, identification and quantification. Sequence editor. | mzML, mzXLM, mzDATA, MGF, ASCII | http://www.bioinfor.com/ (accessed on 19 July 2021) | Peaks® software (Bioinformatics Solutions Inc., Waterloo, ON, Canada) |
| Platform | Database | What Is | Applications | URL | Reference |
|---|---|---|---|---|---|
| AureoLib/Aurewiki | Staphylococcus. aureus | AureoLib is a library to provide easy and intuitive access to protein synthesis data derived from various proteomics experiments | Aureolib provides protein synthesis data derived from various proteomic experiments (adaptation processes); Aurewiki provides functional and expression data (pangenome). | http://www.aureolib.de (accessed on 19 July 2021) | Fuchs et al. [129] |
| BioGrid | Multiplex | The Biological General Repository for Interaction Datasets (BioGRID) is a public database focused on specific biological processes with disease relevance. | Repository of genetic and protein interactions, chemical associations, and post-translational modifications, from model organisms and humans. | http://thebiogrid.org/ (accessed on 19 July 2021) | Oughtred et al. [130,131] |
| Cytoscape | Multiplex | Cytoscape is an open source software platform | Visualization of the molecular interaction networks and biological pathways, and integration of these networks with annotations, gene expression profiles and other data. | http://www.cytoscape.org/index.html (accessed on 19 July 2021) | Shannon et al. [132], Smoot ME et al. [133] |
| PeptideShaker | Multiplex | PeptideShaker is a search engine platform for the identification of proteins from multiple searches and de novo engines. | Protein identification with functional data. | http://compomics.github.io/projects/peptide-shaker.html (accessed on 19 July 2021) | Vaudel et al. [134] |
| PeptideAtlas | Multiplex | PeptideAtlas is a multi-organism, publicly accessible compendium of peptides identified in a large set of tandem MS proteomics experiments. | Protein identification, full annotation, database, data repository, peptides sequence, mapping and storing among others | http://www.peptideatlas.org/ (accessed on 19 July 2021) | Desiere et al. [135] |
| REACTOME | Multiplex | REACTOME is an open-access, manually curated and peer-reviewed pathway database. | Visualization, interpretation and analysis of pathways and interactions in the human biological system. | http://www.reactome.org/ (accessed on 19 July 2021) | Fragegat et al. [136] |
| STRING | Multiplex | STRING is a database of well-known protein interactions, including direct (physical) and indirect (functional) associations, aggregated from other (primary) databases. | Protein–protein interaction identification, pathway analysis, network connectivity, functional prioritazation. | https://string-db.org/ (accessed on 19 July 2021) | Snel et al. [137] Szklarczyk et al. [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Sangiao, E.; Leal Rodriguez, C.; García-Riestra, C. Application and Perspectives of MALDI–TOF Mass Spectrometry in Clinical Microbiology Laboratories. Microorganisms 2021, 9, 1539. https://doi.org/10.3390/microorganisms9071539
Torres-Sangiao E, Leal Rodriguez C, García-Riestra C. Application and Perspectives of MALDI–TOF Mass Spectrometry in Clinical Microbiology Laboratories. Microorganisms. 2021; 9(7):1539. https://doi.org/10.3390/microorganisms9071539
Chicago/Turabian StyleTorres-Sangiao, Eva, Cristina Leal Rodriguez, and Carlos García-Riestra. 2021. "Application and Perspectives of MALDI–TOF Mass Spectrometry in Clinical Microbiology Laboratories" Microorganisms 9, no. 7: 1539. https://doi.org/10.3390/microorganisms9071539
APA StyleTorres-Sangiao, E., Leal Rodriguez, C., & García-Riestra, C. (2021). Application and Perspectives of MALDI–TOF Mass Spectrometry in Clinical Microbiology Laboratories. Microorganisms, 9(7), 1539. https://doi.org/10.3390/microorganisms9071539